

Abstract
AIM: To assess the effect of iron reduction after phlebotomy in rats with “normal” hepatic iron concentration (HIC) on the progression of hepatic fibrosis, as a result of bile duct ligation (BDL).
METHODS: Rats underwent phlebotomy before or after sham operation or BDL. Animals undergone only BDL or sham operation served as controls. Two weeks after surgery, indices of hepatic damage and fibrosis were evaluated.
RESULTS: Phlebotomy lowered HIC. Phlebotomy after BDL was associated with body weight increase, lower hepatic weight, less portal hypertension, less periportal necrosis, less portal inflammation, lower hepatic activity index score and higher albumin levels. On the other hand, phlebotomy before BDL was associated with body weight decrease and hepatic activity index score increase. Phlebotomy after sham operation was not associated with any hepatic or systemic adverse effects.
CONCLUSION: Reduction of HIC after induction of liver damage may have beneficial effects in BDL rats. However, iron deficiency could induce impairment of liver function and may make the liver more susceptible to insults like BDL.
Keywords: Iron, Phlebotomy, Bile duct ligation, Hepatic activity index, Rat.
INTRODUCTION
Chronic liver injury due to various causes may result in hepatic fibrosis, which can lead to cirrhosis and end-stage liver failure[1]. Currently, there are no proven therapeutic options designed to delay or reverse the progression of hepatic fibrosis. Iron overload is a relatively common finding in many patients with a variety of end-stage liver diseases, who have no evidence of genetic hemochromatosi[2,3]. In such patients the presence of hepatic siderosis is associated with more advanced liver dysfunction, more rapid decompensation and decreased survival[4].
The pathogenesis of hepatic fibrosis in patients with iron overload is not well understood. Four possible mechanisms have been suggested. (1) Iron per se is as an inducer of fibrosis even in the absence of necrosis and inflammation, and may act as a profibrogenic agent that stimulates the deposition of collagen by hepatic stellate cells[5]. (2) Iron, as a mediator of hepatocellular necrosis and local inflammation, may activate the peroxidative process, produce oxygen-free radicals, lipid peroxidation damage to protein and DNA and stimulation of fibrosis[6-11]. (3) Iron, as an inducer of fibrosis, may act in conjunction with other hepatotoxins[6]. It has been suggested that mild to moderate hepatic iron concentration (HIC) increase in patients and animals with various liver diseases, like non-alcoholic steatohepatitis, hepatitis C virus (HCV) infection and alcoholic liver disease, may exacerbate liver injury and accelerate the development of hepatic fibrosis[12-20]. Moreover in HCV positive patients undergoing liver transplantation, HIC increase in the donor liver (graft) is associated with fibrosis progression[21]. (4) Iron overload may alter hepatic extracellular matrix degradation[22]. In patients with hemochromatosis, the ratio between serum concentration of matrix metalloproteinases (MMP’s) 1-3 and tissue inhibitor of metalloproteinase 1 is reduced as compared to controls. Iron reduction therapy causes increase in MMP’s 2 and 3 serum concentration[22].
In the liver, iron is present in a few forms: ferritin, hemosiderin, heme and as low molecular weight iron (LMWI). It is thought that part of LMWI bound to weak chelators like ATP and AMP, is readily available to induce tissue damage[23]. This fraction of iron is probably the most susceptible to reduction after iron reduction therapy.
Iron depletion has been reported to improve metabolic derangement in patients with non-alcoholic fatty liver disease, liver damage and response to drug therapy in patients with chronic HCV infection[13,24-27]. Menstruating women with chronic HCV infection who are iron deficient have lower aminotransferase levels and improved liver histology when compared to age-matched menstruating females who are not iron deficient[28]. The possibility of altering the natural history and clinical course of HCV infection and to improve the response to anti-viral therapy (interferon monotherapy) has led to the recommendation to consider iron reduction therapy (phlebotomy) in patients with HCV infection as an adjuvant therapy to the other therapeutic modalities, even if HIC is normal or only mildly elevated[29,30]. Plebotomy is safe in most HCV-infected patients who are usually not iron deficient[31]. In HCV-infected patients in whom ribavirin is combined with interferon, HIC does not influence the response to therapy[32]. However, it is not yet known whether phlebotomy may improve the sustained virological response or histological score in these patients. Patients with chronic cholestatic liver disease do not usually have hepatic iron overload[4]. Moreover, no information exists as to the effect of iron reduction in subjects with cholestatic liver diseases with no increase (“normal levels”) of HIC.
The aim of the present work was to assess the effect of iron reduction after phlebotomy on the HIC in rats with “normal” iron stores and to evaluate whether reduction of HIC may decrease the hepatic damage and slow the progression of hepatic fibrosis induced by bile duct ligation (BDL).
MATERIALS AND METHODS
Forty male Sprague Dawley rats (Harlan, Israel), weighing 180-223 g, were studied. Rats were housed in regular cages situated in an animal room at 24 ºC with a 12-h light-dark cycle. Rats were maintained on standard rat chow (Kofflok, Tel Aviv, Israel) and with free access to tap water. The Ethics Review Committee for Animal Experimentation of the Hebrew University Hadassah Medical School approved all the animal studies described herein. BDL was performed as previously described[33]. Under ketamine anesthesia (100 mg/kg, intramuscular) a median laparotomy was done, the common bile duct was exposed, doubly ligated with 3/0 silk and sectioned between ligatures. The abdominal cavity was closed with two suture lines. In sham operated (SO) rats the portal area and liver hilum were exposed, the common bile duct was isolated and gently mobilized.
Three groups of animals were studied: (1) Control group of BDL (n = 9) and SO (n = 5) rats; (2) Post-procedure phlebotomy group of BDL (n = 8) and SO (n = 4) rats. These rats were phlebotomized on a daily basis for five consecutive days, from the fourth to the ninth post-operative days. Phlebotomy was done through a tail vein with 21-gauge needles under light ether anesthesia; (3) Pre-procedure phlebotomy group of BDL (n = 8) and SO (n = 6) rats. These rats were phlebotomized on a daily basis for five consecutive days from the ninth to the fourth days before the surgical procedure (BDL or sham operation).
Two weeks after the surgical procedure rats were anesthetized with ether. The abdominal cavity was opened by midline incision, and blood was drawn from the inferior vena cava, centrifuged, aliquoted and frozen. Serum albumin, bilirubin, calcium, phosphor, liver enzymes, kidney function tests and electrolytes were measured by the dry chemistry method (Kodak, Rochester, NY, USA). Serum iron (SI) as determined by Guanidine/FerroZine method (Cobas Integra 700). Transferin was determined using the immunoturbidimetric method (Cobas Integra 700). The liver and spleen were removed and weighed. Sections from the liver of each animal included in the study were taken for histological evaluation, determination of HIC, myeloperoxidase (MPO) activity, prostaglandin E2 (PGE2) generation and hydroxyproline (HP) content.
Liver histology
Sections from the liver of each animal were fixed in phosphate-buffered formaldehyde, embedded in paraffin, and 5-μm thick sections were prepared. Sections were stained with hematoxylin and eosin for evaluation of necroinflammatory grading and Masson’s trichrome stains for fibrosis and architectural changes. Histological grading of BDL-induced liver damage was assessed by a modification of the Knodell score[34]. Each specimen was examined for the following features and scored by points of increment from 0-4, according to the severity of the findings as follows: periportal necrosis: none = 0, mild = 1, moderate = 2, marked = 3, severe = 4; portal inflammation: none = 0, mild = 1, moderate = 2, marked = 3, severe = 4; lobular necrosis: none = 0, mild = 1, moderate = 2, marked = 3, severe = 4; fibrosis: none = 0, portal expansion = 1, septal formation = 2, marked bridging fibrosis = 3, cirrhosis = 4. Hepatic activity index (HAI) was calculated for each rat by summation of the points of these four parameters. In addition, bile duct proliferation was assessed in each specimen by allocating points of increment from 0-4, according to the severity of the findings. Histological evaluation of the pathological specimens was done on a blind basis by our pathologist. Reliable correlation was seen in repeated and blinded histological evaluation of the specimens.
Determination of PGE2
One hundred and fifty mg of liver tissue was placed in preweighed tubes containing 1.0 mL of phosphate buffer (50 mmol/L, pH 7.4). The liver was minced with scissors and centrifuged in an Eppendorf centrifuge for 10 s. The pellet was resuspended in 1.0 mL of the above buffer, incubated for 1 min in a vortex mixer. Indomethacin (10 μg) was added and the tubes were centrifuged for 60 s. The supernatant was kept at -20 ºC until radioimmuno-assay was performed. The ability of liver tissue to generate PGE2 was expressed as ng/g wet tissue weight. PGE2 generation was determined by radioimmunoassay as previously described[33].
Determination of MPO activity
Liver tissue (200 mg) was homogenized three times for 30 s each time at 4 ºC with a polytron (Kinematica GmbH, Krierz-Lucern, Switzerland) in 1.0 mL of ice-cold 0.5% hexadecyltrimethylammonium bromide in 50 mmol/L phosphate buffer, pH 6.0. The polytron probe was rinsed twice with 1.0 mL of the buffer and the washing was added to the homogenate. The homogenate was then sonicated for 10 s, freeze-thawed three times and centrifuged for 15 min at 40 000 r/min. An aliquot of the supernatant was taken for determination of enzyme activity according to Bradley et al [35]. The correlation coefficient based on 10 standard curves was r = 0.98 and inter assay variation was 0±1.65% (mean ± SE, n = 57).
Determination of hepatic iron content
Hepatic non-heme iron concentration was measured by the method of Torrance and Bothwell[36].
Determination of hepatic hydroxyproline content
Hepatic hydroxyproline content was measured by the method described by Woessner[37].
Syatistical analysis
Data were expressed as mean ± SE. Group variance was analyzed by the Kruskal-Wallis test, followed by the Mann-Whitney test for multiple comparisons to allow pair-wise testing for significant differences between groups. P ≤ 0.05 was considered statistically significant.
RESULTS
Effect of bile duct ligation
Two weeks after BDLall rats were jaundiced, had lesser weight gain, greater liver and spleen weight than SO rats (Table 1). BDL rats also had higher serum levels of liver enzymes, higher bilirubin and lower albumin levels, higher fibrosis score, higher hepatic HP content, higher hepatic activity index and higher bile duct proliferation score than SO rats (Table 2, Tables 3 and 4 and Figure 1). No change in HIC and content or SI and transferrin were observed as a result of BDL (Table 5 and Table 6).
Table 1.
Weights and phlebotomy volume in different groups of rats (mean±SE)
| Subgroup | n | Weight gain (% from baseline) | Mean phlebotomy Volume1(mL) | Liver weight (% of total body weight) | Splenic weight(% of total body weight) |
| Sham only | 5 | 15.03 ± 18.70a | 0 ± 0 | 3.97 ± 0.16a | 0.41 ± 0.07a |
| Sham and phlebotomy later | 4 | 9.4 ± 7.0 | 0.70 ± 0.13 | 3.62 ± 0.32 | 0.37 ± 0.04 |
| Phlebotomy and sham later | 6 | -5.7 ± 17.3 | 0.86 ± 0.18 | 3.47 ± 0.19 | 0.38 ± 0.03 |
| BDL only | 9 | -4.99 ± 3.90 | 0 ± 0 | 6.89 ± 1.29 | 0.66 ± 0.10 |
| BDL and phlebotomy later | 8 | 8.01 ± 3.1a | 0.71 ± 0.16 | 5.97 ± 0.45 | 0.56 ± 0.07 |
| Phlebotomy and BDL later | 8 | -3.29 ± 20.1 | 0.80 ± 0.15 | 7.02 ± 1.68 | 0.69 ±0.13 |
Mean phlebotomy volume for one phlebotomy session per rat;
P ≤ 0.01 vs BDL only.
Table 2.
Liver function tests and enzymes in different groups of rats (mean±SE)
| Subgroup | n | GGT (IU/L) | ALK- P (IU/L) | Bilirubin (µmol/L) | GPT (IU/L) | Albumin (G/L) |
| Sham only | 5 | 7.0 ± 2.01 | 254 ± 321 | 4.6 ± 1.81 | 53 ± 112 | 28.0 ± 1.11 |
| Sham and phlebotomy later | 4 | 6.0 ± 1.4 | 305 ± 58 | 4.0 ± 0.8 | 66 ± 8 | 29.2 ± 2.2 |
| Phlebotomy and sham later | 6 | 14.3 ± 10.3 | 305 ± 61 | 41.7 ± 39.0 | 55 ± 20 | 28.4 ± 2.3 |
| BDL only | 9 | 74.0 ± 63.4 | 580 ± 174 | 106.0 ± 57.0 | 115 ± 50 | 24.3 ± 2.2 |
| BDL and phlebotomy later | 8 | 65.9 ± 17.9 | 578 ± 75 | 104.0 ± 16.2 | 125 ± 22 | 29.0 ± 2.01 |
| Phlebotomy and BDL later | 8 | 64.3 ± 17.6 | 665 ± 166 | 109.0 ± 39.0 | 144 ± 40 | 23.7 ± 2.1 |
P ≤ 0.007,
P ≤ 0.02 vs BDL only GGT = Gamma-glutamyl transpeptidase, ALK-P = Alkaline phosphatase, GPT = Alanine aminotransferase. No difference in the levels of serum parameters like calcium, phosphor, urea, creatinine and electrolytes were found between the different groups (data not shown).
Table 3.
Histological grading and staging and bile duct proliferation score in different groups of rats (mean±SE)
| Subgroup | n | Periportal necrosis | Portal inflammation | Lobular necrosis | Fibrosis | Bile duct proliferation |
| Sham only | 5 | 0.00 ± 0.00 | 0.40 ± 0.55 | 1.00 ± 1.22 | 0.00 ± 0.001 | 0.00 ± 0.001 |
| Sham and phlebotomy later | 4 | 0.00 ± 0.00 | 1.25 ± 1.26 | 0.25 ± 0.50 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Phlebotomy and sham later | 6 | 0.00 ± 0.00 | 1.00 ± 0.00 | 0.50 ± 0.55 | 0.67 ± 0.52 | 0.00 ± 0.00 |
| BDL only | 9 | 0.89 ± 1.27 | 1.33 ± 1.00 | 2.11 ± 1.83 | 2.89 ± 0.78 | 2.67 ± 0.50 |
| BDL and phlebotomy later | 8 | 0.13 ± 0.35 | 1.00 ± 0.00 | 3.00 ± 1.85 | 2.75 ± 0.71 | 2.88 ± 0.35 |
| Phlebotomy and BDL later | 8 | 2.17 ± 1.33 | 2.67 ± 0.82 | 4.00 ± 0.00 | 3.33 ± 0.52 | 2.83 ± 0.41 |
P ≤ 0.003 vs BDL only.
Table 4.
Hepatic prostaglandin E2 (PGE2) generation, myeloperoxidase (MPO) activity and hydroxyproline (HP) content in different groups of rats (mean±SE)
| Subgroup | n | PGE2 generation (ng/g liver) | HP content (ng/g liver) | MPO activity (units/g liver) |
| Sham only | 5 | 14.8 ± 9.9 | 8.59 ± 0.551 | 0.45 ± 0.14 |
| Sham and phlebotomy later | 4 | 14.7 ± 5.2 | 8.11 ± 1.03 | 0.31 ± 0.12 |
| Phlebotomy and sham later | 6 | 6.9 ± 3.2 | 8.02 ± 1.00 | 0.18 ± 0.05 |
| BDL only | 9 | 8.5 ± 4.5 | 11.00 ± 0.97 | 0.59 ± 0.21 |
| BDL and phlebotomy later | 8 | 3.9 ± 2.11 | 9.80 ± 1.68 | 0.52 ± 0.10 |
| Phlebotomy and BDL later | 8 | 3.4 ± 2.31 | 11.90 ± 1.40 | 0.27 ± 0.171 |
P ≤ 0.018 vs BDL only.
Figure 1.
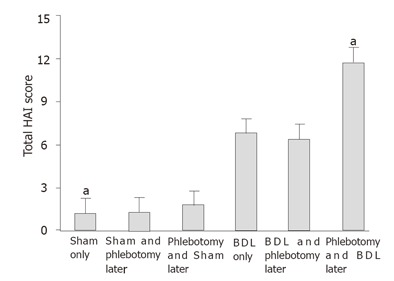
Hepatic activity index (HAI) in the study groups; aP < 0.02 vs BDL only.
Table 5.
Hepatic iron content and concentration in different groups of rats (mean±SE)
| Subgroup | n | Iron concentration (µcg/g/liver) | Iron content (µcg/total liver) |
| Sham only | 5 | 91.8 ± 12.9 | 861.3 ± 158.9 |
| Sham and phlebotomy later | 4 | 40.1 ± 8.83 | 315.1 ± 63.33 |
| Phlebotomy and sham later | 6 | 55.0 ± 19.7 | 358.9 ± 104.4 |
| BDL only | 9 | 80.9 ± 17.2 | 1122.1 ± 358.6 |
| BDL and phlebotomy later | 8 | 37.4 ± 6.91 | 477.2 ± 85.11 |
| Phlebotomy and BDL later | 8 | 49.0 ± 29.6 | 628.0 ± 224.72 |
P ≤ 0.0001,
P ≤ 0.012 vs BDL only;
P ≤ 0.016 vs sham only.
Table 6.
Serum iron and transferrin in different groups of rats (mean±SE)
| Subgroup | n | Serum iron (µg/dL) | Transferrin (mg/dL) |
| Sham only | 5 | 163 ± 21 | 446 ± 46 |
| Sham and phlebotomy later | 4 | 144 ± 38 | 459 ± 17 |
| Phlebotomy and sham later | 6 | 160 ± 20 | 482 ± 103 |
| BDL only | 9 | 205 ± 71 | 640 ± 128 |
| BDL and phlebotomy later | 8 | 197 ± 57 | 732 ± 65 |
| Phlebotomy and BDL later | 8 | 215 ± 116 | 701 ± 83 |
Effect of phlebotomy
Phlebotomy after surgical procedure (SO or BDL) resulted in a decrease of HIC and content. The change was more pronounced in BDL rats than in SO rats. Rats that were phlebotomized before the surgical procedure had higher HIC than those undergone phlebotomy after the surgical procedure (Table 5). No change in SI and transferrin was noted in any group as a result of phlebotomy (Table 6).
Phlebotomy after BDL was associated with body weight in crease, higher albumin levels and lower hepatic PGE2 generation at sacrifice compared to BDL only rats (Tables 1-3). Moreover, periportal necrosis, portal inflammation, fibrosis scores, total HAI and hepatic HP content decreased in rats that were phlebotomized after BDL, although the results did not reach statistical significance. Phlebotomy before BDL was associated (when compared to BDL only rats) with a similar weight loss. Periportal necrosis, portal inflammatory activity and fibrosis score were higher in this group than in the BDL only group. However, while the difference in value for each variable was not statistically significant, the combination of variables (HAI) was significant (12.17 ± 1.93 vs 7.22 ± 3.49, P ≤ 0.018, Figure 1). Surprisingly, hepatic PGE2 generation and MPO activity in the rats that had phlebotomy before BDL were lower than those in the BDL only group.
When both phlebotomy groups undergone BDL were compared, the group that had phlebotomy after the procedure had a better outcome: weight gain, higher albumin level, lower hepatic weight (P = 0.029), less portal hypertension (determined as lower spleen weight) (P = 0.029), less periportal necrosis (P = 0.008) and portal inflammation (P = 0.008) and lower total HAI score (P = 0.008, Tables 1-3, Figure 1).
DISCUSSION
The role of iron in the progression of hepatic damage in various clinical and experimental conditions has usually been studied by iron loading[14,16,18-20], or by iron depletion in situations where mild to moderate iron overload is present[13,24,25,30]. In the present study, we examined the effect of reduction of “normal” hepatic iron stores on liver function and histology in rats subjected to BDL. This situation may be more relevant and analogous to and may mimic more closely the common clinical conditions where most of the patients with non-hemochromatosis and non-end-stage liver diseases do not have iron overloadd[13].
In the present study we were able to demonstrate that it was possible to reduce HIC by phlebotomy and that such a procedure after sham operation was not associated with hepatic adverse events. Liver disease developed in the rats after two weeks of BDL was not associated with HIC increase. Phlebotomy before the surgical procedure was associated with HIC reduction but not with SI or transferrin reduction. The changes in liver histology (mainly necrosis and inflammation) following the HIC reduction indicated that hepatic iron might not have a role in the fibrosis process after BDL[38]. Alternatively, we may also speculate that further reduction of HIC by more aggressive phlebotomy would have an effect on fibrosis. Since SI and transferrin levels did not change following phlebotomy, we assume that the phlebotomy process performed in our rats was not sufficient to alter the course of the fibrosis process. Although no change was observed in fibrosis score, phlebotomized rats after BDL exhibited favorable effects. These rats had body weight increase, higher albumin levels and lower hepatic necroinflammatory activity. Iron deficiency induced by iron chelation may reduce the severity of inflammation in joints and in the gastrointestinal tract[39,40].
HIC was only mildly reduced in the rats undergone phlebotomy before the surgical procedure compared to the BDL only rats. Only the total amount of iron per whole liver was reduced significantly. This could be due to the ability of rats to absorb iron during the time interval between the last phlebotomy session and the sacrifice of the rats (18 d). This may indicate that phlebotomy has a transient effect and that continuous phlebotomy sessions over time should be done in order to sustain a beneficial effect. In cirrhotic subjects an increased iron absorption due to increased duodenal expression of iron transporter, divalent metal transporter 1 has been reported [41]. However this phenomenon does not exist in rats with cholestatic liver disease.
Phlebotomy had a favorable effect on the rats after BDL, but a definite negative effect before BDL. The latter rats lost weight and had a worse HAI (a worse score in periportal necrosis and portal inflammation) as compared to the other BDL groups. A similar trend of changes was observed in the rats subjected to phlebotomy before sham operation. Iron deficiency may affect various organs including eccentric cardiac hypertrophy in rats[42] and various hepatic enzyme functions[43]. Anemic patients with early cirrhosis may have worse hemodynamic parameters (lower systemic vascular resistance, increased cardiac index and hepatic venous pressure gradient) than non-anemic control patients [44]. Increased blood hemoglobin may attenuate splanchnic vasodilatation in portal hypertensive subjects by nitric oxide inactivation[44]. In our study, the rats undergone phlebotomy before BDL had the highest splenic weight (a marker of portal hypertension). Based on this finding we may speculate that deterioration of hemodynamic parameters in these rats is associated with activation of the renin-angiotensin system. This system may have a role in hepatic fibrosis in BDL rats[45].
Hepatic PGE2 generation and MPO activity were used as adjuvant markers for hepatic inflammatory activity in our study, and decreased PGE2 generation was observed in the BDL groups undergone phlebotomy. This is unexpected as iron is known to down-regulate PGE2 generation in various tissues[46,47]. Higher PGE2 levels were also expected in the BDL groups with increased hepatic inflammatory changes. This is similar to the results in our previous study of rats to BDL and colitis, in which colonic PGE2 generation was reduced as compared with a control group[33]. One may speculate that cycloxygenase activity or even phospholipase A2 activity may be defective in rats with experimental liver disease[33,48].
As MPO is found primarily in neutrophil granules, it is a marker of neutrophil content and influx into tissues[49]. Our previous study in BDL rats with colitis found that mucosal inflammation is correlated with MPO activity[33]. In the present study, however no correlation was found between the hepatic inflammatory changes and hepatic MPO activity. Decreased MPO activity in the Carrageenan model of inflammation in rats with acute cholestasis due to bile duct resection has been reported[50]. Additional defects of neutrophil function in BDL rats, like impairment of adhesion, defective phagocytosis, increased superoxide generation and chemotaxis, and increased number of neutrophil counts, have been reported [50-53]. Iron deficiency per se, may also impair neutrophil function and MPO activity and cause a selective defect in the process of inflammation[54,55]. Iron repletion improves some of the above functions[54]. In this study, hepatic MPO activity was the lowest in the rats undergone phlebotomy before the surgical procedure. However there was no clear correlation between HIC, hepatic MPO activity and hepatic inflammation.
In summary, hepatic iron depletion in rats after a liver insult like BDL, may have beneficial effects. “Hepatic iron deficiency” may make the liver more vulnerable to insults such as laparotomy or BDL. Hepatic PGE2 generation and hepatic MPO activity are not reliable markers for hepatic inflammation in rats with cholestatic liver disease.
Footnotes
S- Editor Wang XL and Guo SY L- Editor Elsevier HK E- Editor Cao L
References
- 1.Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig J, Hashimoto E, Porayko MK, Moyer TP, Baldus WP. Hemosiderosis in cirrhosis: a study of 447 native livers. Gastroenterology. 1997;112:882–888. doi: 10.1053/gast.1997.v112.pm9041250. [DOI] [PubMed] [Google Scholar]
- 3.Cotler SJ, Bronner MP, Press RD, Carlson TH, Perkins JD, Emond MJ, Kowdley KV. End-stage liver disease without hemochromatosis associated with elevated hepatic iron index. J Hepatol. 1998;29:257–262. doi: 10.1016/s0168-8278(98)80011-1. [DOI] [PubMed] [Google Scholar]
- 4.Kayali Z, Ranguelov R, Mitros F, Shufelt C, Elmi F, Rayhill SC, Schmidt WN, Brown KE. Hemosiderosis is associated with accelerated decompensation and decreased survival in patients with cirrhosis. Liver Int. 2005;25:41–48. doi: 10.1111/j.1478-3231.2005.01022.x. [DOI] [PubMed] [Google Scholar]
- 5.Gardi C, Arezzini B, Fortino V, Comporti M. Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem Pharmacol. 2002;64:1139–1145. doi: 10.1016/s0006-2952(02)01257-1. [DOI] [PubMed] [Google Scholar]
- 6.Pietrangelo A. Metals, oxidative stress, and hepatic fibrogenesis. Semin Liver Dis. 1996;16:13–30. doi: 10.1055/s-2007-1007215. [DOI] [PubMed] [Google Scholar]
- 7.Bacon BR, Britton RS. The pathology of hepatic iron overload: a free radical--mediated process. Hepatology. 1990;11:127–137. doi: 10.1002/hep.1840110122. [DOI] [PubMed] [Google Scholar]
- 8.Poli G, Parola M. Oxidative damage and fibrogenesis. Free Radic Biol Med. 1997;22:287–305. doi: 10.1016/s0891-5849(96)00327-9. [DOI] [PubMed] [Google Scholar]
- 9.Bedossa P, Houglum K, Trautwein C, Holstege A, Chojkier M. Stimulation of collagen alpha 1(I) gene expression is associated with lipid peroxidation in hepatocellular injury: a link to tissue fibrosis. Hepatology. 1994;19:1262–1271. [PubMed] [Google Scholar]
- 10.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Farinati F, Cardin R, De Maria N, Della Libera G, Marafin C, Lecis E, Burra P, Floreani A, Cecchetto A, Naccarato R. Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J Hepatol. 1995;22:449–456. doi: 10.1016/0168-8278(95)80108-1. [DOI] [PubMed] [Google Scholar]
- 12.George DK, Goldwurm S, MacDonald GA, Cowley LL, Walker NI, Ward PJ, Jazwinska EC, Powell LW. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311–318. doi: 10.1016/s0016-5085(98)70482-2. [DOI] [PubMed] [Google Scholar]
- 13.Sherrington CA, Olynyk JK. Iron as a cofactor in chronic hepatitis C infection. Liver. 2002;22:187–189. doi: 10.1046/j.0106-9543.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 14.Bonkovsky HL, Lambrecht RW, Shan Y. Iron as a co-morbid factor in nonhemochromatotic liver disease. Alcohol. 2003;30:137–144. doi: 10.1016/s0741-8329(03)00127-7. [DOI] [PubMed] [Google Scholar]
- 15.Tung BY, Emond MJ, Bronner MP, Raaka SD, Cotler SJ, Kowdley KV. Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology. 2003;124:318–326. doi: 10.1053/gast.2003.50046. [DOI] [PubMed] [Google Scholar]
- 16.Bassett SE, Di Bisceglie AM, Bacon BR, Sharp RM, Govindarajan S, Hubbard GB, Brasky KM, Lanford RE. Effects of iron loading on pathogenicity in hepatitis C virus-infected chimpanzees. Hepatology. 1999;29:1884–1892. doi: 10.1002/hep.510290623. [DOI] [PubMed] [Google Scholar]
- 17.Martinelli AL, Ramalho LN, Zucoloto S. Hepatic stellate cells in hepatitis C patients: relationship with liver iron deposits and severity of liver disease. J Gastroenterol Hepatol. 2004;19:91–98. doi: 10.1111/j.1440-1746.2004.03255.x. [DOI] [PubMed] [Google Scholar]
- 18.Arezzini B, Lunghi B, Lungarella G, Gardi C. Iron overload enhances the development of experimental liver cirrhosis in mice. Int J Biochem Cell Biol. 2003;35:486–495. doi: 10.1016/s1357-2725(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 19.Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, Brittenham GM. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olynyk J, Hall P, Reed W, Williams P, Kerr R, Mackinnon M. A long-term study of the interaction between iron and alcohol in an animal model of iron overload. J Hepatol. 1995;22:671–676. doi: 10.1016/0168-8278(95)80222-3. [DOI] [PubMed] [Google Scholar]
- 21.Toniutto P, Fabris C, Bortolotti N, Minisini R, Avellini C, Fumo E, Pirisi M. Evaluation of donor hepatic iron concentration as a factor of early fibrotic progression after liver transplantation. J Hepatol. 2004;41:307–311. doi: 10.1016/j.jhep.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 22.George DK, Ramm GA, Powell LW, Fletcher LM, Walker NI, Cowley LL, Crawford DH. Evidence for altered hepatic matrix degradation in genetic haemochromatosis. Gut. 1998;42:715–720. doi: 10.1136/gut.42.5.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson GJ. Non-transferrin-bound iron and cellular toxicity. J Gastroenterol Hepatol. 1999;14:105–108. doi: 10.1046/j.1440-1746.1999.01828.x. [DOI] [PubMed] [Google Scholar]
- 24.Facchini FS, Hua NW, Stoohs RA. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology. 2002;122:931–939. doi: 10.1053/gast.2002.32403. [DOI] [PubMed] [Google Scholar]
- 25.Carlo C, Daniela P, Giancarlo C. Iron depletion and response to interferon in chronic hepatitis C. Hepatogastroenterology. 2003;50:1467–1471. [PubMed] [Google Scholar]
- 26.Fontana RJ, Israel J, LeClair P, Banner BF, Tortorelli K, Grace N, Levine RA, Fiarman G, Thiim M, Tavill AS, et al. Iron reduction before and during interferon therapy of chronic hepatitis C: results of a multicenter, randomized, controlled trial. Hepatology. 2000;31:730–736. doi: 10.1002/hep.510310325. [DOI] [PubMed] [Google Scholar]
- 27.Yano M, Hayashi H, Wakusawa S, Sanae F, Takikawa T, Shiono Y, Arao M, Ukai K, Ito H, Watanabe K, et al. Long term effects of phlebotomy on biochemical and histological parameters of chronic hepatitis C. Am J Gastroenterol. 2002;97:133–137. doi: 10.1111/j.1572-0241.2002.05436.x. [DOI] [PubMed] [Google Scholar]
- 28.Sartori M, Andorno S, Rigamonti C, Grossini E, Nicosia G, Boldorini R. Chronic hepatitis C is mild in menstruating women. J Gastroenterol Hepatol. 2000;15:1411–1417. doi: 10.1046/j.1440-1746.2000.02368.x. [DOI] [PubMed] [Google Scholar]
- 29.Shedlofsky SI. Role of iron in the natural history and clinical course of hepatitis C disease. Hepatogastroenterology. 1998;45:349–355. [PubMed] [Google Scholar]
- 30.Fargion S, Fracanzani AL, Rossini A, Borzio M, Riggio O, Belloni G, Bissoli F, Ceriani R, Ballarè M, Massari M, et al. Iron reduction and sustained response to interferon-alpha therapy in patients with chronic hepatitis C: results of an Italian multicenter randomized study. Am J Gastroenterol. 2002;97:1204–1210. doi: 10.1111/j.1572-0241.2002.05705.x. [DOI] [PubMed] [Google Scholar]
- 31.Streiff MB, Mehta S, Thomas DL. Peripheral blood count abnormalities among patients with hepatitis C in the United States. Hepatology. 2002;35:947–952. doi: 10.1053/jhep.2002.32486. [DOI] [PubMed] [Google Scholar]
- 32.Pianko S, McHutchison JG, Gordon SC, Heaton S, Goodman ZD, Patel K, Cortese CM, Brunt EM, Bacon BR, Blatt LM. Hepatic iron concentration does not influence response to therapy with interferon plus ribavirin in chronic HCV infection. J Interferon Cytokine Res. 2002;22:483–489. doi: 10.1089/10799900252952271. [DOI] [PubMed] [Google Scholar]
- 33.Ackerman Z, Karmeli F, Cohen P, Rachmilewitz D. Experimental colitis in rats with portal hypertension and liver disease. Inflamm Bowel Dis. 2003;9:18–24. doi: 10.1097/00054725-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 34.Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, Wollman J. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 35.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 36.Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci. 1968;33:9–11. [PubMed] [Google Scholar]
- 37.Woessner JF. The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 38.Sadrzadeh SM, Hallaway PE, Nanji AA. The long-acting parenteral iron chelator, hydroxyethyl starch-deferoxamine, fails to protect against alcohol-induced liver injury in rats. J Pharmacol Exp Ther. 1997;280:1038–1042. [PubMed] [Google Scholar]
- 39.Andrews FJ, Morris CJ, Kondratowicz G, Blake DR. Effect of iron chelation on inflammatory joint disease. Ann Rheum Dis. 1987;46:327–333. doi: 10.1136/ard.46.4.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ablin J, Shalev O, Okon E, Karmeli F, Rachmilewitz D. Deferiprone, an oral iron chelator, ameliorates experimental colitis and gastric ulceration in rats. Inflamm Bowel Dis. 1999;5:253–261. doi: 10.1097/00054725-199911000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Stuart KA, Anderson GJ, Frazer DM, Murphy TL, Powell LW, Fletcher LM, Crawford DH. Increased duodenal expression of divalent metal transporter 1 and iron-regulated gene 1 in cirrhosis. Hepatology. 2004;39:492–499. doi: 10.1002/hep.20038. [DOI] [PubMed] [Google Scholar]
- 42.Medeiros DM, Beard JL. Dietary iron deficiency results in cardiac eccentric hypertrophy in rats. Proc Soc Exp Biol Med. 1998;218:370–375. doi: 10.3181/00379727-218-44306. [DOI] [PubMed] [Google Scholar]
- 43.Hercberg S, Galan P. Biochemical effects of iron deprivation. Acta Paediatr Scand Suppl. 1989;361:63–70. doi: 10.1111/apa.1989.78.s361.63. [DOI] [PubMed] [Google Scholar]
- 44.Lee WC, Lin HC, Hou MC, Lee FY, Chang FY, Tsai YT, Lee SD. Effect of anaemia on haemodynamics in patients with cirrhosis. J Gastroenterol Hepatol. 1999;14:370–375. doi: 10.1046/j.1440-1746.1999.01853.x. [DOI] [PubMed] [Google Scholar]
- 45.Jonsson JR, Clouston AD, Ando Y, Kelemen LI, Horn MJ, Adamson MD, Purdie DM, Powell EE. Angiotensin-converting enzyme inhibition attenuates the progression of rat hepatic fibrosis. Gastroenterology. 2001;121:148–155. doi: 10.1053/gast.2001.25480. [DOI] [PubMed] [Google Scholar]
- 46.Hisakawa N, Nishiya K, Tahara K, Matsumori A, Hashimoto K. Down regulation by iron of prostaglandin E2 production by human synovial fibroblasts. Ann Rheum Dis. 1998;57:742–746. doi: 10.1136/ard.57.12.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakuma S, Fujimoto Y, Miyata Y, Ohno M, Nishida H, Fujita T. Effects of Fe(2+), Zn(2+), Cu(2+) and Se(4+) on the synthesis and catabolism of prostaglandins in rabbit gastric antral mucosa. Prostaglandins Leukot Essent Fatty Acids. 1996;54:193–197. doi: 10.1016/s0952-3278(96)90016-2. [DOI] [PubMed] [Google Scholar]
- 48.Laffi G, Carloni V, Baldi E, Rossi ME, Azzari C, Gresele P, Marra F, Gentilini P. Impaired superoxide anion, platelet-activating factor, and leukotriene B4 synthesis by neutrophils in cirrhosis. Gastroenterology. 1993;105:170–177. doi: 10.1016/0016-5085(93)90023-6. [DOI] [PubMed] [Google Scholar]
- 49.Smith JW, Castro GA. Relation of peroxidase activity in gut mucosa to inflammation. Am J Physiol. 1978;234:R72–R79. doi: 10.1152/ajpregu.1978.234.1.R72. [DOI] [PubMed] [Google Scholar]
- 50.Swain MG, Tjandra K, Kanwar S, Kubes P. Neutrophil adhesion is impaired in rat model of cholestasis. Gastroenterology. 1995;109:923–932. doi: 10.1016/0016-5085(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 51.Roughneen PT, Drath DB, Kulkarni AD, Rowlands BJ. Impaired nonspecific cellular immunity in experimental cholestasis. Ann Surg. 1987;206:578–582. doi: 10.1097/00000658-198711000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roughneen PT, Drath DB, Kulkarni AD, Kumar SC, Andrassy RJ, Rowlands BJ. Inflammatory cell function in young rodents with experimental cholestasis: investigations of functional deficits, their etiology, and their reversibility. J Pediatr Surg. 1989;24:668–673. doi: 10.1016/s0022-3468(89)80716-x. [DOI] [PubMed] [Google Scholar]
- 53.Levy R, Schlaeffer F, Keynan A, Nagauker O, Yaari A, Sikuler E. Increased neutrophil function induced by bile duct ligation in a rat model. Hepatology. 1993;17:908–914. [PubMed] [Google Scholar]
- 54.Murakawa H, Bland CE, Willis WT, Dallman PR. Iron deficiency and neutrophil function: different rates of correction of the depressions in oxidative burst and myeloperoxidase activity after iron treatment. Blood. 1987;69:1464–1468. [PubMed] [Google Scholar]
- 55.Andrews FJ, Morris CJ, Lewis EJ, Blake DR. Effect of nutritional iron deficiency on acute and chronic inflammation. Ann Rheum Dis. 1987;46:859–865. doi: 10.1136/ard.46.11.859. [DOI] [PMC free article] [PubMed] [Google Scholar]