

Abstract
AIM: To characterize the IFN-response and its modul-ation by the antiviral compound lamivudine in HBV-transfected HepG2.2.15 cells.
METHODS: HepG2.2.15 and HepG2 cells were stimulated with various concentrations of IFN-α2a in the presence or absence of lamivudine. Then, total RNA was extracted and analysed by customised cDNA arrays and northern blot for interferon-inducible genes (ISGs). In addition, cellular proteins were extracted for EMSA and western blot. HBV replication was assessed by southern blot or ELISAs for HBsAg and HBeAg.
RESULTS: Two genes (MxA, Cig5) with completely abolished and 4 genes (IFITM1, -2, -3, and 6-16) with partially reduced IFN-responses were identified in HepG2.2.15 cells. In 2 genes (IFITM1, 6-16), the response to IFN-α could be restored by treatment with lamivudine. This effect could not be explained by a direct modulation of the Jak/Stat signalling pathway since EMSA and western blot experiments revealed no suppression of Stat1 activation and ISGF3 formation after stimulation with IFN-α in HepG2.2.15 compared to HepG2 cells.
CONCLUSION: These results are consistent with the assumption that chronic hepatitis B may specifically modulate the cellular response to IFN by a selective blockage of some ISGs. Antiviral treatment with lamivudine may partially restore ISG expression by reducing HBV gene expression and replication.
Keywords: Hepatitis B, IFN-α, Gene expression, Lamivudine
INTRODUCTION
Hepatitis B (HBV) is a hepatotropic DNA virus capable of causing both acute and chronic hepatitis in humans. It is estimated that over 350 million people are chronically infected with HBV worldwide. Currently approved therapeutic strategies for treatment of HBV include interferon-alpha (IFN-α), the nucleoside analogue lamivudine and the nucleotide analogue adefovir[1,2]. However, only a minority of patients treated with IFN-α has a long-term sustained response with ‘eradication’ of the virus. Patients with a high viral load, in particular, rarely respond to IFN therapy. Treatment with lamivudine, on the other hand, is complicated by a high rate of viral resistance and a high relapse rate after cessation of therapy, respectively[3]. Both the emergence of viral resistance and relapse after therapy are often associated with a hepatitis flare, which can sometimes be fatal. Thus, novel strategies are needed to improve treatment for this disease.
To develop new regimens it is necessary to gain further insights into the interactions between HBV and the main antiviral system of the host, the IFN-system. It has been shown that typeIand type II interferons are able to suppress HBV-replication in livers from HBV-transgenic mice[4-6]. This could also be demonstrated in vitro by using immortalized hepatocyte cell lines from these animals[7] and involves elimination of pregenomic RNA-containing capsids, inhibition of DNA replication and reduction of steady-state levels of HBV transcripts. The effector mechanisms that have been associated with IFN-induced suppression of HBV-replication include MxA[8] and proteasome mediated activities[9,10]. Additional data suggest a role for GTP-binding proteins, signalling and various other molecules in the control of HBV replication[11]. HBV can counteract these antiviral effector mechanisms by inhibiting proteasome activities in an HBX-dependent manner[12] and by suppressing MxA expression at the promoter level[13]. Furthermore, it has been shown that HBV replicated at higher levels in HBV-transgenic mice crossed with IRF-1 or PKR deficient mice while replication was unchanged in transgenic mice crossed with RNase L deficient mice[14].
Assuming that HBV may interfere with the expression of ISGs, one would predict that the ISG expression in cell lines with and without HBV may be different and this would be modulated by inhibition of HBV gene expression and replication. The present study was performed to test this hypothesis. Using customized cDNA arrays for ISGs, we could identify 2 ISGs (MxA and Cig5) that are completely abolished in HBV-transfected HepG2.2.15 cells and 4 genes (IFITM1, -2, -3 and 6-16) with partially reduced responses. This suppression could partially be restored in 2 genes (IFITM1, 6-16) by treatment with the nucleoside analogue lamivudine suggesting an additional therapeutic mechanism for this drug.
MATERIALS AND METHODS
Cell culture
HepG2.2.15 cells were kindly provided by G. Acs (Mount Sinai Medical Cancer, New York, NY) and maintained in Dulbecco’s Modified Eagle’s Medium, supplemented with 2 mmol/L L-glutamine 50 IU/mL of penicillin, 50 mg/L of streptomycin, 500 mg/L of G418, 5% (vol/vol) fetal bovine serum, at 37°C in humidified incubators at 5% CO2. The cells were seeded at a density of 8 × 105 cells and maintained in a confluent state for 2 to 3 d before being treated with antiviral compounds. At first, various concentrations from 0.04 μmol/L to 100 μmol/L of lamivudine were used to reach the suitable drug concentration, which profoundly suppressed HBV replication without cytotoxicity. At the same time, a time course of drug action also was evaluated. Over a period of 10 d lamivudine was added to the medium daily, then the cells were stimulated by addition of IFN-α for 6 h. Thereafter, the media were collected and DNA or RNA was extracted for further analysis.
Analysis of secreted HBV particles
Detection of HBsAg and HBeAg was carried out by using a commercially available kit (Dade Behring) according to the manufacturer’s instructions. Medium samples collected from HepG2.2.15 cells were centrifuged at 1200 rpm for 10 min to remove cellular debris, transferred to clean tubes and stored at -20°C until analysed. HBsAg and HBeAg amounts were evaluated from absorbance reading values (450 nm) compared to the constructed controls.
HBV DNA analysis
Extracellular virion HBV-DNA analysis: Medium of HepG2.2.15 cells was collected and centrifuged (10 min, 2000 × g), and polyethylene glycol (Mr, 8000) was added to the supernatant at a concentration of 10% (wt/vol) followed by overnight precipitation at 4°C. The virions were pelletted (30 min, 10 000 × g), and the pellet was resuspended in lysis buffer (10 mmol/L Tris-Cl, 5 mmol/L EDTA, 150 mmol/L NaCl, 1% SDS) at room temperature for 15 min. Proteinase K was added at a concentration of 500 μg/mL and the suspension incubated for 2 h at 56°C. The digest was extracted with phenol/chloroform, 1:1 (vol/vol) or chloroform, respectively, and the DNA was precipitated with 2.5 vol. of ethanol. The DNA pellet was dissolved in TE solution and then spotted onto Hybond-N+ membranes. Alternatively, the DNA was electrophoresed in 1.2% agarose gel followed by blotting onto Hybond-N+ membranes. The bolt was hybridized with a 32P-labeled HBV DNA probe (digested by NsiIfrom plasmids that contained the full length HBV genome sequence dimer and labelled with a RediprimeTM II Random prime labelling system), washed with 2 × SSC/0.1% SDS at room temperature for 20 min, twice, and 0.1 × SSC/0.1% SDS at 60°C for 45 min, and then autoradiographed. The intensity of the autoradiographic dots or bands was quantitated using the Cyclone Storage Phosphor System (Packard Instrument Company, Median, Conn.). All drug concentrations were tested in duplicate or triplicate, with antiviral effects being scored as the amount of HBV DNA present in the media relative to that in untreated controls.
Intracellular HBV replicative intermediates (RI) analysis: HepG2.2.15 cells were consecutively treated with various concentrations of lamivudine for 10 d. The cytoplasmic preparations containing HBV core particles were isolated from the treated cells. Cells were lysed with lysis buffer (50 mmol/L Tris-Cl, PH 7.4, 150 mmol/L NaCl, 5 mmol/L MgCl2, 0.5% NP-40) at room temperature for 5-10 min. The cytoplasmic fraction was separated from the nuclear fraction by centrifugation. Unprotected DNA was removed by adjusting cytoplasmic preparations so that they contained 10 mmol/L MgCl2 and 500 μg/mL of DNaseI(Roche, Germany) followed by a 1 h incubation at 37°C. To extract replicative intermediates (RI), EDTA, sodium dodecyl sulfate (SDS), NaCl and proteinase K (QIAGEN) were added separately and sequentially to final concentrations of 10 mmol/L EDTA, 1% SDS, 100 mmol/L NaCl and 500 mg/L of proteinase K. The sample was incubated for 1.5 h at 56°C and then subjected to sequential phenol and chloroform extraction and isopropanol precipitation. Precipitated nucleic acids were resuspended in a small volume of TE solution and digested with 100 mg/L of RNase (Roche, Germany) for 1 h at 37°C. Twenty micrograms of cytoplasmic preparations containing HBV replicative intermediates DNA (RI) were then analysed by electrophoresis in 1.2% agarose gels, followed by blotting onto Hybond-N+ membranes. The bolt was hybridized with a 32P-labeled HBV DNA probe (digested by NsiIfrom plasmids which contain full length HBV genome sequence dimer, and labelled with a RediprimeTM II Random prime labelling system), washed with 2 × SSC/0.1% SDS at room temperature for 20 min, twice, and 0.1 × SSC/0.1% SDS at 60°C for 45 min, and then autoradiographed as described above.
RNA extraction
Total RNA was isolated from cells using Trizol according to the manufacturer’s instructions. RNA quantity and quality was assessed by determination of the optical density at 260 and 280 nm using spectrophotometry and additional visualisation by agarose gel electrophoresis.
Gene expression profiling by customized cDNA macroarrays
Radiolabelled cDNA was generated from 20 μg total RNA by reverse transcription with SuperscriptII(Gibco, MD) in the presence of 32P-dCTP. Residual RNA was hydrolysed by alkaline treatment at 70°C for 20 min and the cDNA was purified using G-50 columns (Amersham Pharmacia, UK). Before hybridisation to the macroarrays the labelled cDNA was mixed with 50 μg COT-DNA (Gibco) and 10 μg Poly-A DNA (Sigma), denatured at 95°C for 5 min and hybridised for 1h to minimise non-specific binding. Preparation of the macroarrays (representing 150 known ISGs), hybridisation of the radioactive cDNAs and scanning and analysis of the macroarrays were carried out as described previously[15].
Northern blot analysis
5 μg of total RNA was electrophoresed through a 1.2% agarose gel containing formaldehyde and then transferred to Hybond-N+ membranes. The immobilized RNA was hybridized with a 32P-labeled DNA probe (IMAGE clones PCR products, purified with Gel Extract kit, QIAGEN).
Electrophoretic Mobility Shift Assay
At 80% to 90% confluence, cells were stimulated with IFN-α for 6 h. Preparations of nuclear extracts were performed according to the instruction of the manufacturer (PIERCE, NE-PERTM Nuclear Extraction Reagent). Nuclear extracts/DNA binding reactions were performed in 20 μL containing 15 μg nuclear extract protein and 4 μL Gel Shift Binding 5 × Buffer (20% glycerol, 5 mmol/L MgCl2, 2.5 mmol/L EDTA, 2.5 mmol/L DTT, 250 mmol/L Tris-Cl, PH 7.5, 0.25 mg/mL Poly (dI-dC)·Poly (dI-dC)). ISRE/GAS consensus oligonucleotides (5’-AAG TAC TTT CAG TTT CAT ATT ACT CTA-3’) from the promoter region of the IFN-α responsive genes were used. Mutant oligonucleotides (5’-AAG TAC TTT CAG TGG TCT ATT ACT CTA-3’) were used as control. The probes were end-labeled with γ-32P-ATP (U K, 3000 Ci/mol) at room temperature for 20 min. Complexes were separated from the probe in 4% naive poly-acrylamide gel in 0.5 × TBE buffer. The gels were subsequently dried and autoradiographed.
Western blot analysis
After interferon treatment, cells were washed once with ice-cold phosphate-buffered saline. Cells were lysed on ice for 30 min in 0.5 mL lysis buffer containing 50 mmol/L Tris, pH 8.0, 10% Glycerol, 0.5% NP40, 150 mmol/L NaCl, 1 mmol/L DTT, 1 mmol/L EDTA, 1 mmol/L Sodiumorthovanadate, 170 mg/L phenylmethylsulfonyl fluoride, 2 mg/L Aprotinin, 1 mg/L Leupeptin. Lysates were cleared by centrifugation in a microcentrifuge at high speed for 30 min at 4°C. Protein concentration of the supernatant was measured with Bradford reagent. Equal amounts (100 μg) of proteins were suspended in sodium-dodecyl sulphate (SDS)-sample buffer, boiled for 5 min and separated by electrophoresis (NuPAGE 4%-12% Bis-Tris Gel, Invitogen). The separated proteins were transferred to a polyvinylidene difluoride membrane (Hybond-PTM, Amersham Biosciences). After blocking for 1 h at room temperature in 10% non-fat dry milk in Tris-buffered saline with 0.1% Tween-20 (TBST) or 1% BSA for antibodies specific for phosphorylated epitopes, membranes were incubated with anti-p38, anti-pp38 (Santa Cruz), anti-Stat1, anti-Stat1(pY701) and anti-ERK1, anti-ERK1/2(pT202/pY204) (BD Biosciences) overnight at 4°C, and thereafter with horseradish peroxidase-conjugated anti-rabbit or anti-Mouse IgG (1:5000) (Amersham Biosciences) for 1 h at room temperature. The proteins were detected with enhanced chemiluminescence reagent (ECL, Amersham).
Southern blot analysis
Twenty micrograms of cytoplasmic preparations containing HBV replicative intermediates (RI) DNA were analysed by Southern blotting as above.
RESULTS
Differential expression of ISGs in HepG2.2.15 and HepG2 upon stimulation with IFN-α
Type 1 IFNs are known to induce an intracellular antiviral state against many viruses. Therefore, we developed a customized cDNA array methodology to study the expression of IFN stimulated genes (ISGs). At present, this system permits the analysis of several hundred genes of interest. A substantial spectrum of known ISGs is analysed with this macroarray (Table 1). The sensitivity of this method has also been assessed previously[15]. Conventionally, in most micro- and macroarray systems a 2-fold change in the expression level is regarded as being significant.
Table 1.
Complete list of genes investigated in this study
| Gene Name | Acc. No. | Gene Name | Acc. No. | Gene Name | Acc. No. |
| 101F6 | AA544950 | IFI 16 | M63838 | Mdm2 | Z12020 |
| 2-5 OAS | X02875 | IFI 41 | L22342 | MEN1 | U93237 |
| 2-5 OAS | D00068 | IFI 44 | D28915 | Met | AA410591 |
| 5' nucleotidase | X55740 | IFI 6-16 | BC015603 | Mig | X72755 |
| 60S Ribosomal protein L11 | U43522 | IFI27 | X67325 | MIP-1b/CCL4 | NM_002984 |
| 72 kDa type IV collagenase | J03210 | IFI4 | X79448 | MLK 2 | X90846 |
| 9-27 | J04164 | IFIT 1 | M24594 | MMP-1 | M13509 |
| ADAM-10 | AF009615 | IFIT4 | U72882 | MxA | M33882 |
| ADAM-17 | U69611 | IFIT4 | AF083470 | MxA | M33882 |
| akt-1 | NM_005163 | IFITM2 | X57351 | MxB | M30818 |
| akt-2 | M77198 | IFITM3 | X57352 | MxB | M30818 |
| Alpha-1-antiproteinase | K01396 | IFN omega 1 | X58822 | NCAM | M74387 |
| Alpha-crystallin | U05569 | IFN-AR1 | J03171 | NF-IL-6 | X52560 |
| ATF-2 | X15875 | IFN-AR2 | L42243 | NFkB | M58603 |
| Auto Ag SS-A/Ro | NM_003141 | IFN-g | M29383 | NKC-4 | M59807 |
| bad | U66879 | IFN-GR1 | J03143 | n-myc | Y00664 |
| BAK1 | X84213 | IFN-GR2 | U05875 | p19 | U40343 |
| BAX | U19599 | IFI 17 | J04164 | p48/ISGF3g | M87503 |
| Bax | L22474 | IFP 35 | U72882 | p53 | M14694 |
| bcl-2 | M14745 | IFP-53 | X62570 | p57Kip2 | U22398 |
| BRCA1 | U14680 | IFRG28 | AJ251832 | p70 S6 kinase | M60724 |
| BST2 | D28137 | ikBa | M69043 | PAI-1 | M16006 |
| BTG1 | X61123 | IL-1 α | M28983 | PCBP | M80563 |
| Calcyclin | J02763 | IL-10 | M57627 | PDGF-alpha | X06374 |
| Calretiulin | M84739 | IL-10 R α | U00672 | PDK1 | Y15056 |
| CASP | AJ006470 | IL-10 R β | Z17227 | PDK2 | NM_002611 |
| Caspase 7 | U67319 | IL-12R β | U64198 | Phosph. Scram. 1 | AF098642 |
| Caspase 8 | X98172 | IL13RA | U81379 | Phosph.glycerate kin. | V00572 |
| Caspase-1 | M87507 | IL13RA 2 | U70981 | Pi3-kinase | NM_006219 |
| Caspase-9 | U60521 | IL-15 | U14407 | PIAS x-beta | AF077954 |
| Cat. o-methyltransferase | M58525 | IL-15RA | U31628 | pig7 | AF010312 |
| CBFA | NM_004349 | IL-18 | D49950 | pim-1 | M16750 |
| CBP | U85962 | IL-18 bprot | AB019504 | PK R | AF072860 |
| CCR1 | L09230 | IL2 | U25676 | PKR | U50648 |
| CCR5 | U54994 | IL-2R α | K03122 | plectin (PLEC1) | U53204 |
| CD5 | X04391 | IL2RG | D11086 | PLOD2 | U84573 |
| cdk inhibitor p27KIP1 | U10909 | IL6 | X04602 | PML-1 | M79462 |
| C-fox | NM_005252 | IL-8 | M28130 | PPP3CA | L14778 |
| CG12-1 | AF070675 | IL8RB | L19593 | Pro. 4-hydroxyl. | M24486 |
| C-jun | J04111 | iNOS | L09210 | Prot.-ATPase-like pr. | D89052 |
| C-myc | L00058 | Int-6 | U62962 | PTEN | U96180 |
| C-myc | V00568 | Integrin β 7 | M62880 | pyridoxal kinase | U89606 |
| Collagen α1 (I) | Z74615 | integrin-β-6 | NM_000888 | raf (c-raf-1) | X03484 |
| Collagen α2 (I) | J03464 | IP-10 | X02530 | RAP46/Bag-1 | Z35491 |
| Collagen, type XVI, alpha 1 | M92642 | IP-30 | J03909 | RbAp48 | X74262 |
| Complement compound C1r | J04080 | IRF 1 | X14454 | Reticulocalbin | D42073 |
| COX17 | L77701 | IRF 4 | U52682 | RGS2 | NM_002923 |
| Cpp32 | NM_004346 | IRF 5 | U51127 | RHO | NM_000539 |
| CREB | NM_004379 | IRF-1 | L05072 | RHO GDP-dis.inh. 2 | L20688 |
| CTRL-1 | X71877 | IRF-2 | X15949 | RING 10 | NM_004159 |
| CXCR4 | AF005058 | Irf-7 | U73036 | RING4 | X57522 |
| Cyclin D1 | M64349 | ISG15 | AA406020 | Smad1 | U59423 |
| Cyp19 (aromata) | M28420 | ISG15 | M13755 | Smad2 | AF027964 |
| Cys-X-Cys,member 11 | AF030514 | ISG-56K | M24594 | Smad4 | U44378 |
| DEAD box binding protein 1 | AF077951 | KIAA0129 | D50919 | Smad5 | U73825 |
| DEAD-box protein p72 | U59321 | KIAA0235 | D87078 | Smad7 | AF015261 |
| Destrin | S65738 | KIAA0284 | AB006622 | SnoN | X15219 |
| DP (β1) | M83664 | LIPA | U04285 | SOCS 3/ssi-3 | AB004904 |
| DR-α | J00194 | LMP-2 | X66401 | SOCS 4/CIS 4 | AB006968 |
| E2F-1 | U47677 | L-selectin | M25280 | SOCS1 | N91935 |
| egr-1 | X52541 | Mad 4 | X03541 | SOCS-1 | NM_003745 |
| Elastase 2 | M34379 | MAP2K1 | NM_002755 | SOCS2 | AF020590 |
| ERM | X76184 | MAP2K1IP1 | NM_021970 | SOCS-3 | NM_003955 |
| F-actin capping protein | U56637 | MAP2K2 | L11285 | Stannin | NM_003498 |
| Farn. pyro. syn. | J05262 | MAP2K3 | NM_002756 | STAT 6 | U16031 |
| FAS/Apo-1 | M67454 | MAP2K4 | L36870 | STAT1 (91kDa) | M97935 |
| fas-ligand | U08137 | MAP2K5 | NM_002757 | STAT1 (91kDa) | M97935 |
| Fibronectin-1 | X02761 | MAP2K6 | U39657 | STAT2 | M97934 |
| FK506 binding protein 6 | AF038847 | MAP2K7 | AF022805 | STAT4 | L78440 |
| FKHRL1 | AF041336 | MAP3K1 | AF042838 | STAT5A | L41142 |
| Folate receptor | X62753 | MAP3K11 | NM_002419 | STAT5B | U47686 |
| gadd45 | M60974 | MAP3K14 | NM_003954 | Succinyl CoA Ligase | AF058953 |
| Galectin-1 | J04456 | MAP3K2 | NM_006609 | TAP1 (Ring4) | L21204 |
| Gamma actin | X04098 | MAP3K3 | U78876 | TFE3 | X96717 |
| Gamma2-adaptin (G2AD) | AF068706 | MAP3K4 | NM_005922 | TGF-bR1 | L11695 |
| GAPDH | X01677 | MAP3K5 | NM_005923 | TGF-bR2 | D50683 |
| GATA 3 | X58072 | MAP3K7 | NM_003188 | TGF-bR3 | L07594 |
| GBP-1 | M55542 | MAP4K1 | NM_007181 | TGIF | X89750 |
| GBP-2 | M55543 | MAP4K3 | NM_003618 | TIMP-1 | M59906 |
| Granzyme B | M17016 | MAPK10 | NM_002753 | TIMP-2 | J05593 |
| GSK3 | NM_002093 | MAPK11 | NM_002751 | TIMP-3 | U14394 |
| HCV-ass. p44 | D28915 | MAPK12 | NM_002969 | TIMP-4 | U76456 |
| HLA-A (MHCI Ag B27) | NM_002116 | MAPK13 | AF004709 | TNF-alpha | X01394 |
| HLA-E | X56841 | MAPK14 | NM_001315 | TRAF6 | U78798 |
| Homo sapins STAT | M97936 | MAPK3 | X60188 | Transferrin | M12530 |
| Hou | U32849 | MAPK6 | NM_002748 | Transthyretin | D00096 |
| HPAST protein | AF001434 | MAPK7 | NM_002749 | TRIP14 | L40387 |
| hsf1 (tcf5) | M64673 | MAPK8 | NM_002750 | trk oncogene | X03541 |
| hsp90 (CDw52) | X15183 | MAPK8IP2 | NM_012324 | TTF-2 | AF073771 |
| Hypoxia-ind. Factor-1 | U22431 | MAPK9 | U35003 | UBE2L6 | AA292074 |
| ICAM-1 | M24283 | MAPKAPK2 | NM_004759 | VCAM -1 | M30257 |
| ICSB 1 | M91196 | MAPKAPK3 | NM_004635 | VEGF-C | U43142 |
| IDO | M34455 | MCP-1/CCL2 | X14768 | Virpirin (Cig5) | AF026941 |
Genes of interest were selected from the UniGene database. These genes comprise known ISGs and genes of intrinsic interest which might or might not be induced by IFNs in different cell systems. They include genes involved in cell proliferation, immune responses and the responses to a variety of cytokines. 5’ IMAGE clones with 0.5-0.8 kb length were chosen and obtained from RZPD, Berlin, Germany.
In the established hepatoma cell line, hepG2.2.15 with stably transfected HBV genomes[16], ISG expression was examined using the cDNA macroarrays (Table 2). While many ISGs, e.g., 2-5 OAS, IFI 17, and RING4, were normally stimulated by IFN-α, several other ISGs were expressed at a lower level compared with the ISG expression in HepG2 cells. The induction of 2 ISGs, MxA and Cig5, was completely inhibited in HepG2.2.15 cells, while a partial inhibition was observed for 4 ISGs, IFITM1, IFITM2, IFITM3, and 6-16 (Table 1, Figure 1). Thus, only a subgroup of ISGs was down regulated in HepG2.2.15.
Table 2.
Suppression of ISG induction in HepG2.2.15 cells, effect of lamivudine treatment
| Gene | Acc. No. | HepG2 | 2.2.15 | HepG2 /lam | 2.2.15 /lam |
| I complete inhibition | |||||
| MxA | M33882 | 5.9 | 0.6 | 4.0 | 0.8 |
| cig5 | AF026941 | 2.2 | 0.9 | 2.1 | 1.2 |
| II partial inhibition | |||||
| IFITM3 | X57352 | 3.4 | 1.6 | 3.4 | 1.8 |
| IFITM2 | X57351 | 2.7 | 1.6 | 2.2 | 1.8 |
| III reversible inhibition | |||||
| IFI 6-16 | BC015603 | 7.9 | 4.4 | 7.2 | 6.5 |
| IFITM1 | M24594 | 4.9 | 2.5 | 4.8 | 4.6 |
| IV no inhibition | |||||
| 2-5OAS | D00068 | 3.8 | 4.0 | 4.4 | 5.8 |
| MxB | M30818 | 1.4 | 2.0 | 1.4 | 1.9 |
| Caspase 7 | U67319 | 2.3 | 2.4 | 2.2 | 2.1 |
| IFI 17 | J04164 | 3.3 | 2.9 | 2.8 | 2.7 |
| IFI 27 | X67325 | 1.9 | 2.1 | 1.8 | 2.2 |
| IFI T4 | U72882 | 2.7 | 1.9 | 2.6 | 1.8 |
| RING4 | X57522 | 2.4 | 2.3 | 2.5 | 3.5 |
Cells were stimulated with 100 U/mL IFN-α for 6 h with or without pre-treatment with 2 μmol/L lamivudine for 10 d. Then, RNA was isolated and assayed by cDNA macroarray. Data are shown as fold induction compared to the untreated control. Lam: Lamivudine.
Figure 1.
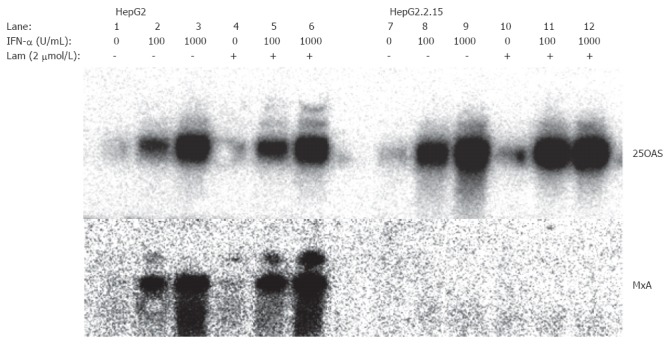
Northern blot analysis of ISG expression and its modulation by lamivudine in HepG2 and HepG2.2.15 cells. HepG2 and HepG2.2.15 cells were cultured in the absence or presence of 2 μmol/L of lamivudine for 10 d. Then, the cells were stimulated with 100 or 1000 IU/mL of IFN-α for 6 h. Total cellular RNAs were isolated for Northern blotting hybridization. Lam: lamivudine.
Analysis of the IFN response in HepG2.2.15 and HepG2 cells
The results above suggested that the IFN-signalling pathway is only partially inhibited in HepG2.2.15. Western blotting and EMSA and were carried out to analyse Stat1 activation and ISGF3 formation in HepG2 and HepG2.2.15 cells. The phosphorylated form of Stat1 was detected by western blot in IFN-α treated cells (Figure 2). The phosphorylation of Stat1 was enhanced in HepG2.2.15, compared with HepG2. Furthermore, Figure 3 showed that the formation of ISGF3 in HepG2.2.15 cells occurred after IFN-α stimulation, as occured in HepG2 cells. These data clearly show that the IFN-signalling pathway is generally not blocked in HepG2.2.15 cells. The results consistently show that both steps were evenly enhanced in HepG2.2.15. In addition, activation of ERK and p38 MAPKinase was not altered in HepG2.2.15 cells (data not shown).
Figure 2.
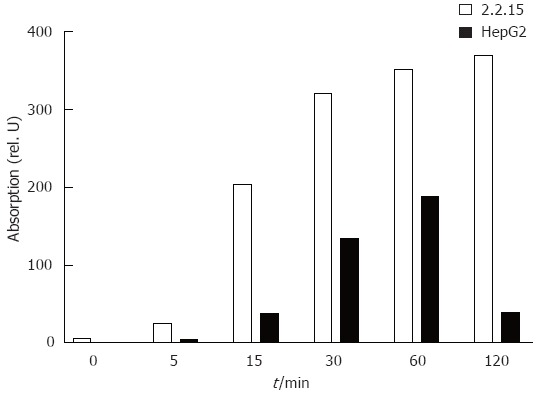
Analysis of Stat1 phosphorylation after IFN-α stimulation in HepG2 and HepG2.2.15 cells. Cells were stimulated with 100 U/mL of IFN-α for the indicated time points. Then, nuclear proteins were extracted and analysed by western blot. Data were quantified using Imagequant and are shown as relative units.
Figure 3.
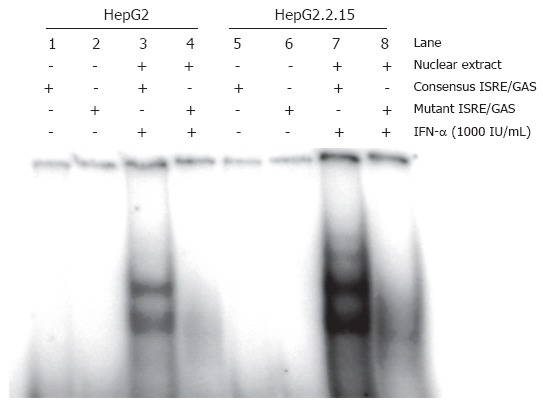
Analysis of ISGF3 formation after IFN-α stimulation in HepG2 and HepG2.2.15 cells. HepG2 and HepG2.2.15 cells were stimulated with 1000 IU/mL of IFN-α for 6 h followed by isolation of nuclear extracts (NE-PERTM reagent kits) for EMSA analysis.
Reduction of the production of HBV proteins and the HBV replication by lamivudine treatment
The difference in ISG expression in HepG2 and HepG2.2.15 cell lines may be partly due to the presence of HBV replication in the later one. Consequently, the ISG expression in HepG2.2.15 would change if the HBV gene expression or replication is suppressed. To test this hypothesis, we determined the optimal condition to reduce HBV gene expression and replication using the nucleoside analogue lamivudine. HepG2.2.15 cells were treated with lamivudine at various concentrations from 0.04 μmol/L to 100 μmol/L. The antiviral activity was determined by quantitation of secreted HBsAg and HBeAg particles, extracellular virions and intracellular HBV replicative intermediates (RI). Figure 4A shows that treatment with lamivudine led to a significant reduction of secreted HBsAg and HBeAg in the supernatant of HepG2.2.15 cells. Parallel to the reduction of HBsAg and HBeAg production, the extracellular virion DNA in the culture supernatant of HepG2.2.15 cells and intracellular HBV replicative intermediates (RI) decreased after treatment with 2 μmol/L or 20 μmol/L of lamivudine for 10 d (Figure 4B and C). Maximal levels of suppression of HBV were observed after 10 d of lamivudine treatment. At that time, levels of RI were not more than 1.5% of controls in cultures of the 2 μmol/L treatment group. Based on these results, we chose a concentration of 2 μmol/L and duration of 10 d to suppress HBV replication in our system to study the modulatory effects of lamivudine on the IFN-response.
Figure 4.
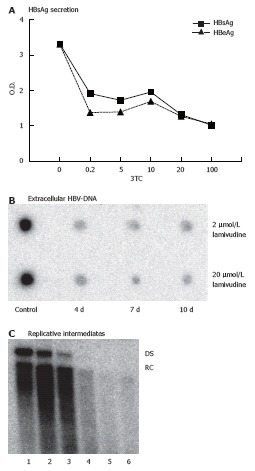
Antiviral effects of lamivudine in HepG2.2.15 cells. A: HepG2.2.15 cells were cultivated with various concentrations (0 to 100 μmol/L) of 3 TC for 10 d. Then, supernatants were harvested and assayed for the presence of HBsAg and HBeAg by ELISA; B: HepG2.2.15 cells were treated with 2 or 20 μmol/L of lamivudine for 4, 7 and 10 d, respectively. Then, supernatants were collected and extracellular HBV-DNA was analyzed by dot blot hybridization; C: HepG2.2.15 cells were treated with various concentrations of lamivudine for 10 d. Then, intracellular HBV replicative intermediates were isolated for southern blotting. Lane 1: control, lane 2: 0.04 μmol/L, lane 3: 0.2 μmol/L, lane 4: 5 μmol/L, lane 5: 25 μmol/L, lane 6: 125 μmol/L, RC, relaxed circular HBV-DNA; DS, double stranded linear HBV-DNA.
The IFN response in HepG2.2.15 and HepG2 cells after lamivudine treatment
The effect of lamivudine on ISG expression in HepG2.2.15 and HepG2 was investigated by using gene macroarrays. No effect was observed for the stimulation of MxA and Cig5 expression by lamivudine treatment (Table 2). Both genes did not respond with an increased expression upon IFN-α stimulation. An increase of the IFN-α concentration to 1000 units per mL or a prolonged incubation with IFN-α did not change the expression of MxA and cig5. The reduced induction of IFITM 2 and IFITM 3 expression could not be enhanced by lamivudine treatment. In contrast, IFITM1 and 6-16 expression could be restored by lamivudine treatment of HepG2.2.15 cells (Table 1, Figure 1). This indicates that lamivudine can only partially normalize the IFN-response in HBV-transfected HepG2.2.15 cells at concentrations that profoundly inhibit viral replication and secretion of viral particles. Lamivudine had no effect on ISG expression in HepG2 cells and did not enhance the induction of many other ISGs, such as 2.5 OAS and MxB.
DISCUSSION
In the present work, we found that HepG2.2.15 and HepG2 respond differently to IFN-α. Several ISGs were not induced in HepG2.2.15 while they were expressed in HepG2 cells after IFN-α. There may be multiple reasons for the different ISG expression profiles in these cell lines, though HepG2.2.15 was derived from HepG2[16]. Previous data indicated that the expression of the IFN-inducible gene MxA was specifically inhibited by HBV proteins in HBV-transfected HepG2 or HuH7 cells[13], and this was accompanied by diminished antiviral activity of IFN[17]. In our study, we confirmed this finding with MxA expression being completely diminished in HBV-transfected HepG2.2.15 cells. In addition, we showed that additional ISG (Cig5, IFITM1, -2, -3 and 6-16) expression was completely abolished or partially reduced by HBV. The majority of ISGs, however, are expressed and inducible in both HepG2 and HepG2.2.15 cells, indicating that the HBV gene expression and replication had no effect on these ISGs. Consistently, Rosmorduc et al[17] demonstrated that 2´5OAS expression is not affected by HBV. Our results support the view that the HBV-mediated inhibition of the IFN-response, if any, represents a specific rather than global effect. The Stat1 activation or ISGF3 formation in HepG2.2.15 cells appeared to be normal, indicating that the Jak/Stat signalling pathway is intact and functional. These findings are corroborated by the data from Fernandez et al[13] who demonstrated that the inhibition of MxA induction in HepG2 cells occurs at the promoter level.
We then asked the question whether the HBV-mediated suppressive effect on the IFN-response could be reverted by treatment with the nucleoside analogue lamivudine, which is an effective inhibitor of HBV replication in vitro[18] and in vivo[19,20]. Lamivudine is phosphorylated within the cell and then incorporated into nascent viral DNA by the HBV polymerase during replication[21] resulting in the termination of HBV DNA elongation. Lamivudine also inhibits reverse transcriptase activity directly through competitive inhibition. Although some reports indicate that lamivudine exerts synergistic effects with IFN, the underlying mechanisms are not clear[22,23]. To answer this question we first established the optimal conditions for in vitro treatment of HepG2.2.15 cells with lamivudine. The results indicated that lamivudine exerted potent antiviral activities in our system as it strongly suppressed the formation of HBV replicative intermediates and extracellular HBV DNA at concentrations that correspond well to plasma levels found in patients that are treated with this drug. However, HBsAg and HBeAg secretion was only down regulated and not completely blocked. After treatment with lamivudine for 10 d, the induction of IFITM1 and 6-16 expression could be enhanced while MxA, Cig5, IFITM2 and IFITM3 induction remained unchanged. This indicates that lamivudine can at least partially improve the impaired IFN response in HBV-transfected cells. IFITM 1 to 3 and 6-16 belong to a family called small ISGs[24]. IFITM 1 to 3 are classified as members of the 1-8 group while 6-16 is a member of the ISG12 group. These genes were under the control of multiple elements responding to IFN-α stimulation including ISGF3 and interferon. It is likely that the lamivudine treatment partially reduces HBV gene expression and therefore contributes to the improved ISG expression. On the other hand, the continuing HBV protein production may still dominantly interfere with the expression of many ISGs, such as cig5 and IFITM3.
These findings are corroborated by our study that shows an improved IFN response of PBMC from HBV patients after treatment with adefovir. Some reports have also suggested a restoration of weak T helper cell and CTL responses after initiation of lamivudine therapy[25,26]. Although it is certainly a possibility, it still remains to be determined whether this effect can be explained by an enhanced responsiveness to IFNs.
In conclusion, our results suggest that HBV specifically modulates the IFN response in HepG2 cells by a selective suppression of certain ISGs. This suppression is at least partially reversible by antiviral treatment with the nucleoside analogue lamivudine.
Footnotes
Supported by grants from the Deutsche Forschungsgemeinschaft (DFG SCHL 377/2-2, LU 669/2-1 and GRK 1045/1)
S- Editor Wang J L- Editor Lutze M E- Editor Bai SH
References
- 1.Hadziyannis SJ, Papatheodoridis GV, Vassilopoulos D. Treatment of HBeAg-negative chronic hepatitis B. Semin Liver Dis. 2003;23:81–88. doi: 10.1055/s-2003-37584. [DOI] [PubMed] [Google Scholar]
- 2.Heathcote J. Treatment of HBe antigen-positive chronic hepatitis B. Semin Liver Dis. 2003;23:69–80. doi: 10.1055/s-2003-37588. [DOI] [PubMed] [Google Scholar]
- 3.Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, Dienstag JL, Heathcote EJ, Little NR, Griffiths DA, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714–1722. doi: 10.1053/j.gastro.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 4.Guidotti LG, Ando K, Hobbs MV, Ishikawa T, Runkel L, Schreiber RD, Chisari FV. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc Natl Acad Sci USA. 1994;91:3764–3768. doi: 10.1073/pnas.91.9.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guidotti LG, Guilhot S, Chisari FV. Interleukin-2 and alpha/beta interferon down-regulate hepatitis B virus gene expression in vivo by tumor necrosis factor-dependent and -independent pathways. J Virol. 1994;68:1265–1270. doi: 10.1128/jvi.68.3.1265-1270.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wieland SF, Guidotti LG, Chisari FV. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J Virol. 2000;74:4165–4173. doi: 10.1128/jvi.74.9.4165-4173.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pasquetto V, Wieland SF, Uprichard SL, Tripodi M, Chisari FV. Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J Virol. 2002;76:5646–5653. doi: 10.1128/JVI.76.11.5646-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordien E, Rosmorduc O, Peltekian C, Garreau F, Bréchot C, Kremsdorf D. Inhibition of hepatitis B virus replication by the interferon-inducible MxA protein. J Virol. 2001;75:2684–2691. doi: 10.1128/JVI.75.6.2684-2691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robek MD, Boyd BS, Wieland SF, Chisari FV. Signal transduction pathways that inhibit hepatitis B virus replication. Proc Natl Acad Sci USA. 2004;101:1743–1747. doi: 10.1073/pnas.0308340100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robek MD, Wieland SF, Chisari FV. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J Virol. 2002;76:3570–3574. doi: 10.1128/JVI.76.7.3570-3574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wieland SF, Vega RG, Müller R, Evans CF, Hilbush B, Guidotti LG, Sutcliffe JG, Schultz PG, Chisari FV. Searching for interferon-induced genes that inhibit hepatitis B virus replication in transgenic mouse hepatocytes. J Virol. 2003;77:1227–1236. doi: 10.1128/JVI.77.2.1227-1236.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Protzer U, Hu Z, Jacob J, Liang TJ. Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J Virol. 2004;78:4566–4572. doi: 10.1128/JVI.78.9.4566-4572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernández M, Quiroga JA, Carreño V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J Gen Virol. 2003;84:2073–2082. doi: 10.1099/vir.0.18966-0. [DOI] [PubMed] [Google Scholar]
- 14.Guidotti LG, Morris A, Mendez H, Koch R, Silverman RH, Williams BR, Chisari FV. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J Virol. 2002;76:2617–2621. doi: 10.1128/JVI.76.6.2617-2621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlaak JF, Hilkens CM, Costa-Pereira AP, Strobl B, Aberger F, Frischauf AM, Kerr IM. Cell-type and donor-specific transcriptional responses to interferon-alpha. Use of customized gene arrays. J Biol Chem. 2002;277:49428–49437. doi: 10.1074/jbc.M205571200. [DOI] [PubMed] [Google Scholar]
- 16.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosmorduc O, Sirma H, Soussan P, Gordien E, Lebon P, Horisberger M, Bréchot C, Kremsdorf D. Inhibition of interferon-inducible MxA protein expression by hepatitis B virus capsid protein. J Gen Virol. 1999;80(Pt 5):1253–1262. doi: 10.1099/0022-1317-80-5-1253. [DOI] [PubMed] [Google Scholar]
- 18.Doong SL, Tsai CH, Schinazi RF, Liotta DC, Cheng YC. Inhibition of the replication of hepatitis B virus in vitro by 2',3'-dideoxy-3'-thiacytidine and related analogues. Proc Natl Acad Sci USA. 1991;88:8495–8499. doi: 10.1073/pnas.88.19.8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nevens F, Main J, Honkoop P, Tyrrell DL, Barber J, Sullivan MT, Fevery J, De Man RA, Thomas HC. Lamivudine therapy for chronic hepatitis B: a six-month randomized dose-ranging study. Gastroenterology. 1997;113:1258–1263. doi: 10.1053/gast.1997.v113.pm9322520. [DOI] [PubMed] [Google Scholar]
- 20.Honkoop P, de Man RA, Zondervan PE, Schalm SW. Histological improvement in patients with chronic hepatitis B virus infection treated with lamivudine. Liver. 1997;17:103–106. doi: 10.1111/j.1600-0676.1997.tb00789.x. [DOI] [PubMed] [Google Scholar]
- 21.Cammack N, Rouse P, Marr CL, Reid PJ, Boehme RE, Coates JA, Penn CR, Cameron JM. Cellular metabolism of (-) enantiomeric 2'-deoxy-3'-thiacytidine. Biochem Pharmacol. 1992;43:2059–2064. doi: 10.1016/0006-2952(92)90162-c. [DOI] [PubMed] [Google Scholar]
- 22.Korba BE, Cote P, Hornbuckle W, Schinazi R, Gangemi JD, Tennant BC, Gerin JL. Enhanced antiviral benefit of combination therapy with lamivudine and alpha interferon against WHV replication in chronic carrier woodchucks. Antivir Ther. 2000;5:95–104. [PubMed] [Google Scholar]
- 23.Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, Simon C, So TM, Gerken G, de Man RA, Niesters HG, Zondervan P, Hansen B, Schalm SW. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet. 2005;365:123–129. doi: 10.1016/S0140-6736(05)17701-0. [DOI] [PubMed] [Google Scholar]
- 24.Martensen PM, Justesen J. Small ISGs coming forward. J Interferon Cytokine Res. 2004;24:1–19. doi: 10.1089/107999004772719864. [DOI] [PubMed] [Google Scholar]
- 25.Boni C, Bertoletti A, Penna A, Cavalli A, Pilli M, Urbani S, Scognamiglio P, Boehme R, Panebianco R, Fiaccadori F, et al. Lamivudine treatment can restore T cell responsiveness in chronic hepatitis B. J Clin Invest. 1998;102:968–975. doi: 10.1172/JCI3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boni C, Penna A, Ogg GS, Bertoletti A, Pilli M, Cavallo C, Cavalli A, Urbani S, Boehme R, Panebianco R, et al. Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology. 2001;33:963–971. doi: 10.1053/jhep.2001.23045. [DOI] [PubMed] [Google Scholar]