

Abstract
Infection with hepatitis B virus (HBV) leads to a wide spectrum of clinical presentations ranging from an asymptomatic carrier state to self-limited acute or fulminant hepatitis to chronic hepatitis with progression to cirrhosis and hepatocellular carcinoma. Infection with HBV is one of the most common viral diseases affecting man. Both viral factors as well as the host immune response have been implicated in the pathogenesis and clinical outcome of HBV infection. In this review, we will discuss the impact of virus-host interactions for the pathogenesis of HBV infection and liver disease. These interactions include the relevance of naturally occurring viral variants for clinical disease, the role of virus-induced apoptosis for HBV-induced liver cell injury and the impact of antiviral immune responses for outcome of infection.
Keywords: Host response, Viral hepatitis, Mutants, Pathogenesis, Resistance
MOLECULAR VIROLOGY OF HEPATITIS B VIRUS INFECTION
Hepatitis B virus (HBV) is a small DNA virus and belongs to a group of hepatotropic DNA viruses (hepadnaviruses)[1,2]. The virus consists of a nucleocapsid and an outer envelope composed mainly of three hepatitis B surface antigens (HBsAgs) that play a central role in the diagnosis of HBV infection. The nucleocapsid contains hepatitis B core antigen (HBcAg), a DNA polymerase-reverse transcriptase, the viral genome as well as cellular proteins[1,2].
The genome consists of a partially double-stranded circular DNA molecule of about 3200 base pairs in length with known sequence as well as genetic organization. The pre-surface 1 [pre-S1]/pre-surface 2 [pre-S2]/and surface genes [S] code for the various HBsAgs. The protein encoded by the pre-core [pre-C]/core gene [C] undergoes post-translational modification to yield hepatitis B e antigen (HBeAg), which is a seromarker for high viral replication[2]. The core gene codes for HBcAg, the major structural protein of the nucleocapsid. Finally, the X gene codes for the hepatitis B x antigen (HBxAg). HBxAg has been shown to be a potent transactivator of cellular and viral genes. A variety of interactions with cellular proteins has been proposed as potential targets of HBxAg. The precise functions of HBxAg, however, in the viral life cycle and the natural course of HBV infection remain to be established. In addition to the known viral genes, several cis- and trans-acting genetic elements involved in the fine control of gene expression, RNA packaging and viral replication have been identified[1,2].
The viral DNA polymerase-reverse transcriptase is encoded by the polymerase gene [P] and is of central importance for viral replication. Different from all known mammalian DNA viruses, hepadnaviruses replicate via reverse transcription of a RNA intermediate[3,4], the pregenomic RNA, which is a strategy central to the life cycle of RNA retroviruses. Similarities and differences between retroviral and hepadnaviral replication have been defined[1]. Based on the unique replication cycle of HBV, antiviral therapeutic strategies aimed at the reverse transcription of HBV RNA or at HBV reverse transcriptase have been successfully used as antivirals to treat HBV infection[5-13].
VIRAL VARIANTS AND PATHOGENESIS OF INFECTION
Evidence has been accumulating that certain HBV mutants are associated with unique clinical manifestations, may affect the natural course of the infection and confer resistance to antiviral agents (Table 1)[14-17]. Naturally occurring mutations in the context of various genotypes have been identified in the structural and non-structural genes as well as regulatory elements of the virus. The best characterized mutants are the pre-core (pre-C) stop codon mutations resulting in a loss of hepatitis B e antigen[18], defined clusters of mutations in the core promoter resulting in enhanced viral replication[19-21], and mutations in the reverse transcriptase/polymerase genes conferring resistance to antivirals[16,22]. Furthermore, several mutations in the HBV surface gene have been identified which alter the antigenicity of the viral surface proteins (HBsAg) and structure of the viral envelope[15,23].
Table 1.
HBV variants and their potential impact for pathogenesis of HBV infection
| HBV region | Mutation | Molecular phenotype | Clinical relevance |
| Pre-S/S | Pre-S1/ pre-S2/ | Misassembly | Fibrosing cholestatic |
| S-promotor | hepatitis | ||
| S | Alteration of B- and | Vaccine escape | |
| S splicing | T-cell epitopes | Immune escape Diagnostic escape | |
| Pre-C | Pre-C-stop | Loss of HBeAg | Severe hepatitis HBeAg-deficiency |
| Core | Core | Alteration of T-cell epitopes | Viral persistence Severe hepatitis |
| RT/Pol | Pol | Replication deficiency | Viral latency Viral latency Pol |
| Resistance to antivirals | Therapy escape | ||
| Regulatory Core promotor Elements | Enhanced replication and core expression Decreased HBeAg synthesis | Severe hepatitis Modulation of drug resistance HBeAg seronegativity | |
| Enhancer I | Decreased replication | Chronic hepatitis |
HBV genomic region, mutation, molecular phenotype and clinical relevance are indicated.
HBeAg variants and HBeAg-seronegativity
One of the first HBV mutations clinically recognized and functionally characterized was the pre-C stop codon mutation, resulting in a loss of HBeAg. Not all patients with chronic hepatitis B become HBV DNA negative, despite seroconversion from HBeAg to anti-HBe. Numerous studies have shown that these patients are infected with a pre-C/C mutant[24,25]. This mutant has a translation stop codon at the end of the pre-C gene. Thus, the pre-C/C fusion protein, a precursor of HBeAg, cannot be synthesized. In these patients, therefore, viral replication may persist despite elimination of HBeAg and seroconversion to anti-HBe. While the loss of HBeAg appears irrelevant for the biology of the virus, it may play an important role in the interaction of the virus with the immune system. Secreted HBeAg has been proposed to have an immunoregulatory function in utero by establishing T-cell tolerance to HBeAg and HBcAg that may predispose neonates born to HBV-infected mothers to develop persistent HBV infection[26]. Recent studies have further demonstrated an immunomodulatory role of HBeAg in antigen presentation and recognition by CD4+ T-cells[27]. The selection of HBeAg mutants in the host may be due in part to immunomodulatory properties of HBeAg resulting in a survival advantage for the virus[28]. Whether and how this mutation - either alone or in combination with other mutations - affects the clinical course of HBV infection is still unclear.
Of interest in this respect is the observation that pre-C stop codon mutants are found not only in patients with fulminant hepatitis[18,29-32] or chronic active hepatitis B[24,25,33-35], but also in asymptomatic HBV carriers[32] or acute, self-limited hepatitis [36]. In the woodchuck model, the pre-C stop codon mutation was found to exert no effect on viral replication or the severity of liver disease. Infections with the pre-C stop codon mutant, however, did not take a chronic course[37]. Interestingly, in the duck hepatitis B virus model the pre-C stop codon mutant replicates less well and is overgrown by wild-type virus during the natural course of coinfection[38].
Core promoter variants and enhanced viral replication
During the last couple of years mutations have been identified in regulatory genetic elements of the HBV genome. Several independent studies have identified and functionally characterized distinct mutations clustered in enhancer II of the HBV core promotor. Core promoter mutations are predominantly found in patients with a more aggressive course of disease such as fulminant[19,39-41] or chronic hepatitis B[21,33,42-45]. Some of the patients have a decrease or loss of HBeAg[39,43].
A common hallmark of core promoter mutations is the biological phenotype of enhanced viral replication in transfected hepatoma cell lines[19,21,33,39-44] and primary hepatocytes[20]. The most prevalent mutant comprises a double mutation (A to T at nucleotide 1764 and G to A at nucleotide 1766, nucleotide numbering according to[46] located at the 3`end of enhancer II of the basal core promotor being present in up to 80% of individuals chronically infected with HBV[47].
Several other core promotor mutations in immuncom-promized patients and severe or fulminant liver disease have been identified[41,42,45]. A common phenotype of these mutations seems to be the enhanced viral encapsidation by altering the balance between pre-C and C RNA transcript levels[42]. Several of these mutations have been shown to create additional transcription factor (HNF-1, HNF-3, HNF-4 or C/EBP) binding sites[41,42,48]. The magnitude of mutant-induced enhancement of viral replication seems to be dependent on the HBV subtype/genotype[40,49].
The phenotype of enhanced viral replication may be the reason why core promoter mutants seem to be selected in immunosuppressed patients or patients with chronic hepatitis. Interestingly, one study suggested the presence of core promotor mutations was significantly associated with the development of hepatocellular carcinoma (HCC)[50]. Furthermore, the high-replication phenotype of viral strains containing core promotor mutations may play a role in the pathogenesis of more aggressive or severe disease associated with these mutations. Interestingly, the transmission of a high-replication strain containing two core promotor mutations and the pre-C mutation to HBV-naive patients has resulted in an outbreak of fulminant fatal hepatitis[19]. Enhanced viral replication with the concomitant increase in viral protein expression may result in a differential immune response as well as a more rapid and widespread infection in naive patients[19,20,40]. Altered viral kinetics accompanied by a more vigorous cellular immune response may be important mechanisms resulting in more severe liver injury and potentially fulminant hepatitis. Another factor contributing to fulminant hepatic failure associated with defined core promoter mutations may be hepatocyte apoptosis induced by the HBV variant[20]. Since not all patients with fulminant hepatitis are infected with HBV strains exhibiting a high-replication phenotype[51] additional mechanisms for the pathogenesis of fulminant hepatitis likely exist[52].
Variants of HBsAg and immune escape
A variety of mutations have been identified in the HBV structural genes resulting in differential antigen recognition and immune response[53]. During the course of a passive-active HBV immunization program in southern Italy, several children were infected with HBV despite a primary response to the HBsAg vaccine. Molecular analyses showed that one of these children was infected with a HBV mutant[54,55]. This mutant exhibited a defect in the S region of the HBV genome (glycine (Gly) to arginine (Arg) at amino acid position 145) with loss of the group-specific antigenic determinant a, which is the main target of the vaccine response. Further biological characterization of this mutant in the context of replication-competent viral genomes revealed that this mutation also results in impaired S secretion and decreased virion stability[56]. This viral mutant was able to escape the immune surveillance and thereby resulted in an infection despite the presence of anti-HBs antibodies (‘vaccine escape mutant’). Similar mutants have been detected in Japan[57] and the Gambia[58] and presumably occur world-wide[53]. Consistent with these initial observations, a recent study demonstrated the accumulation of HBsAg a determinant mutants during the implementation of universal vaccination programs in Taiwan[59]. In contrast to these epidemiological observations, a study in the chimpanzee model using currently available US-licensed HBV vaccines demonstrated protection against the ‘classical’ vaccine escape (Gly-145-Arg) mutant in vivo. This study provides strong evidence that immunization with recombinant HBV vaccines stimulates anti-HBs that is broadly reactive and protects the host efficiently from infection with HBV strains containing the Gly-145-Arg mutant[60]. Since this study only evaluated the protective properties of licensed HBV vaccines against the Gly-145-Arg mutant, it does not exclude the possibility that other genuine vaccine escape mutants result in immunization failure[60]. Further studies are needed to address this issue. In addition to immune escape on the B cell level, vaccine escape mediated by defined HBsAg epitopes can occur on the T cell level[61]. Taken together, these findings indicate, that careful epidemiologic monitoring of vaccine failure caused by infection with HBV mutants may be crucial for the success of global immunization strategies.
‘Immune escape mutants’ have also been reported in patients after liver transplantation for HBV-related chronic liver disease who had received monoclonal or polyclonal anti-HBs antibodies to prevent reinfection of the graft[62-65]. ‘Immune escape mutants’ have also been identified in anti-HBs positive individuals[57,66].
In addition to ‘vaccine escape mutants’ and ‘immune escape mutants’, ‘diagnosis escape mutants’ have been described, since most of the HBsAg detection assays are based on anti-HBs antibodies[57,67]. Diagnostic escape may also be the result of a posttranscriptional effect of viral mutations on HBsAg expression as described by Hass et al in an elegant study isolating a naturally occurring mutation targeting splicing of subgenomic RNA[68,69]. The emergence of these variants may potentially contribute to occult HBsAg-negative HBV infection[70].
Mutations have further been detected in the pre-S1 or pre-S2 regions of the HBV genome. Their clinical significance is as yet unknown, however. Since the preS1 region appears to be important for the very first steps of viral infection[71,72], it is conceivable that variants affecting this region may also affect the biological phenotype of the virus by altering its ability to bind or infect hepatocytes. A pre-S2 defective HBV variant has been associated with fulminant hepatitis B[73]. A cluster of mutations in the S promotor (two deletions and a point mutation in the regulatory element CCAAT) isolated from a patient with fibrosing cholestatic hepatitis after HBV reinfection of the transplanted liver has been shown to result in virus retention and misassembly[74,75]. Furthermore, a recent study demonstrates evidence that patients with progressive liver disease have a higher frequency of pre-S deletion[76]. HBsAg variants resulting in a defect or impairment in virion secretion have also been described in severe and fulminant hepatitis B[56,77].
Mutations conferring resistance to antiviral therap
In recent years polymerase gene mutants have been identified in patients treated with antiviral drugs, resulting in drug resistance, respectively[78]. Lamivudine, a nucleoside analogue, is a potent inhibitor of HBV replication and is clinically used as an antiviral for the treatment of chronic hepatitis B and advanced HBV-induced liver disease[11,12,79,80]. Adefovir is an alternative nucleotide analogue previously licensed for the treatment of chronic hepatitis B[9,10]. Other nucleoside analogues currently under clinical investigations include entecavir, emtricitabine, clevudine and telbivudine[16,78]. Several of these drugs act as chain-terminators during the synthesis of the nascent DNA strand, thereby terminating viral replication[16]. Other nucleoside or nucleotide analogues such as adefovir and entecavir interfere with priming and minus-strand DNA elongation[16]. Adefovir and-to a lesser extent-lamivudine also target initial plus-strand DNA repair[81].
The selection of drug resistant mutants depends on several factors. As the viral polymerase is subjected to a spontaneous error rate, viral mutants accumulate during the natural course of the disease. When an antiviral pressure is applied, the mutations exhibiting the best replication capacity in the presence of the drug are selected. The mutant spread depends on its level of intrinsic resistance and on its replicative fitness[16]. This may explain in part why the peaks of drug resistance observed with lamivudine (23% at year one and up to 65% at year five of treatment[82,83]) and with adefovir dipivoxil (2% at two years and 3.9% at three years of treatment[84,85]) are different.
These mutant viral genomes are characterized by selective amino acid changes in various domains of the HBV reverse transcriptase/polymerase. In particular, lamivudine resistance has been extensively studied. The reverse transcriptase polymerase of HBV and HIV share a common and highly conserved tyrosine, methionine, aspartate, aspartate (YMDD) nucleotide-binding motif in the catalytic domain of the enzyme. Similar to the development of lamivudine-resistant HIV mutants, lamivudine treatment of patients with chronic HBV infection results in drug-resistant strains, characterized by YMDD to YIDD (tyrosine, isoleucin, aspartate, aspartate; “M204I”) or YVDD (tyrosine, valine, aspartate, aspartate; “M204V”) mutations in the catalytic C-domain of the reverse transcriptase[16,86]. Interestingly, the mutant genomes exhibited lower levels of replication when compared with wild-type genomes[87]. Further studies identified additional mutations in the neighboring B domain (V173L and L180M[88]). Interestingly, these mutations can restore partially the replication capacity of the C-domain mutations[88].
In contrast to lamivudine, viral resistance to adefovir appears to be mediated pre-dominantly by a mutation in the D-domain[84]. The N236T mutation confers only a 5-10-fold resistance to adefovir[84], which may explain the delayed emergence of this mutant[16]. Another mutation A181V located in the B-domain has been described[16]. Three recently isolated cases of primary adefovir resistance were due to a mutation comprising I233V. Interestingly, the viral variants containing this mutation were sensitive to tenofovir[22]. Drug resistance due to viral mutations in the reverse transcriptase/polymerase represents an important clinical issue in the management of chronic hepatitis B and may ultimately require the development of novel treatment approaches including the combination of various antiviral strategies[16,78].
In summary, a large variety of HBV mutations associated with various pathological conditions as well as drug resistance have been isolated and described in detail[15]. Studies in vitro as well as in vivo have defined the functional relevance of several viral mutants. Furthermore, these studies provided important clues for the understanding of the impact of these mutants on the pathogenesis of disease and the molecular characterization of drug resistance. Functional studies of mutants in established and emerging HBV in vivo models[89] may ultimately allow confirmation of the relationship between defined mutations and their clinical relevance[15].
MECHANISMS OF HBV-INDUCED LIVER DISEASE: APOPTOSIS
The induction of apoptosis is a hallmark of many viruses infecting humans. Although HBV is considered as a non-cytopathic virus[14], hepadnavirus-induced apoptosis and cytopathic effects have been described in several experimental model systems: First, a duck hepatitis B variant containing a single amino acid change in the large surface antigen resulting in accumulation of cccDNA resulted in a strong cytopathic effect in hepatocytes in vitro and in vivo[90-92]. In this system, the level of viral replication and cccDNA formation correlated with cytopathic effects in infected hepatocytes[90]. Second, intracellular retention of the HBV large surface protein has been shown to induce apoptosis in cell lines[93,94]. In this model overexpression of the large surface antigen resulted in cellular vacuolization and apoptosis of transfected hepatoma cells[94]. Third, the HBX protein has been suggested to induce apoptosis in both a p53-dependent and p53-independent manner[95-97]. Exploring the mechanism of these previous observations, a recent study has elegantly demonstrated that HBX interacts with c-FLIP, a key regulator of the death-inducing signaling complex[96]. Recruitment of c-FLIP to the death-inducing signaling complex is inhibited by HBX resulting in hyperactivation of caspase-8 and caspase-3 by death signals[96]. Finally, a viral variant containing two core promoter mutations associated with fulminant hepatitis has been shown to induce apoptosis in primary Tupaia hepatocytes[20]. Interestingly, in the latter model induction of apoptosis was independent of viral replication suggesting that viral protein synthesis was sufficient for the virus-induced hepatocyte cell death. Since the two core promoter mutations resulted in two amino acid changes of the HBX protein, the HBX protein may be a potential candidate mediating this effect[20]. Further studies in animal model systems are needed to elucidate the impact of HBV-induced apoptosis for HBV-induced liver injury[20].
IMMUNOPATHOGENESIS OF HBV INFECTION
Apart from direct biological effects of viral variants (Table 1) there is growing consensus that the host immune response, especially the virus-specific T cell response[98], is the key determinant influencing the course of disease and the onset of liver disease.
Successful immune response
A genomic array analysis using liver RNA obtained at multiple time points after HBV infection in chimpanzees demonstrated that HBV did not induce any genes during entry and expansion[99], suggesting it is a stealth virus early in the infection. In contrast, a large number of T cell-derived IFN-gamma-regulated genes were induced in the liver during viral clearance[99], reflecting the impact of an adaptive T cell response that inhibits viral replication and kills infected cells, thereby terminating the infection. These results are in agreement with several studies performed in acutely infected patients suggesting an important role of the adaptive T cell response[98,100-105]. Indeed, several studies have shown that the peripheral blood cytotoxic T lymphocyte (CTL) response to HBV is polyclonal and multispecific in patients with acute viral hepatitis and that it persists indefinitely after recovery, when it is maintained by continued antigenic stimulation by residual virus that persists, apparently harmlessly, in healthy convalescent individuals[106]. In contrast, the CTL response to HBV is relatively weak in patients with chronic HBV infection, except during spontaneous disease flares or interferon (IFN) induced recovery, when it is readily detectable[101,104]. These earlier studies have been confirmed by using new techniques, such as the tetramer technology. By using this approach, Maini et al could, for example, show that multispecific HBV-specific CD8+ T cell responses are detectable during the acute phase of self-limited infection and decline thereafter[100]. Of note, a recent study could also demonstrate that HBV-specific CD8+ T cells accumulate in the infected organ, i.e., the liver, where they remain detectable at high frequencies even after HBsAg seroconversion[107].
Studies in the HBV transgenic mouse model revealed that, in addition to causing viral hepatitis, virus-specific T cells as well as NK and NKT cells can abolish HBV expression and replication without killing the hepatocytes and that this antiviral activity is mediated by interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα)[104,108,109] (Figure 1). Importantly, studies in the chimpanzee model indicated that the same events are operative during natural infection. Indeed, in these studies it was shown that the early phase of HBV clearance was temporally associated with the appearance of CD3, CD8 and IFNγ in the liver, which reflects the influx of T cells into the liver[110]. Of note, there was only limited liver disease during this time although almost 100% of hepatocytes were infected, clearly suggesting that noncytopathic mechanisms were active during this early phase of viral clearance. In contrast, the final elimination of the virus that occurred several weeks later occurred in the presence of significant liver disease, indicating the presence of cytopathic effector functions that are probably mediated by virus-specific T cells[110].
Figure 1.
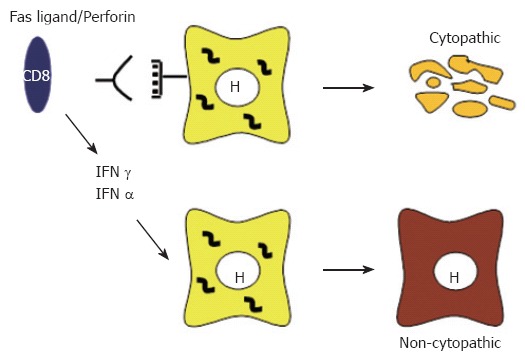
Cytopathic and non-cytopathic T cell responses against HBV infection.
To determine which subset of T cells is responsible for viral clearance and liver disease, the course of HBV infection in chimpanzees that were depleted of its CD4+ or CD8+ cells was monitored[111]. Depletion of CD4+ cells did not significantly alter the course of infection. In contrast to the minor effects observed during CD4-depletion, CD8-depletion greatly prolonged the infection and delayed the onset of viral clearance and liver disease until CD8+ T cells reappeared in the circulation and virus-specific CD8+ T cells entered the liver[111]. Indeed, in the absence of CD8+ cells, the duration of peak infection was prolonged and the time of onset of the initial decrease in HBV DNA levels and increase in serum ALT activity was delayed. In addition, the time required for both the first phase of viral clearance and for the final elimination of the virus was markedly delayed and prolonged in the CD8-depleted animal. It is important to note that the reappearance of CD8+ cells correlated with the appearance of IFNγ producing virus-specific CD8+ T cells in the liver, the onset of a mild liver disease, the appearance of IFNγ mRNA in the liver and a 50 fold reduction in total liver HBV DNA[111]. These results suggest that HBV replication is inhibited early and non-cytopathically in a CD8 dependent and probably IFNγ associated manner. The final elimination of the virus occurred several months later and was associated with a rebound of CD8+ cells to baseline levels, a surge of the intrahepatic CD8+ T cell response, a surge in intrahepatic IFNγ mRNA and a surge in sALT activity. Thus, these results demonstrate that intrahepatic HBV-specific CD8+ T cells are required for rapid viral clearance during acute HBV infection. In addition, the data suggest the existence of dual antiviral functions that overlap temporally during natural infection but can be clearly separated by CD8 depletion: a primarily noncytolytic CD8+ dependent mechanism that may be mediated by IFNγ and a primarily cytolytic mechanism that clears the remaining infected cells. It is also important to note that virus-specific CD8+ T cells display oscillating effector functions during the course of infection that seem primarily influenced by the interaction of the T cells with the antigen-presenting cells[112,113].
T cell failure
In contrast to the strong and multispecific T cell responses observed during acute self-limited HBV infection, patients with chronic hepatitis B tend to have weak and narrowly focused immune responses[98]. The mechanisms that contribute to the failure of the virus-specific T cell response in chronically infected patients are only poorly understood and may include T cell deletion, anergy, exhaustion, ignorance and T cell dysfunction. HBV establishes chronic hepatitis mainly by vertical transmission from HBV infected mothers to neonates. The immunomodulatory effects of the HBeAg might play a role in this setting since it has been shown to be tolerable to T cells in transgenic mice. An important mechanism for the development of viral persistence in adults may be the development of viral escape mutations. Of note, mutational inactivation of B-cell and T cell epitopes has been demonstrated in chronic HBV infection but it seems to occur much less compared to the hepatitis C virus[101,104,114]. Little information is currently available about the intrahepatic T cell response during chronic HBV infection. Of note, one study showed functional, tetramer positive CD8+ T cells in the blood and the liver of chronically HBV infected patients. However, the number of intrahepatic HBV-specific, tetramer-positive T cells did not differ between HBeAg negative patients with normal ALT levels and HBeAg positive patients with increased ALT levels, even though the intrahepatic cellular infiltrate was greater in the latter group[115]. These results suggest a differential contribution of HBV-specific and HBV non-specific bystander lymphocytes in the pathogenesis of chronic hepatitis. T cell dysfunction might also contribute to viral persistence. For example, Reignat et al have shown that HBsAg-specific CD8+ T cells display abnormal HLA/ peptide tetramer binding properties in contrast to HBcAg-positive CD8+ T cells[116]. A high viral load may be one important factor that contributes to T cell failure. In this regard, it is important to note that antiviral treatment can overcome CD8+ T cell hypo-responsiveness in subjects with chronic HBV infection, suggesting that the T cells are present but suppressed[117,118]. First evidence suggests that next to high viral loads and a lack of virus-specific CD4+ T cell help, regulatory T cells may contribute to this T cell suppression[119,120]. Clearly, additional studies are required to better understand the complex host-virus interactions that determine the outcome of HBV infection.
CONCLUSION
Virus-host interactions, especially the virus-specific T cell response, are the key factors accounting for the pathogenesis of HBV infection. Viral variants may influence the course of disease and deserve special attention in the setting of antiviral therapy, immune escape and reactivation during immunosuppressive therapy.
Footnotes
S- Editor Liu Y L- Editor Lutze M E- Editor Ma WH
References
- 1.Nassal M. Hepatitis B virus replication: novel roles for virus-host interactions. Intervirology. 1999;42:100–116. doi: 10.1159/000024970. [DOI] [PubMed] [Google Scholar]
- 2.Hollinger FB, Liang TJ. Hepatitis B virus. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams and Wilkins; 2001. pp. 2971–3036. [Google Scholar]
- 3.Summers J, Mason WS. Replication of the genome of a hepatitis B--like virus by reverse transcription of an RNA intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 4.Bartenschlager R, Schaller H. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked protein) believed to prime reverse transcription. EMBO J. 1988;7:4185–4192. doi: 10.1002/j.1460-2075.1988.tb03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feld J, Locarnini S. Antiviral therapy for hepatitis B virus infections: new targets and technical challenges. J Clin Virol. 2002;25:267–283. doi: 10.1016/s1386-6532(02)00107-5. [DOI] [PubMed] [Google Scholar]
- 6.Tong MJ, Tu SS. Treatment of patients with chronic hepatitis B with adefovir dipivoxil. Semin Liver Dis. 2004;24 Suppl 1:37–44. doi: 10.1055/s-2004-828677. [DOI] [PubMed] [Google Scholar]
- 7.Wright TL. Clinical trial results and treatment resistance with lamivudine in hepatitis B. Semin Liver Dis. 2004;24 Suppl 1:31–36. doi: 10.1055/s-2004-828676. [DOI] [PubMed] [Google Scholar]
- 8.van Bömmel F, Wünsche T, Mauss S, Reinke P, Bergk A, Schürmann D, Wiedenmann B, Berg T. Comparison of adefovir and tenofovir in the treatment of lamivudine-resistant hepatitis B virus infection. Hepatology. 2004;40:1421–1425. doi: 10.1002/hep.20464. [DOI] [PubMed] [Google Scholar]
- 9.Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman ML, Jeffers L, Goodman Z, Wulfsohn MS, Xiong S, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med. 2003;348:808–816. doi: 10.1056/NEJMoa020681. [DOI] [PubMed] [Google Scholar]
- 10.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, Marcellin P, Lim SG, Goodman Z, Wulfsohn MS, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N Engl J Med. 2003;348:800–807. doi: 10.1056/NEJMoa021812. [DOI] [PubMed] [Google Scholar]
- 11.Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, Tanwandee T, Tao QM, Shue K, Keene ON, Dixon JS, Gray DF, Sabbat J. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521–1531. doi: 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- 12.Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z, Crowther L, Condreay LD, Woessner M, Rubin M, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med. 1999;341:1256–1263. doi: 10.1056/NEJM199910213411702. [DOI] [PubMed] [Google Scholar]
- 13.Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 14.Baumert TF, Blum HE. Hepatitis B virus mutations: molecular biology and clinical relevance. Vir Hep Rev. 2000;6:177–192. [Google Scholar]
- 15.Baumert TF, Barth H, Blum HE. Genetic variants of hepatitis B virus and their clinical relevance. Minerva Gastroenterol Dietol. 2005;51:95–108. [PubMed] [Google Scholar]
- 16.Zoulim F. Mechanism of viral persistence and resistance to nucleoside and nucleotide analogs in chronic hepatitis B virus infection. Antiviral Res. 2004;64:1–15. doi: 10.1016/j.antiviral.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Pawlotsky JM. The concept of hepatitis B virus mutant escape. J Clin Virol. 2005;34 Suppl 1:S125–S129. doi: 10.1016/s1386-6532(05)80021-6. [DOI] [PubMed] [Google Scholar]
- 18.Liang TJ, Hasegawa K, Rimon N, Wands JR, Ben-Porath E. A hepatitis B virus mutant associated with an epidemic of fulminant hepatitis. N Engl J Med. 1991;324:1705–1709. doi: 10.1056/NEJM199106133242405. [DOI] [PubMed] [Google Scholar]
- 19.Baumert TF, Rogers SA, Hasegawa K, Liang TJ. Two core promotor mutations identified in a hepatitis B virus strain associated with fulminant hepatitis result in enhanced viral replication. J Clin Invest. 1996;98:2268–2276. doi: 10.1172/JCI119037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumert TF, Yang C, Schürmann P, Köck J, Ziegler C, Grüllich C, Nassal M, Liang TJ, Blum HE, von Weizsäcker F. Hepatitis B virus mutations associated with fulminant hepatitis induce apoptosis in primary Tupaia hepatocytes. Hepatology. 2005;41:247–256. doi: 10.1002/hep.20553. [DOI] [PubMed] [Google Scholar]
- 21.Buckwold VE, Xu Z, Chen M, Yen TS, Ou JH. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J Virol. 1996;70:5845–5851. doi: 10.1128/jvi.70.9.5845-5851.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schildgen O, Sirma H, Funk A, Olotu C, Wend UC, Hartmann H, Helm M, Rockstroh JK, Willems WR, Will H, et al. Variant of hepatitis B virus with primary resistance to adefovir. N Engl J Med. 2006;354:1807–1812. doi: 10.1056/NEJMoa051214. [DOI] [PubMed] [Google Scholar]
- 23.Kann M, Gerlich WH. Hepatitis B. in: Collier L, Balows, A. Sussmann, M. Topley Wilson's Microbiology and Microbial Infections. London: Edward Arnold Ltd; 2005. [Google Scholar]
- 24.Brunetto MR, Stemler M, Bonino F, Schodel F, Oliveri F, Rizzetto M, Verme G, Will H. A new hepatitis B virus strain in patients with severe anti-HBe positive chronic hepatitis B. J Hepatol. 1990;10:258–261. doi: 10.1016/0168-8278(90)90062-v. [DOI] [PubMed] [Google Scholar]
- 25.Akahane Y, Yamanaka T, Suzuki H, Sugai Y, Tsuda F, Yotsumoto S, Omi S, Okamoto H, Miyakawa Y, Mayumi M. Chronic active hepatitis with hepatitis B virus DNA and antibody against e antigen in the serum. Disturbed synthesis and secretion of e antigen from hepatocytes due to a point mutation in the precore region. Gastroenterology. 1990;99:1113–1119. doi: 10.1016/0016-5085(90)90632-b. [DOI] [PubMed] [Google Scholar]
- 26.Milich DR, Jones JE, Hughes JL, Price J, Raney AK, McLachlan A. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci USA. 1990;87:6599–6603. doi: 10.1073/pnas.87.17.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milich DR. Do T cells "see" the hepatitis B core and e antigens differently? Gastroenterology. 1999;116:765–768. doi: 10.1016/s0016-5085(99)70203-9. [DOI] [PubMed] [Google Scholar]
- 28.Diepolder HM, Ries G, Jung MC, Schlicht HJ, Gerlach JT, Gr ner N, Caselmann WH, Pape GR. Differential antigen-processing pathways of the hepatitis B virus e and core proteins. Gastroenterology. 1999;116:650–657. doi: 10.1016/s0016-5085(99)70187-3. [DOI] [PubMed] [Google Scholar]
- 29.Kosaka Y, Takase K, Kojima M, Shimizu M, Inoue K, Yoshiba M, Tanaka S, Akahane Y, Okamoto H, Tsuda F. Fulminant hepatitis B: induction by hepatitis B virus mutants defective in the precore region and incapable of encoding e antigen. Gastroenterology. 1991;100:1087–1094. doi: 10.1016/0016-5085(91)90286-t. [DOI] [PubMed] [Google Scholar]
- 30.Omata M, Ehata T, Yokosuka O, Hosoda K, Ohto M. Mutations in the precore region of hepatitis B virus DNA in patients with fulminant and severe hepatitis. N Engl J Med. 1991;324:1699–1704. doi: 10.1056/NEJM199106133242404. [DOI] [PubMed] [Google Scholar]
- 31.Igaki N, Nakaji M, Moriguchi R, Akiyama H, Tamada F, Oimomi M, Goto T. An outbreak of fulminant hepatitis B in immunocompromised hemodialysis patients. J Gastroenterol. 2003;38:968–976. doi: 10.1007/s00535-003-1180-1. [DOI] [PubMed] [Google Scholar]
- 32.Liu CJ, Kao JH, Lai MY, Chen PJ, Chen DS. Precore/core promoter mutations and genotypes of hepatitis B virus in chronic hepatitis B patients with fulminant or subfulminant hepatitis. J Med Virol. 2004;72:545–550. doi: 10.1002/jmv.20024. [DOI] [PubMed] [Google Scholar]
- 33.Tacke F, Gehrke C, Luedde T, Heim A, Manns MP, Trautwein C. Basal core promoter and precore mutations in the hepatitis B virus genome enhance replication efficacy of Lamivudine-resistant mutants. J Virol. 2004;78:8524–8535. doi: 10.1128/JVI.78.16.8524-8535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 35.Naoumov NV, Schneider R, Grötzinger T, Jung MC, Miska S, Pape GR, Will H. Precore mutant hepatitis B virus infection and liver disease. Gastroenterology. 1992;102:538–543. doi: 10.1016/0016-5085(92)90101-4. [DOI] [PubMed] [Google Scholar]
- 36.Mphahlele MJ, Shattock AG, Boner W, Quinn J, McCormick PA, Carman WF. Transmission of a homogenous hepatitis B virus population of A1896-containing strains leading to mild resolving acute hepatitis and seroconversion to hepatitis B e antigen antibodies in an adult. Hepatology. 1997;26:743–746. doi: 10.1002/hep.510260329. [DOI] [PubMed] [Google Scholar]
- 37.Chen HS, Kew MC, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. J Virol. 1992;66:5682–5684. doi: 10.1128/jvi.66.9.5682-5684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chuang WL, Omata M, Ehata T, Yokosuka O, Hosoda K, Imazeki F, Ohto M. Coinfection study of precore mutant and wild-type hepatitis B-like virus in ducklings. Hepatology. 1994;19:569–576. doi: 10.1002/hep.1840190305. [DOI] [PubMed] [Google Scholar]
- 39.Sato S, Suzuki K, Akahane Y, Akamatsu K, Akiyama K, Yunomura K, Tsuda F, Tanaka T, Okamoto H, Miyakawa Y, et al. Hepatitis B virus strains with mutations in the core promoter in patients with fulminant hepatitis. Ann Intern Med. 1995;122:241–248. doi: 10.7326/0003-4819-122-4-199502150-00001. [DOI] [PubMed] [Google Scholar]
- 40.Baumert TF, Marrone A, Vergalla J, Liang TJ. Naturally occurring mutations define a novel function of the hepatitis B virus core promoter in core protein expression. J Virol. 1998;72:6785–6795. doi: 10.1128/jvi.72.8.6785-6795.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pult I, Chouard T, Wieland S, Klemenz R, Yaniv M, Blum HE. A hepatitis B virus mutant with a new hepatocyte nuclear factor 1 binding site emerging in transplant-transmitted fulminant hepatitis B. Hepatology. 1997;25:1507–1515. doi: 10.1002/hep.510250633. [DOI] [PubMed] [Google Scholar]
- 42.Günther S, Piwon N, Iwanska A, Schilling R, Meisel H, Will H. Type, prevalence, and significance of core promoter/enhancer II mutations in hepatitis B viruses from immunosuppressed patients with severe liver disease. J Virol. 1996;70:8318–8331. doi: 10.1128/jvi.70.12.8318-8331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamoto H, Tsuda F, Akahane Y, Sugai Y, Yoshiba M, Moriyama K, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J Virol. 1994;68:8102–8110. doi: 10.1128/jvi.68.12.8102-8110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parekh S, Zoulim F, Ahn SH, Tsai A, Li J, Kawai S, Khan N, Trépo C, Wands J, Tong S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol. 2003;77:6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Preikschat P, Günther S, Reinhold S, Will H, Budde K, Neumayer HH, Krüger DH, Meisel H. Complex HBV populations with mutations in core promoter, C gene, and pre-S region are associated with development of cirrhosis in long-term renal transplant recipients. Hepatology. 2002;35:466–477. doi: 10.1053/jhep.2002.30698. [DOI] [PubMed] [Google Scholar]
- 46.Raney AK, Johnson JL, Palmer CN, McLachlan A. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J Virol. 1997;71:1058–1071. doi: 10.1128/jvi.71.2.1058-1071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Buckwold VE, Hon MW, Ou JH. Mechanism of suppression of hepatitis B virus precore RNA transcription by a frequent double mutation. J Virol. 1999;73:1239–1244. doi: 10.1128/jvi.73.2.1239-1244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng Y, Li J, Ou JH. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J Virol. 2004;78:6908–6914. doi: 10.1128/JVI.78.13.6908-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lamberts C, Nassal M, Velhagen I, Zentgraf H, Schröder CH. Precore-mediated inhibition of hepatitis B virus progeny DNA synthesis. J Virol. 1993;67:3756–3762. doi: 10.1128/jvi.67.7.3756-3762.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kao JH, Chen PJ, Lai MY, Chen DS. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327–334. doi: 10.1053/gast.2003.50053. [DOI] [PubMed] [Google Scholar]
- 51.Sterneck M, Kalinina T, Günther S, Fischer L, Santantonio T, Greten H, Will H. Functional analysis of HBV genomes from patients with fulminant hepatitis. Hepatology. 1998;28:1390–1397. doi: 10.1002/hep.510280530. [DOI] [PubMed] [Google Scholar]
- 52.Bartholomeusz A, Locarnini S. Hepatitis B virus mutants and fulminant hepatitis B: fitness plus phenotype. Hepatology. 2001;34:432–435. doi: 10.1053/jhep.2001.26764. [DOI] [PubMed] [Google Scholar]
- 53.Zuckerman JN, Zuckerman AJ. Mutations of the surface protein of hepatitis B virus. Antiviral Res. 2003;60:75–78. doi: 10.1016/j.antiviral.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 54.Carman WF, Zanetti AR, Karayiannis P, Waters J, Manzillo G, Tanzi E, Zuckerman AJ, Thomas HC. Vaccine-induced escape mutant of hepatitis B virus. Lancet. 1990;336:325–329. doi: 10.1016/0140-6736(90)91874-a. [DOI] [PubMed] [Google Scholar]
- 55.Waters JA, Kennedy M, Voet P, Hauser P, Petre J, Carman W, Thomas HC. Loss of the common "A" determinant of hepatitis B surface antigen by a vaccine-induced escape mutant. J Clin Invest. 1992;90:2543–2547. doi: 10.1172/JCI116148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalinina T, Iwanski A, Will H, Sterneck M. Deficiency in virion secretion and decreased stability of the hepatitis B virus immune escape mutant G145R. Hepatology. 2003;38:1274–1281. doi: 10.1053/jhep.2003.50484. [DOI] [PubMed] [Google Scholar]
- 57.Yamamoto K, Horikita M, Tsuda F, Itoh K, Akahane Y, Yotsumoto S, Okamoto H, Miyakawa Y, Mayumi M. Naturally occurring escape mutants of hepatitis B virus with various mutations in the S gene in carriers seropositive for antibody to hepatitis B surface antigen. J Virol. 1994;68:2671–2676. doi: 10.1128/jvi.68.4.2671-2676.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fortuin M, Karthigesu V, Allison L, Howard C, Hoare S, Mendy M, Whittle HC. Breakthrough infections and identification of a viral variant in Gambian children immunized with hepatitis B vaccine. J Infect Dis. 1994;169:1374–1376. doi: 10.1093/infdis/169.6.1374. [DOI] [PubMed] [Google Scholar]
- 59.Hsu HY, Chang MH, Liaw SH, Ni YH, Chen HL. Changes of hepatitis B surface antigen variants in carrier children before and after universal vaccination in Taiwan. Hepatology. 1999;30:1312–1317. doi: 10.1002/hep.510300511. [DOI] [PubMed] [Google Scholar]
- 60.Ogata N, Cote PJ, Zanetti AR, Miller RH, Shapiro M, Gerin J, Purcell RH. Licensed recombinant hepatitis B vaccines protect chimpanzees against infection with the prototype surface gene mutant of hepatitis B virus. Hepatology. 1999;30:779–786. doi: 10.1002/hep.510300309. [DOI] [PubMed] [Google Scholar]
- 61.Bauer T, Weinberger K, Jilg W. Variants of two major T cell epitopes within the hepatitis B surface antigen are not recognized by specific T helper cells of vaccinated individuals. Hepatology. 2002;35:455–465. doi: 10.1053/jhep.2002.30903. [DOI] [PubMed] [Google Scholar]
- 62.McMahon G, Ehrlich PH, Moustafa ZA, McCarthy LA, Dottavio D, Tolpin MD, Nadler PI, Ostberg L. Genetic alterations in the gene encoding the major HBsAg: DNA and immunological analysis of recurrent HBsAg derived from monoclonal antibody-treated liver transplant patients. Hepatology. 1992;15:757–766. doi: 10.1002/hep.1840150503. [DOI] [PubMed] [Google Scholar]
- 63.Carman WF, Trautwein C, van Deursen FJ, Colman K, Dornan E, McIntyre G, Waters J, Kliem V, Müller R, Thomas HC, et al. Hepatitis B virus envelope variation after transplantation with and without hepatitis B immune globulin prophylaxis. Hepatology. 1996;24:489–493. doi: 10.1002/hep.510240304. [DOI] [PubMed] [Google Scholar]
- 64.Protzer-Knolle U, Naumann U, Bartenschlager R, Berg T, Hopf U, Meyer zum Büschenfelde KH, Neuhaus P, Gerken G. Hepatitis B virus with antigenically altered hepatitis B surface antigen is selected by high-dose hepatitis B immune globulin after liver transplantation. Hepatology. 1998;27:254–263. doi: 10.1002/hep.510270138. [DOI] [PubMed] [Google Scholar]
- 65.Ghany MG, Ayola B, Villamil FG, Gish RG, Rojter S, Vierling JM, Lok AS. Hepatitis B virus S mutants in liver transplant recipients who were reinfected despite hepatitis B immune globulin prophylaxis. Hepatology. 1998;27:213–222. doi: 10.1002/hep.510270133. [DOI] [PubMed] [Google Scholar]
- 66.Lu M, Lorentz T. De novo infection in a renal transplant recipient caused by novel mutants of hepatitis B virus despite the presence of protective anti-hepatitis B surface antibody. J Infect Dis. 2003;187:1323–1326. doi: 10.1086/373902. [DOI] [PubMed] [Google Scholar]
- 67.Jongerius JM, Wester M, Cuypers HT, van Oostendorp WR, Lelie PN, van der Poel CL, van Leeuwen EF. New hepatitis B virus mutant form in a blood donor that is undetectable in several hepatitis B surface antigen screening assays. Transfusion. 1998;38:56–59. doi: 10.1046/j.1537-2995.1998.38198141499.x. [DOI] [PubMed] [Google Scholar]
- 68.Hass M, Hannoun C, Kalinina T, Sommer G, Manegold C, Günther S. Functional analysis of hepatitis B virus reactivating in hepatitis B surface antigen-negative individuals. Hepatology. 2005;42:93–103. doi: 10.1002/hep.20748. [DOI] [PubMed] [Google Scholar]
- 69.Baumert TF, Köck J, Blum HE. A novel target of hepatitis B virus mutations: splicing of surface RNA. Hepatology. 2005;42:21–23. doi: 10.1002/hep.20791. [DOI] [PubMed] [Google Scholar]
- 70.Chemin I, Trépo C. Clinical impact of occult HBV infections. J Clin Virol. 2005;34 Suppl 1:S15–S21. doi: 10.1016/s1386-6532(05)80005-8. [DOI] [PubMed] [Google Scholar]
- 71.Glebe D, Urban S, Knoop EV, Cag N, Krass P, Grün S, Bulavaite A, Sasnauskas K, Gerlich WH. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology. 2005;129:234–245. doi: 10.1053/j.gastro.2005.03.090. [DOI] [PubMed] [Google Scholar]
- 72.Engelke M, Mills K, Seitz S, Simon P, Gripon P, Schnölzer M, Urban S. Characterization of a hepatitis B and hepatitis delta virus receptor binding site. Hepatology. 2006;43:750–760. doi: 10.1002/hep.21112. [DOI] [PubMed] [Google Scholar]
- 73.Pollicino T, Zanetti AR, Cacciola I, Petit MA, Smedile A, Campo S, Sagliocca L, Pasquali M, Tanzi E, Longo G, et al. Pre-S2 defective hepatitis B virus infection in patients with fulminant hepatitis. Hepatology. 1997;26:495–499. doi: 10.1002/hep.510260235. [DOI] [PubMed] [Google Scholar]
- 74.Bock CT, Tillmann HL, Maschek HJ, Manns MP, Trautwein C. A preS mutation isolated from a patient with chronic hepatitis B infection leads to virus retention and misassembly. Gastroenterology. 1997;113:1976–1982. doi: 10.1016/s0016-5085(97)70018-0. [DOI] [PubMed] [Google Scholar]
- 75.Bock CT, Tillmann HL, Manns MP, Trautwein C. The pre-S region determines the intracellular localization and appearance of hepatitis B virus. Hepatology. 1999;30:517–525. doi: 10.1002/hep.510300206. [DOI] [PubMed] [Google Scholar]
- 76.Chen BF, Liu CJ, Jow GM, Chen PJ, Kao JH, Chen DS. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology. 2006;130:1153–1168. doi: 10.1053/j.gastro.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 77.Kalinina T, Riu A, Fischer L, Will H, Sterneck M. A dominant hepatitis B virus population defective in virus secretion because of several S-gene mutations from a patient with fulminant hepatitis. Hepatology. 2001;34:385–394. doi: 10.1053/jhep.2001.26516. [DOI] [PubMed] [Google Scholar]
- 78.Durantel D, Brunelle MN, Gros E, Carrouée-Durantel S, Pichoud C, Villet S, Trepo C, Zoulim F. Resistance of human hepatitis B virus to reverse transcriptase inhibitors: from genotypic to phenotypic testing. J Clin Virol. 2005;34 Suppl 1:S34–S43. doi: 10.1016/s1386-6532(05)80008-3. [DOI] [PubMed] [Google Scholar]
- 79.Lai CL, Chien RN, Leung NW, Chang TT, Guan R, Tai DI, Ng KY, Wu PC, Dent JC, Barber J, et al. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N Engl J Med. 1998;339:61–68. doi: 10.1056/NEJM199807093390201. [DOI] [PubMed] [Google Scholar]
- 80.Marcellin P, Lau GK, Bonino F, Farci P, Hadziyannis S, Jin R, Lu ZM, Piratvisuth T, Germanidis G, Yurdaydin C, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2004;351:1206–1217. doi: 10.1056/NEJMoa040431. [DOI] [PubMed] [Google Scholar]
- 81.Köck J, Baumert TF, Delaney WE, Blum HE, von Weizsäcker F. Inhibitory effect of adefovir and lamivudine on the initiation of hepatitis B virus infection in primary tupaia hepatocytes. Hepatology. 2003;38:1410–1418. doi: 10.1016/j.hep.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 82.Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, Dienstag JL, Heathcote EJ, Little NR, Griffiths DA, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714–1722. doi: 10.1053/j.gastro.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 83.Lau DT, Khokhar MF, Doo E, Ghany MG, Herion D, Park Y, Kleiner DE, Schmid P, Condreay LD, Gauthier J, et al. Long-term therapy of chronic hepatitis B with lamivudine. Hepatology. 2000;32:828–834. doi: 10.1053/jhep.2000.17912. [DOI] [PubMed] [Google Scholar]
- 84.Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C, Brosgart C, Colledge D, Edwards R, Ayres A, et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003;125:292–297. doi: 10.1016/s0016-5085(03)00939-9. [DOI] [PubMed] [Google Scholar]
- 85.Marcellin P, Chang TT, Lim SG, Sievert W, Tong M, Arterburn S, Xiong S, Brosgart CL, Currie G. Long term efficacy and safety of adefovir dipivoxil in HBeAg chronic hepatitis B patients: increasing serologic, virologic and biochemical response over time. Hepatology. 2004;41:A1135. [Google Scholar]
- 86.Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA, Tyrrell DL, Brown N, Condreay LD. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology. 1998;27:1670–1677. doi: 10.1002/hep.510270628. [DOI] [PubMed] [Google Scholar]
- 87.Melegari M, Scaglioni PP, Wands JR. Hepatitis B virus mutants associated with 3TC and famciclovir administration are replication defective. Hepatology. 1998;27:628–633. doi: 10.1002/hep.510270243. [DOI] [PubMed] [Google Scholar]
- 88.Delaney WE, Yang H, Westland CE, Das K, Arnold E, Gibbs CS, Miller MD, Xiong S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol. 2003;77:11833–11841. doi: 10.1128/JVI.77.21.11833-11841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dandri M, Volz TK, Lütgehetmann M, Petersen J. Animal models for the study of HBV replication and its variants. J Clin Virol. 2005;34 Suppl 1:S54–S62. doi: 10.1016/s1386-6532(05)80011-3. [DOI] [PubMed] [Google Scholar]
- 90.Lenhoff RJ, Summers J. Construction of avian hepadnavirus variants with enhanced replication and cytopathicity in primary hepatocytes. J Virol. 1994;68:5706–5713. doi: 10.1128/jvi.68.9.5706-5713.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lenhoff RJ, Luscombe CA, Summers J. Competition in vivo between a cytopathic variant and a wild-type duck hepatitis B virus. Virology. 1998;251:85–95. doi: 10.1006/viro.1998.9394. [DOI] [PubMed] [Google Scholar]
- 92.Lenhoff RJ, Luscombe CA, Summers J. Acute liver injury following infection with a cytopathic strain of duck hepatitis B virus. Hepatology. 1999;29:563–571. doi: 10.1002/hep.510290236. [DOI] [PubMed] [Google Scholar]
- 93.Bruss V. Revisiting the cytopathic effect of hepatitis B virus infection. Hepatology. 2002;36:1327–1329. doi: 10.1053/jhep.2002.37351. [DOI] [PubMed] [Google Scholar]
- 94.Foo NC, Ahn BY, Ma X, Hyun W, Yen TS. Cellular vacuolization and apoptosis induced by hepatitis B virus large surface protein. Hepatology. 2002;36:1400–1407. doi: 10.1053/jhep.2002.36819. [DOI] [PubMed] [Google Scholar]
- 95.Su F, Theodosis CN, Schneider RJ. Role of NF-kappaB and myc proteins in apoptosis induced by hepatitis B virus HBx protein. J Virol. 2001;75:215–225. doi: 10.1128/JVI.75.1.215-225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim KH, Seong BL. Pro-apoptotic function of HBV X protein is mediated by interaction with c-FLIP and enhancement of death-inducing signal. EMBO J. 2003;22:2104–2116. doi: 10.1093/emboj/cdg210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chirillo P, Pagano S, Natoli G, Puri PL, Burgio VL, Balsano C, Levrero M. The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc Natl Acad Sci USA. 1997;94:8162–8167. doi: 10.1073/pnas.94.15.8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Invest. 1997;99:1472–1477. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci USA. 2004;101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maini MK, Boni C, Ogg GS, King AS, Reignat S, Lee CK, Larrubia JR, Webster GJ, McMichael AJ, Ferrari C, et al. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology. 1999;117:1386–1396. doi: 10.1016/s0016-5085(99)70289-1. [DOI] [PubMed] [Google Scholar]
- 101.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 102.Thimme R, Chang KM, Pemberton J, Sette A, Chisari FV. Degenerate immunogenicity of an HLA-A2-restricted hepatitis B virus nucleocapsid cytotoxic T-lymphocyte epitope that is also presented by HLA-B51. J Virol. 2001;75:3984–3987. doi: 10.1128/JVI.75.8.3984-3987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Urbani S, Boni C, Missale G, Elia G, Cavallo C, Massari M, Raimondo G, Ferrari C. Virus-specific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. J Virol. 2002;76:12423–12434. doi: 10.1128/JVI.76.24.12423-12434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Webster GJ, Reignat S, Maini MK, Whalley SA, Ogg GS, King A, Brown D, Amlot PL, Williams R, Vergani D, et al. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology. 2000;32:1117–1124. doi: 10.1053/jhep.2000.19324. [DOI] [PubMed] [Google Scholar]
- 106.Penna A, Artini M, Cavalli A, Levrero M, Bertoletti A, Pilli M, Chisari FV, Rehermann B, Del Prete G, Fiaccadori F, et al. Long-lasting memory T cell responses following self-limited acute hepatitis B. J Clin Invest. 1996;98:1185–1194. doi: 10.1172/JCI118902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sprengers D, van der Molen RG, Kusters JG, De Man RA, Niesters HG, Schalm SW, Janssen HL. Analysis of intrahepatic HBV-specific cytotoxic T-cells during and after acute HBV infection in humans. J Hepatol. 2006;45:182–189. doi: 10.1016/j.jhep.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 108.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 109.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 110.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 111.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boettler T, Panther E, Bengsch B, Nazarova N, Spangenberg HC, Blum HE, Thimme R. Expression of the interleukin-7 receptor alpha chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J Virol. 2006;80:3532–3540. doi: 10.1128/JVI.80.7.3532-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Isogawa M, Furuichi Y, Chisari FV. Oscillating CD8(+) T cell effector functions after antigen recognition in the liver. Immunity. 2005;23:53–63. doi: 10.1016/j.immuni.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 114.Rehermann B, Pasquinelli C, Mosier SM, Chisari FV. Hepatitis B virus (HBV) sequence variation of cytotoxic T lymphocyte epitopes is not common in patients with chronic HBV infection. J Clin Invest. 1995;96:1527–1534. doi: 10.1172/JCI118191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, King AS, Herberg J, Gilson R, Alisa A, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–1280. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reignat S, Webster GJ, Brown D, Ogg GS, King A, Seneviratne SL, Dusheiko G, Williams R, Maini MK, Bertoletti A. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med. 2002;195:1089–1101. doi: 10.1084/jem.20011723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Boni C, Bertoletti A, Penna A, Cavalli A, Pilli M, Urbani S, Scognamiglio P, Boehme R, Panebianco R, Fiaccadori F, et al. Lamivudine treatment can restore T cell responsiveness in chronic hepatitis B. J Clin Invest. 1998;102:968–975. doi: 10.1172/JCI3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boni C, Penna A, Ogg GS, Bertoletti A, Pilli M, Cavallo C, Cavalli A, Urbani S, Boehme R, Panebianco R, et al. Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology. 2001;33:963–971. doi: 10.1053/jhep.2001.23045. [DOI] [PubMed] [Google Scholar]
- 119.Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, Janssen HL. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–778. doi: 10.1002/hep.20649. [DOI] [PubMed] [Google Scholar]
- 120.Franzese O, Kennedy PT, Gehring AJ, Gotto J, Williams R, Maini MK, Bertoletti A. Modulation of the CD8+-T-cell response by CD4+ CD25+ regulatory T cells in patients with hepatitis B virus infection. J Virol. 2005;79:3322–3328. doi: 10.1128/JVI.79.6.3322-3328.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]