

Abstract
Objective
Metabolic stress primes monocytes for accelerated chemokine-mediated adhesion, migration and recruitment into vasculature lesions by increasing actin remodeling. The mechanism linking metabolic stress to accelerated actin turnover and enhanced monocyte migration was not known. We tested the hypothesis that in metabolically primed monocytes, the acceleration of MCP-1-induced chemotaxis is mediated by the hyper-activation of cofilin.
Approach and Results
Metabolic priming was induced by exposing human THP-1 monocytes to diabetic conditions, i.e. human native LDL plus high glucose concentrations (LDL+HG). In healthy monocytes, MCP-1 induced the phosphorylation and inactivation of cofilin. This response was completely blocked in metabolically primed monocytes, but restored by overexpression of the thiol transferase, glutaredoxin 1 (Grx1). Cofilin kinase, LIMK1, and cofilin phosphatase, SSH1L, were not affected by metabolic stress. However, metabolic priming increased 3.8-fold the S-glutathionylation of the SSH1L-binding protein 14-3-3zeta, resulting in its caspase-dependent degradation. Grx1 overexpression inhibited LDL+HG-induced S-glutathionylation and degradation of 14-3-3zeta. The C25S mutant of 14-3-3zeta was resistant to both S-glutathionylation and degradation induced by LDL+HG. Overexpression of the C25S mutant restored MCP-1-induced cofilin phosphorylation and prevented accelerated migration of metabolically stressed monocytes, suggesting that loss of 14-3-3zeta increases the pool of free SSH1L phosphatase, thereby preventing the phosphorylation and deactivation of cofilin in response to chemokine activation.
Conclusions
By preventing the inactivation of cofilin, metabolic stress-induced degradation of 14-3-3zeta promotes the conversion of blood monocytes into a hyper-migratory, proatherogenic phenotype.
Keywords: atherosclerosis, cell migration, monocytes, redox signaling
INTRODUCTION
Recruitment of monocyte-derived macrophages into the arterial wall is rate-limiting for the initiation and progression of atherosclerotic lesions 1. Monocyte chemoattractant protein-1 (MCP-1) is the main chemoattractant responsible for the recruitment of monocytes into early atherosclerotic lesions. MCP-1 and other chemokines are synthesized by endothelial cells, smooth muscle cells, and macrophages in response to oxidized lipids 2. Monocyte adhesion and transmigration requires efficient remodeling and reorganization of the actin cytoskeleton. Previously, we reported that metabolic priming, the exposure of monocytes to metabolic stress, significantly increases actin remodeling in response to chemokines, increasing monocyte adhesion and chemotaxis, and accelerating atherosclerotic lesion formation in mice 3. However, the molecular mechanisms underlying accelerated actin remodeling remain unclear.
Actin dynamics are regulated by cofilin, a member of the actin-depolymerizing factor (ADF) protein family 4. Cofilin activity is controlled through the phosphorylation of serine residue 3 (Ser-3), whereby phosphorylation inhibits cofilin’s actin depolymerizing activity, and dephosphorylation results in the reactivation of the protein 5, 6. LIM kinases 1 and 2, which specifically phosphorylate cofilin at Ser-3, have been identified as the enzymes responsible for cofilin inactivation 7, 8. The phosphatases responsible for the dephosphorylation and activation of cofilin belong to the family of Slingshot (SSH) protein phosphatases (SSH in Drosophila and SSH1L, SSH2L, and SSH3L in mammals). SSH phosphatases specifically dephosphorylate Ser-3 of inactive cofilin 9, 10. Unlike LIM kinases, whose activities are regulated by direct post-translational modifications, the activity of SSH is governed by their association with regulatory proteins, such as 14-3-3zeta 11–14. SSH1L is activated upon its release from 14-3-3zeta, involving a poorly defined process that results in the oxidation of 14-3-3zeta 12, 15. We now identified S-glutathionylation-induced degradation of 14-3-3zeta as the critical step in SSH-1L release. Hyper-S-glutathionylation and degradation of 14-3-3zeta induced by metabolic stress is responsible for the SSH1L-mediated hyperactivation of cofilin in metabolically “primed” monocytes and contributes to the conversion of metabolically stressed monocytes into a hyperchemotactic and proatherogenic phenotype.
METHODS
A detailed methods section describing all procedures and protocols is available in the Online Data Supplement.
RESULTS
MCP-1-induced cofilin phosphorylation is inhibited by metabolic stress but restored by overexpressing Grx1
Previously, we reported that chronic exposure to a hypercholesterolemic (LDL) and/or hyperglycemic (HG) environment sensitized THP-1 monocytes to the chemoattractant MCP-1, resulting in increase in monocyte migration through a mechanism that involves protein S-glutathionylation and increased actin remodeling 3. Exposure of monocytes to LDL+HG decreases the F-actin/G-actin ratio in response to MCP-1, indicating enhanced actin turnover in metabolically primed monocytes 3. To identify the mechanism that accelerates actin turnover, we first determined if metabolic stress alters the activation state of cofilin, a central regulator of actin turnover 16. In healthy monocytes, MCP-1 induced a gradual increase in cofilin phosphorylation, indicating that cofilin was being inactivated. In contrast, metabolic priming resulted in decreased cofilin phosphorylation following MCP-1 stimulation and completely suppressed the inactivation of cofilin observed in healthy monocytes, indicating cofilin was being hyper-activated (Fig. 1A, Fig.SI). Total cofilin levels, on the other hand, did not change in response to metabolic stress (Fig. SII).
Figure 1. MCP-1-induced cofilin phosphorylation is inhibited by metabolic stress, but restored by overexpressing Grx1.
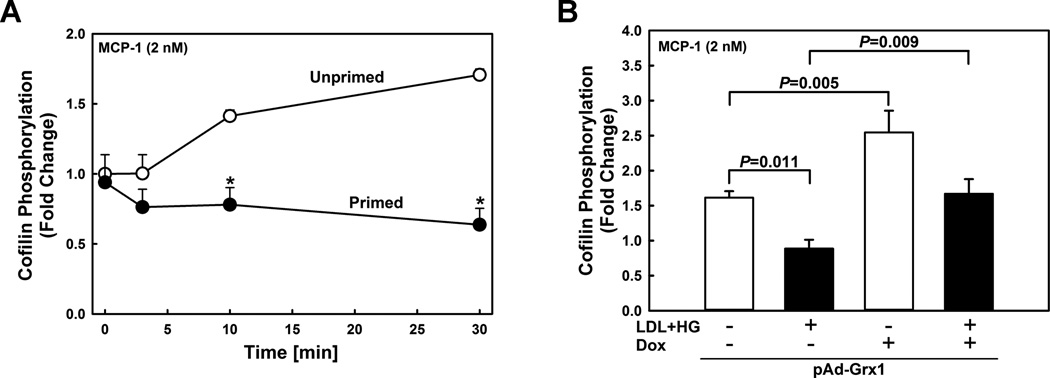
(A) Time course of MCP-1-induced cofilin phosphorylation assessed by Western blot analysis in unprimed (open symbols) and metabolically-primed THP-1 monocytes (closed symbols). *: P < 0.05 versus control (no LDL+HG, no MCP-1, t=0 min). (B) Effect of Grx1 overexpression on MCP-1-induced cofilin phosphorylation in unprimed (open bars) and metabolically-primed THP-1 monocytes (closed bars). THP-1 monocytes were infected with a doxycycline (Dox)-inducible adenoviral vector carrying the sequence for a Grx1-EGFP fusion protein (pAd-Grx1). Grx1 expression was induced by adding doxycycline (Dox; 1 µg/ml). Cofilin phosphorylation was stimulated for 30 min with rMCP-1 (2 nmol/L). Results shown are means ± SE of 4–6 independent experiments.
Overexpression of the thiol transferase Grx1 protects monocytes against metabolic priming and normalizes chemotaxis in response to MCP-1 3. Increasing Grx1 levels also protected monocytes from increased actin turnover (Fig. SIII). However, metabolic stress does not induce any significant change in the Grx1 expression levels 17. Interestingly, compared with wildtype blood monocytes isolated from, Grx1-deficient mice showed no change in their basal 14-3-3zeta expression or level of cofilin phosphorylation (Fig. SIV). It should be noted though that in in contrast to human monocytes, murine cells express Grx2 splice variants that like Grx1 localize to the cytosol 18 and may have compensated for the loss of Grx1 activity in monocytes from Grx1-null mice. Furthermore, there are other enzymes systems, including the thioredoxin family and peroxiredoxins, that are capable of deglutathionylating proteins, albeit less effectively than Grx 19, 20. These findings may explain why Grx1 deficiency alone does not mimic metabolic priming (Fig. SIV), yet overexpression of Grx1 protects monocytes from the effects of metabolic stress.
In the absence of MCP-1 stimulation, Grx1 overexpression had no effect on cofilin phosphorylation in monocytes, irrespective of whether the cells were primed or not (not shown). However, increasing Grx1 expression (Fig. SV) increased MCP-1-induced cofilin phosphorylation by 255% in healthy and by 167% in primed monocytes (Fig. 1B), indicating that cofilin phosphorylation is thiol redox sensitive and regulated by S-glutathionylation-dependent events. Importantly, MCP-1-induced cofilin dephosphorylation detected in metabolically primed monocytes (Fig. 1B, 2nd bar) was significantly blunted by Grx1 overexpression (Fig. 1B, 4th bar). In fact, increasing Grx1 activity in metabolically primed monocytes fully restored the delayed inactivation of cofilin following MCP-1 stimulation observed in healthy monocytes. (Fig. 1B, 1st bar).
Metabolic priming decreases 14-3-3zeta protein levels in monocytes
Cofilin is inactivated by phosphorylation of Ser-3 by LIM-kinase (LIMK) 21 and is reactivated by dephosphorylation of Ser-3 by SSH phosphatases 9. Although other SSH phosphatase isoforms (SSH2L, SSH3L) have also been reported to dephosphorylate and activate cofilin, SSH1L is the major SSH isoform in THP-1 monocytes (Fig. SVI). We therefore explored whether metabolic stress-induced priming altered the protein levels and enzyme activities of the key regulators of cofilin activity. Compared to vehicle-treated cells, metabolically primed THP-1 monocytes showed no change in SSH1L and LIMK1 protein level (Fig. 2A+C, Fig. SVII) or activities (Fig. 2B+D), suggesting an alternative mechanism for the regulation of cofilin phosphorylation.
Figure 2. Metabolic priming does not change protein levels or enzymatic activities of SSH1L and LIMK1 in monocytes.

(A+C) Protein levels of cofilin phosphatase (Slingshot-1L, SSH1L) and cofilin kinase (LIM domain kinase 1, LIMK1) were assessed by Western blot analysis in unprimed (C) and metabolically primed THP-1 monocytes (LDL+HG). (B) Phosphatase activity of SSH1L immunoprecipitates in unprimed and metabolically primed THP-1 monocytes was assayed toward phospho-cofilin peptide. Catalytic activity of SSH1L was assayed spectrophotometrically as the amount of inorganic phosphate released. (D) Unprimed and metabolically primed THP-1 monocytes were lysed and endogenous LIMK1 was immunoprecipitated and subjected to an in vitro kinase reaction, using His6-cofilin as a substrate. Reaction mixtures were subjected to SDS-PAGE and analyzed by Western blot with antiphopho-cofilin and anti-histidine tag antibodies. (n=3-4).
SSH phosphatases bind to regulatory proteins, such as 14-3-3 12–14. Binding to 14-3-3 does not alter SSH1L activity 14 but regulates the subcellular distribution of SSH1L by sequestering SSH1L and preventing translocation of the phosphatase to cofilin associated with the actin cytoskeleton 13. We therefore examined how metabolic stress affects the expression levels of 14-3-3 family members in monocytes. THP-1 monocytes primarily expressed 14-3-3zeta and 14-3-3gamma (Fig. SVIII). Metabolic priming of THP-1 cells decreased 14-3-3zeta protein levels by 35% (Fig. 3A), but 14-3-3gamma protein levels were not altered (Fig. SIX), suggesting that metabolic stress leads to the specific degradation of 14-3-3zeta. Surprisingly, the proteasomal inhibitor MG132 did not prevent metabolic stress-induced 14-3-3zeta degradation. However, when THP-1 monocytes were exposed to metabolic stress in the presence of the pan-caspase inhibitor Z-VAD-FMK, the decrease of 14-3-3zeta was completely blocked (Fig. SX).
Figure 3. Metabolic priming decreases 14-3-3zeta protein levels but increases S-glutathionylation of 14-3-3zeta in monocytes.
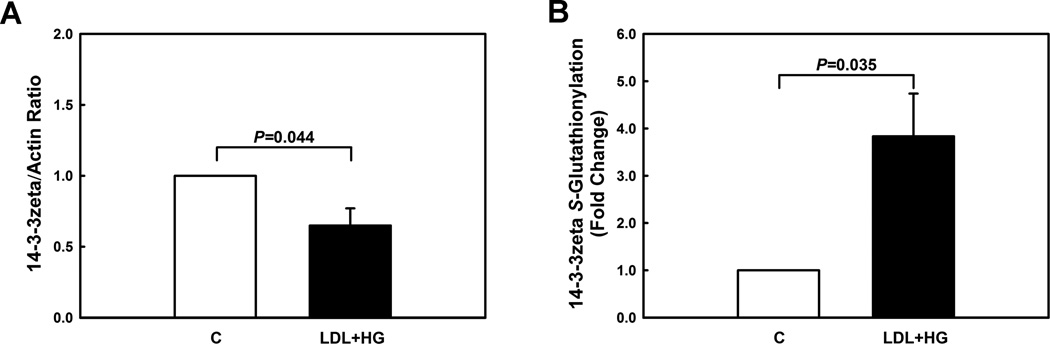
(A) Protein levels of 14-3-3zeta were assessed by Western blot analysis in unprimed (C) and metabolically primed THP-1 monocytes (LDL+HG). (B) THP-1 monocytes were preincubated with BIOGEE (250 µ M) and subsequently primed for 24 h with either vehicle (open bar) or LDL+HG (black bar). After precipitation of Biotin-labeled proteins with streptavidin agarose, 14-3-3zeta S-glutathionylation was assessed by Western blot analysis using an antibody recognizing 14-3-3zeta. (n=3–5)
Metabolic Stress Promotes S-Glutathionylation and Subsequent Degradation of 14-3-3zeta
We showed that metabolic stress in monocytes promotes protein S-glutathionylation, i.e., the formation of mixed disulfides between GSH and reactive protein thiols 3, 22. Furthermore, previous studies by Lind and colleagues showed that 14-3-3zeta is S-glutathionylated in diamide-treated ECV304 endothelial-like cells 23. To explore if 14-3-3zeta is a target of metabolic stress-induced S-glutathionylation in primed monocytes, we performed streptavidin-bead pulldown experiments in THP-1 monocytes preloaded with biotin-labeled glutathione. Compared to healthy THP-1 monocytes, levels of biotin-labeled 14-3-3zeta were increased 3.8-fold in primed monocytes (Fig. 3B, Fig. SXI), confirming that 14-3-3zeta is S-glutathionylated in response to metabolic stress. RNAi-mediated knockdown of 14-3-3zeta protein levels by 60 % (Fig. 4A, Fig. SXII) suppressed MCP-1-induced cofilin phosphorylation (Fig. 4B, Fig. SXIII) and accelerated 2-fold the chemotaxis of THP-1 monocytes in response to MCP-1 (Fig. 4C). Finally, blocking 14-3-3zeta S-glutathionylation in THP-1 monocytes by overexpressing Grx1 prevented the loss of 14-3-3zeta induced by metabolic priming (Fig. 5A, Fig. SXIV). These results, suggest that 1) metabolic stress promotes the S-glutathionylation of 14-3-3zeta, 2) this posttranslational modification results in the caspase-dependent degradation of the protein, and 3) that decreasing 14-3-3zeta protein levels alone is sufficient to recapitulate the effects of metabolic stress on monocyte chemotaxis.
Figure 4. Knockdown of 14-3-3zeta suppresses MCP-1-induced cofilin phosphorylation and accelerates chemotaxis in monocytes.

To assess the effects of 14-3-3zeta deficiency on monocytes, THP-1 monocytes were transfected for 72 h with either non-targeting siRNA (siCont) or two different siRNAs directed against 14-3-3zeta (si#1 and si#2). 14-3-3zeta protein levels (A) and MCP-1 (2 nmol/L)-induced cofilin phosphorylation (B) and chemotaxis (C) were assessed. (n=3-4)
Figure 5. 14-3-3zeta S-glutathionylation on cysteine residue 25 by metabolic stress promotes its degradation.

(A) THP-1 monocytes were infected with a doxycycline (Dox)-inducible adenoviral vector carrying the sequence for a Grx1-EGFP fusion protein (pAd-Grx1), and Grx1 expression was induced by treating cells for 24 h with 1 µ g/ml doxycycline (Dox). THP-1 monocytes were then treated for 24 h with vehicle or primed with LDL+HG, and 14-3-3zeta levels were measured by Western blot analysis (n=3). (B–D) THP-1 monocytes were infected with lentiviral vectors carrying either wild type Flag-tagged 14-3-3zeta (WT) or Flag-tagged 14-3-3zeta in which Cys25 was mutated to a serine residue (C25S). Cells were then treated for 24 h with vehicle or primed with LDL+HG, and 14-3-3zeta protein levels (B), cofilin phosphorylation (C), and chemotactic responses to MCP-1 were assessed. (n=3-4)
Cysteine residue 25 (Cys25) of 14-3-3zeta was previously reported to be S-nitrosylated 24. To test if Cys25 is S-glutathionylated in metabolically primed monocytes, we mutated Cys25 to a serine residue. In contrast to wild type (WT) 14-3-3zeta, the C25S mutant (C25S) of 14-3-3zeta was completely resistant to S-glutathionylation (Fig. SXV), demonstrating that Cys25 is the only cysteine residue in 14-3-3zeta that is S-glutathionylated in response to metabolic stress. Next, we used a lentivirus-based transduction system to overexpress Flag-tagged 14-3-3zeta (WT or C25S mutant) in THP-1 monocytes. In contrast to WT 14-3-3zeta, the C25S mutant of 14-3-3zeta was completely resistant to degradation by metabolic priming (Fig. 5B, Fig. SXVI), supporting the concept that metabolic stress promotes S-glutathionylation of 14-3-3zeta, targeting this regulatory protein for degradation.
Pull-down experiments with anti-Flag antibodies in metabolically primed THP-1 monocytes overexpressing either WT or C25S mutant 14-3-3zeta revealed that S-glutathionylation decreased 14-3-3zeta protein levels but did not change the SSH1L/14-3-3zeta ratio, suggesting that the ability of 14-3-3zeta to bind SSH1L was not affected by metabolic priming (Fig. SXVII). Nevertheless, these results also suggest that 14-3-3zeta levels may be the limiting factor in determining the availability of free SSH1L, and that any reduction in 14-3-3zeta protein induced by metabolic stress increases SSH1L available for cofilin (re)activation. This hypothesis is supported by the finding that THP-1 monocytes expressing the S-glutathionylation-resistant C25S mutant 14-3-3zeta were completely protected from the effects of metabolic priming. Although primed, in response to MCP-1, these cells showed the same delayed phosphorylation and inactivation profile for cofilin observed in healthy monocytes (Fig. 5C) and their chemotactic responses were fully normalized (Fig. 5D). Together these findings support a critical role for S-glutathionylation in regulating 14-3-3zeta expression levels and thus its capacity to sequester SSH1L. Our findings provide evidence for a novel redox sensitive mechanism regulating cofilin activity and monocyte chemotaxis.
To examine whether this mechanism operates in vivo and 14-3-3zeta levels are diminished in monocytes of mice suffering from metabolic disorders, we isolated and purified blood monocytes from LDL-R−/− mice fed either a low-fat diet (LFD) or a high-fat diet (HFD). Feeding LDL-R−/− mice a HFD for 11 weeks increases total plasma cholesterol level 3-fold and plasma triglyceride levels 1.5-fold compared to LDL-R−/− mice fed a low fat diet (LFD) 25. As predicted, 14-3-3zeta protein levels were reduced by 44% in monocytes isolated from dyslipidemic LDL-R−/− mice compared to monocytes from normolipidemic LDL-R−/− mice (Fig. 6A, Fig. SXVIII). Furthermore, cofilin phosphorylation induced by MCP-1 was completely suppressed in monocytes from HFD-mice (Fig. 6B, Fig. SXIX), suggesting that with the loss of 14-3-3zeta, excessive SSH1L was released, preventing the inactivation of cofilin and thus the termination of MCP-1-induced actin remodeling. Importantly, in advanced atherosclerotic lesions of the aortic root of HFD-fed LDLR−/− mice, 14-3-3zeta and phospho-cofilin localized to macrophage-rich regions of the plaque. Staining for both proteins appeared to be less intense in the early aortic root lesion of LFD-fed LDLR−/− mice, but this was likely due to equally less intense macrophage staining (Fig. SXX). Our findings are in agreement with those reported by Umahara et al., demonstrating that 14-3-3zeta localizes to macrophage in the human carotid atherosclerotic lesion and it is the only isoform located in the nuclei of macrophages, in addition to the cytosol 26.
Figure 6. 14-3-3zeta protein levels are reduced and cofilin is hyperactivated in blood monocytes from dyslipidemic atherosclerosis-prone mice.

Blood monocytes were isolated and purified from LDL receptor-deficient mice that were fed for 10 weeks either a low fat diet (LFD) or a high fat diet (HFD). (A) 14-3-3zeta levels were determined by Western blot analysis (n=5). (B) MCP-1-induced cofilin phosphorylation was assessed by Western blot analysis (n=4). (C) Hypothetical model for the hyper-activation of cofilin in metabolically primed monocytes.
DISCUSSION
Cofilin severs filaments and promotes the dissociation of subunits from filament pointed ends, accelerating actin disassembly 27, 28. Severing actin filaments increases the number of free barbed ends of filaments, and, if these ends remain uncapped and actin monomers are available, they undergo rapid growth 29, 30. Metabolic stress accelerates monocyte migration in response to MCP-1 through increased actin remodeling 3, but the mechanisms underlying increased actin turnover were not clear. In the present study, we show that thiol oxidative stress induced by metabolic stress dramatically alters the temporal activity/phosphorylation profile of cofilin observed in response to MCP-1 stimulation, switching from a process characterized by the delayed onset of cofilin phosphorylation and inactivation in healthy monocytes to a state of sustained hyperactivation of cofilin in metabolically primed monocytes.
Two kinase families have been shown to phosphorylate and deactivate cofilin: the LIM Lin-11/Isl-1/Mec-3 (LIM) kinases and the testicular protein (TES) kinases. The LIM kinases, LIMK1 and LIMK2, are ubiquitous in their tissue distribution 31, 32, whereas TESK1 is expressed most abundantly in testicular tissue 33, suggesting that in monocytes LIMK1 and/or LIMK2 are the likely kinases involved in cofilin inactivation. Although earlier studies implicated the involvement of phosphatases with broad substrate specificities, such as PP1, PP2A and PP2B, in cofilin activation, selective inhibitors of these phosphatases largely fail to block cofilin dephosphorylation 34. In mammalian cells, SSH1L, along with SSH2L and SSH3L, dephosphorylates both phospho-ADF and phospho-cofilin at the critical Ser3 residue, thereby suppressing actin filament assembly induced by LIMK1 or TESK1 9. Notably, SSH3L was less effective in dephosphorylating these substrates in comparison with the two other isoforms 10. In agreement with these reports, our data suggest SSH1L is the major cofilin phosphatase in THP-1 monocytes.
Surprisingly, in primed monocyte we found no changes in either the protein level or the activity of the two enzymes that control the phosphorylation and activation state of cofilin, cofilin kinase, LIMK1, and phospho-cofilin phosphatase, SSH1L. We were also unable to demonstrate any changes in the levels of either cofilin S-glutathionylation or cofilin protein levels in response to metabolic stress, suggesting that the redox-sensitive step in the regulation of cofilin activity does not appear to involve cofilin itself. Our findings suggest an alternative, thiol redox-sensitive mechanism for the regulation of cofilin activity and actin turnover in monocytes that appears to contribute to the metabolic stress-induced conversion of monocytes into a hyper-migratory, proatherogenic phenotype associated with metabolic disorders (Figure 6C).
To facilitate actin reorganization at the leading edge, SSH is released from 14-3-3, and translocates to the actin filaments 13, 35. Recent evidence indicates that 14-3-3 proteins associate with cofilin and SSH1L, thereby preventing cofilin dephosphorylation and translocation of SSH to F-actin-rich cortical regions 13, 14, 36. 14-3-3 proteins may therefore play a role in the formation of localized regulatory complexes composed of cofilin, LIMK and F-actin as well as SSH 14, 36, 37. Four isoforms of 14-3-3 proteins, 14-3-3gamma, tau, zeta, and beta, have been shown to interact with SSH1L and form inhibitory complexes by sequestering the phosphocofilin phosphatase in the cytosolic fraction 13. Of the four isoforms interacting with SSH1L, only 14-3-3gamma and zeta are expressed in the monocyte/macrophage linage 38–40, including THP-1 monocytes.
We propose that in monocytes, protein levels of 14-3-3zeta, and thus its ability to sequester SSH1L, is controlled by S-glutathionylation at cysteine residue 25, which targets the protein for caspase-dependent rather than proteasomal degradation (Fig. 6C). 14-3-3zeta S-glutathionylation has been reported in ECV304 endothelial-like cells 23, but the functional relevance of this posttranslational modification was not known. The novel mechanism we propose is supported by our findings that overexpression of Grx1 protects metabolically primed monocytes against hyper-responsiveness to MCP-1 3 and that overexpression of an S-glutathionylation-resistant C25S mutant of 14-3-3zeta both mimicked the protective effects of Grx1 overexpression on monocyte priming and restored the cofilin activation profile.
In summary, our data support a central role for the S-glutathionylation of 14-3-3zeta in the regulation of monocyte chemotaxis. Metabolic stress-induced disruption of monocyte redox homeostasis promotes the degradation of 14-3-3zeta and the conversion of blood monocytes into a hyper-migratory, proatherogenic phenotype. The fact that 14-3-3zeta levels are significantly decreased in monocytes of hyperlipidemic, atherosclerosis-prone mice raises the possibility that 14-3-3zeta deficiency in monocytes may be a biomarker for monocyte dysfunction associated with metabolic disorders and a risk factor for atherosclerosis.
Supplementary Material
SIGNIFICANCE.
Recruitment of monocytes into the arterial wall is a rate-limiting process in the initiation and progression of atherosclerotic lesions which requires the controlled remodeling of the actin cytoskeleton. Actin turnover and monocyte (trans)migration are accelerated in metabolically stressed monocytes, but the underlying molecular mechanisms were unclear. Here we demonstrate that exposure of monocytes to metabolic stress prevents cofilin inactivation in response to MCP-1. This hyper-activation of cofilin is triggered by S-glutathionylation and subsequent caspase-dependent, but proteasome-independent degradation of 14-3-3zeta, resulting in the release of the phospho-cofilin phosphatase, SSH1L, sequestered by 14-3-3zeta. Overexpression of an S-glutathionylation and degradation-resistant (C25S) mutant of 14-3-3zeta prevents the hyper-activation of cofilin and protects monocytes against metabolic stress-induced hyper-reactivity to MCP-1. Our data identified a novel redox-sensitive mechanism for the regulation of actin turnover and cell migration, and suggest that decreased 14-3-3zeta levels in monocyte may be a biomarker for monocyte dysfunction associated with metabolic disorders and a risk factor for atherosclerosis.
ACKNOWLEDGEMENTS
We would like to thank Dr. In Kyoung Lim (Ajou University) for providing the 6-His tagged wild-type mouse cofilin clone. We are grateful to Leigh Ann Piefer for the careful review and editing of the manuscript.
SOURCES OF FUNDING
This work was supported a grant to R.A. from the NIH (RO1 HL115858).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- Ser-3
serine residue 3
- SSH
Slingshot phosphatase
- SSH1L
Slingshot-1L phosphatase
- P-cofilin
phosphorylated cofilin
- LFD
low-fat diet
- HFD
high-fat diet
- Grx1
glutaredoxin1
- EGFP
enhanced green fluorescent protein
- Dox
doxycycline
- LDL+HG
100 µg/ml of LDL plus 25 mmol/L glucose
- LIMK
LIM-kinase
- MCP-1
Monocyte chemoattractant protein-1
Footnotes
DISCLOSURES
There are no conflicts to disclose.
REFERENCES
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 3.Ullevig S, Zhao Q, Lee CF, Seok KH, Zamora D, Asmis R. NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler Thromb Vasc Biol. 2012;32:415–426. doi: 10.1161/ATVBAHA.111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20:187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- 6.Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- 7.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 8.Amano T, Tanabe K, Eto T, Narumiya S, Mizuno K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem J. 2001;354:149–159. doi: 10.1042/0264-6021:3540149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 10.Ohta Y, Kousaka K, Nagata-Ohashi K, Ohashi K, Muramoto A, Shima Y, Niwa R, Uemura T, Mizuno K. Differential activities, subcellular distribution and tissue expression patterns of three members of Slingshot family phosphatases that dephosphorylate cofilin. Genes Cells. 2003;8:811–824. doi: 10.1046/j.1365-2443.2003.00678.x. [DOI] [PubMed] [Google Scholar]
- 11.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell. 2009;20:2650–2660. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagata-Ohashi K, Ohta Y, Goto K, Chiba S, Mori R, Nishita M, Ohashi K, Kousaka K, Iwamatsu A, Niwa R, Uemura T, Mizuno K. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J Cell Biol. 2004;165:465–471. doi: 10.1083/jcb.200401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B, Sampath R, Bamburg JR, Bernard O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 2005;24:473–486. doi: 10.1038/sj.emboj.7600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maheswaranathan M, Gole HK, Fernandez I, Lassegue B, Griendling KK, San MA. Platelet-derived growth factor (PDGF) regulates Slingshot phosphatase activity via Nox1-dependent auto-dephosphorylation of serine 834 in vascular smooth muscle cells. J Biol Chem. 2011;286:35430–35437. doi: 10.1074/jbc.M111.268284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okada K, Ravi H, Smith EM, Goode BL. Aip1 and cofilin promote rapid turnover of yeast actin patches and cables: a coordinated mechanism for severing and capping filaments. Mol Biol Cell. 2006;17:2855–2868. doi: 10.1091/mbc.E06-02-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ullevig SL, Kim HS, Nguyen HN, Hambright WS, Robles AJ, Tavakoli S, Asmis R. Ursolic acid protects monocytes against metabolic stress-induced priming and dysfunction by preventing the induction of Nox4. Redox Biol. 2014;2:259–266. doi: 10.1016/j.redox.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hudemann C, Lonn ME, Godoy JR, Zahedi AF, Capani F, Holmgren A, Lillig CH. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid Redox Signal. 2009;11:1–14. doi: 10.1089/ars.2008.2068. [DOI] [PubMed] [Google Scholar]
- 19.Jung CH, Thomas JA. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch Biochem Biophys. 1996;335:61–72. doi: 10.1006/abbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 20.Nulton-Persson AC, Starke DW, Mieyal JJ, Szweda LI. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry. 2003;42:4235–4242. doi: 10.1021/bi027370f. [DOI] [PubMed] [Google Scholar]
- 21.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 22.Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci U S A. 2012;109:E2803–E2812. doi: 10.1073/pnas.1212596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 24.Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao M, Zhao Q, Lee CF, Tannock LR, Smart EJ, LeBaron RG, Phelix CF, Rangel Y, Asmis R. Thiol oxidative stress induced by metabolic disorders amplifies macrophage chemotactic responses and accelerates atherogenesis and kidney injury in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2009;29:1779–1786. doi: 10.1161/ATVBAHA.109.191759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umahara T, Uchihara T, Koyama S, Hashimoto T, Akimoto J, Haraoka J, Iwamoto T. Isoform-specific immunolocalization of 14-3-3 proteins in atherosclerotic lesions of human carotid and main cerebral arteries. J Neurol Sci. 2012;317:106–111. doi: 10.1016/j.jns.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- 29.DesMarais V, Ghosh M, Eddy R, Condeelis J. Cofilin takes the lead. J Cell Sci. 2005;118:19–26. doi: 10.1242/jcs.01631. [DOI] [PubMed] [Google Scholar]
- 30.Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. doi: 10.1083/jcb.148.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizuno K, Okano I, Ohashi K, Nunoue K, Kuma K, Miyata T, Nakamura T. Identification of a human cDNA encoding a novel protein kinase with two repeats of the LIM/double zinc finger motif. Oncogene. 1994;9:1605–1612. [PubMed] [Google Scholar]
- 32.Ikebe C, Ohashi K, Fujimori T, Bernard O, Noda T, Robertson EJ, Mizuno K. Mouse LIM-kinase 2 gene: cDNA cloning, genomic organization, and tissue-specific expression of two alternatively initiated transcripts. Genomics. 1997;46:504–508. doi: 10.1006/geno.1997.5060. [DOI] [PubMed] [Google Scholar]
- 33.Toshima J, Ohashi K, Okano I, Nunoue K, Kishioka M, Kuma K, Miyata T, Hirai M, Baba T, Mizuno K. Identification and characterization of a novel protein kinase, TESK1, specifically expressed in testicular germ cells. J Biol Chem. 1995;270:31331–31337. doi: 10.1074/jbc.270.52.31331. [DOI] [PubMed] [Google Scholar]
- 34.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 35.Nishita M, Wang Y, Tomizawa C, Suzuki A, Niwa R, Uemura T, Mizuno K. Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase Slingshot and its role for insulin-induced membrane protrusion. J Biol Chem. 2004;279:7193–7198. doi: 10.1074/jbc.M312591200. [DOI] [PubMed] [Google Scholar]
- 36.Gohla A, Bokoch GM. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr Biol. 2002;12:1704–1710. doi: 10.1016/s0960-9822(02)01184-3. [DOI] [PubMed] [Google Scholar]
- 37.Birkenfeld J, Betz H, Roth D. Identification of cofilin and LIM-domain-containing protein kinase 1 as novel interaction partners of 14-3-3 zeta. Biochem J. 2003;369:45–54. doi: 10.1042/BJ20021152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q, Fong CC, Zhang Y, Tzang CH, Fong WF, Yang M. cDNA microarray analysis of the differentially expressed genes involved in murine pre-osteoclast RAW264.7 cells proliferation stimulated by dexamethasone. Life Sci. 2008;82:135–148. doi: 10.1016/j.lfs.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi R, Deavers M, Patenia R, Rice-Stitt T, Halbe J, Gallardo S, Freedman RS. 14-3-3 zeta protein secreted by tumor associated monocytes/macrophages from ascites of epithelial ovarian cancer patients. Cancer Immunol Immunother. 2009;58:247–258. doi: 10.1007/s00262-008-0549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barry EF, Felquer FA, Powell JA, Biggs L, Stomski FC, Urbani A, Ramshaw H, Hoffmann P, Wilce MC, Grimbaldeston MA, Lopez AF, Guthridge MA. 14-3-3:Shc scaffolds integrate phosphoserine and phosphotyrosine signaling to regulate phosphatidylinositol 3-kinase activation and cell survival. J Biol Chem. 2009;284:12080–12090. doi: 10.1074/jbc.M807637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.