

Abstract
AIM: To investigate the efficacy of angiotensin II receptor antagonist on hepatic stellate cells (HSCs) activation in the patients with non-alcoholic steatohepatitis (NASH).
METHODS: Seven patients with NASH were prescribed losartan, a selective angiotensin II type 1 receptor antagonist (50 mg/d) for 48 wk. Liver biopsies were performed both at the entry and end of the study in all patients. Quiescent and activated HSCs were identified by double immunostaining using anti-p75 and α-smooth muscle actin antibodies, and the number of each phenotype was counted. Similarly, the liver specimens obtained from the eight patients with non-alcoholic fatty liver (NAFL) were also examined as controls.
RESULTS: In NASH hepatic tissues, activated HSCs were dominantly distributed as compared with those in NAFL. The 48-wk losartan treatment induced a remarkable decrease in activated HSCs and a mild increase in quiescent phenotypes.
CONCLUSION: Our data suggest the crucial involvement of HSCs in anti-fibrotic effect of angiotensin II receptor antagonist on patients with NASH.
Keywords: NASH, NAFLD, Hepatic fibrosis, HSC, Losartan
INTRODUCTION
Non-alcoholic steatohepatitis (NASH) is a distinct clinical entity characterized by steatosis, varying degrees of lobular inflammation and fibrosis in the liver, and this disease can potentially progress[1,2]. Previous data showed that about 20% of the patients developed to liver cirrhosis[3] and several cases of hepatocellular carcinoma are related to NASH[4]. Although the causes of NASH are not well defined, hepatic stellate cell (HSC) is suggested to play a pivotal role in the progression of hepatic fibrosis as other chronic liver diseases[5,6]. Stimulated HSCs transform into myofibroblast-like cell, and produce a large amount of extracellular matrix components, resulting in the formation of fibrosis[7]. However, to our knowledge, the efficacy of any treatments for NASH on the activation of HSCs has not been reported yet. Recently, we have demonstrated that losartan, a selective angiotensin II type 1 receptor antagonist, improved hepatic necroinflammation and fibrosis in patients with NASH[8]. Based on our results, we hypothesized that an angiotensin II receptor antagonist inhibits the progression of hepatic fibrosis through the inactivation of HSCs in NASH. In this study, we, therefore, investigated the efficacy of long-term losartan treatment on HSCs in patients with NASH.
MATERIALS AND METHODS
Patients
Seven patients with both NASH and hypertension, which was defined as a systolic blood pressure over 140 mmHg and/or a diastolic blood pressure over 90 mmHg, and eight patients with non-alcoholic fatty liver (NAFL) were enrolled into this study. A detailed alcohol history was taken by at least two physicians and confirmed by at least one close relative who lived with the patient. All patients consumed less than 40 g of alcohol per week, and were negative for hepatitis B serological tests, antibody to hepatitis C virus or any autoantibodies, including anti-nuclear antibody, and anti-mitochondrial antibody. Serum ceruloplasmin and α1-antitrypsin levels were normal in all patients. Four patients in NASH group and three in NAFL group were obese (body mass index > 25); four in NASH and one in NAFL had diabetes mellitus; two in NASH and four in NAFL had impaired glucose tolerance; three in NASH and four in NAFL had hyperlipidemia; and one patient in each group had hyperuricemia. Four of eight patients with NAFL group had hypertension.
Study design
At the entry of the study, some of the patients had already been on medication, including benzodiazepines, calcium antagonists or anticoagulants for at least 12 mo. These therapeutic regimens were not changed after an induction of losartan treatment in patients with NASH. No patient was taking any angiotensin converting enzyme inhibitors or angiotensin II receptor antagonists before the study. Laboratory examination showed abnormally high serum transaminase and γ-glutamyl transpeptidase concentrations in all patients. A liver biopsy was performed prior to entry the study, which revealed a moderate to severe lobular steatosis in both groups. On the other hand, various degrees of hepatic necroinflammation and fibrosis were demonstrated only in NASH group. After the estimation at the entry, patients with NASH were prescribed losartan at a dose of 50 mg/d for 48 wk, and a physical and laboratory examination were monitored every 4 wk. A liver biopsy was repeated at the end of the study in the patients with NASH. The Ethical Committee of Asahikawa Medical College for Clinical Study approved all protocols, and informed consents were obtained from all patients.
Liver histology
Hematoxylin & eosin and Azan stainings were performed on fixed histological sections. Two experienced hepatopathologists graded necroinflammation and staged fibrosis in a blinded fashion according to the Grading and Staging system of NASH proposed by the American Association for the Study of Liver Diseases[9]. To identify HSCs, double immunostaining was performed using monoclonal anti-p75 (a low affinity nerve growth factor receptor) antibody (Novocastra, Newcastle, UK) and monoclonal anti-α-smooth muscle actin (α-SMA) antibody (Dako, Kidlington, UK) as previously described[10,11]. Color development was achieved with BCIP/NBT for p75 and DAB for α-SMA, and counter-staining was performed using direct fast red. Activated HSCs were distinguished from quiescent HSCs by their myofibroblast-like form and the disappearance of fat vesicles in cytoplasm[7]. The numbers of both activated and quiescent HSCs were blindly counted by two independent pathologists and averaged in five fields per slide at ×200 magnification.
Statistical analysis
Data were expressed as median values with ranges. Mann-Whitney U-test was used to compare two independent data, and Wilcoxon rank sum test was used for paired data. A P value < 0.05 was considered statistically significant.
RESULTS
Before the study, serum γ-glutamyl transpeptidase, type IV collagen 7 S and fasting blood glucose levels in NASH group were significantly higher as compared with NAFL group. Although body mass index, serum transaminase, hyaluronic acid, ferritin and triglyceride levels in the NASH group tended to be higher as compare with the NAFL group, there was no statistically significant difference (Table 1). No side effect was noted, and the body mass index was unchanged during the losartan treatment in patients with NASH. The necroinflammatory grade and stage of fibrosis diagnosed by liver biopsies were improved after losartan treatment [for grade: pretreatment, 2(1-3), posttreatment, 1(1-2), P < 0.05; for stage: pretreatment, 3(1-4), posttreatment, 2 (1-4), P < 0.05].
Table 1.
Characteristics and biochemical data of patients with non-alcoholic fatty liver (NAFL) and non-alcoholic steatohepatitis (NASH) before losartan treatment
| Paremeters | NAFL (n = 8) | NASH (n = 7) |
| Age (yr) Male/female | 59 (34-76) 4/4 | 59 (41-65) 2/5 |
| BMI (kg/m2) | 24.5 (21.4-31.4) | 26.1 (22.6-36.7) |
| SBP (mmHg) | 130 (92-156) | 148 (127-171) |
| DBP (mmHg) | 78 (54-96) | 90 (71-100) |
| AST (IU/L) | 54 (42-70) | 84 (20-128) |
| ALT (IU/L) | 72 (56-110) | 132 (54-138) |
| γ-GTP (IU/L) | 30 (17-43) | 53 (42-127)a |
| TC (mg/dL) | 209 (169-254) | 222 (160-319) |
| TG (mg/dL) | 128 (86-198) | 124 (65-295) |
| FBS (mg/dL) | 95 (86-112) | 109 (97-132)a |
| Ferritin (ng/mL) | 111 (30-264) | 199 (54-379) |
| HA (ng/mL) | 46.5 (10.0-78.3) | 89.4 (12.3-197.0) |
| IV collagen (ng/mL) | 2.7 (2.0-3.5) | 5.2 (3.1-9.9)a |
Values are expressed as median (range).
P < 0.05 vs NAFL; BMI: body mass index; SBP: systolic blood pressure; DBP: diastolic blood pressure; AST: aspartate aminotransferase; ALT: alanine aminotransferase; γ-GTP: γ-glutamyl transpeptidase; TC: total cholesterol; TG: triglyceride; FBS: fasting blood glucose; HA: hyaluronic acid; IV collagen: type IV collagen 7S.
HSCs were identified in the perisinusoidal area of both NAFL and NASH liver sections (Figure 1). In NAFL liver tissues, quiescent forms of HSC, characterized by small and circular cell bodies containing lipid droplets in the cytoplasm, were dominantly distributed, and active forms of HSC were rarely observed. On the other hand, activated HSCs (myofibroblast-like phenotype), characterized by enlarged cell bodies, fewer lipid droplets and the presence of membranous processes, were conspicuously scattered in NASH liver tissues. These activated HSCs were preferentially associated with areas of fibrosis (Figure 2). The number of activated HSCs and ratio of activated/quiescent HSCs in NASH liver tissues were significantly higher as compared with those in NAFL liver tissues (Figures 3A and 3B). On the other hand, the number of quiescent HSCs was significantly lower in NASH liver tissues as compared with those in NAFL liver tissues (Figure 3C). In patients with NASH, the number of activated HSCs in the liver sections with mild necroinflammation (grade 1) was significantly lower as compared with those with moderate to severe necroinflammation (grades 2 and 3) [grade 1: 57(46-68); grades 2 and 3: 153(117-201), P < 0.05]. Although the number of activated HSCs in the liver sections with mild fibrosis (stages 1 and 2) tended to be lower as compared with those with severe fibrosis (stages 3 and 4), there was no statistically significant difference[stages 1 and 2: 68(46-185); stages 3 and 4: 145(117-201)].
Figure 1.
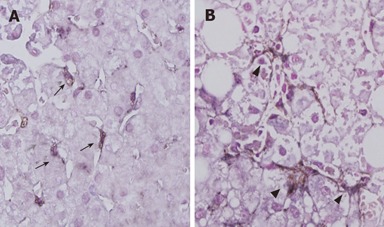
Expression of HSCs in patients with NASH and NAFL (× 400). Liver sections were immunostained with both monoclonal anti-p75 antibody and monoclonal anti-α-SMA antibodies. Although most perisinusoidal HSCs were quiescent form (arrow heads) in NAFL, a number of arrow head were detected in the patient with NASH (arrow heads).
Figure 2.
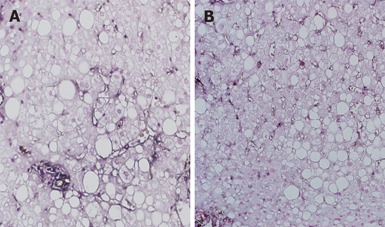
Significant decrease of activated and quiescent HSCs by 48-wk losartan treatment in a patient with NASH (double immunostaining with anti-p75 and α-SMA antibodies, × 200).
Figure 3.
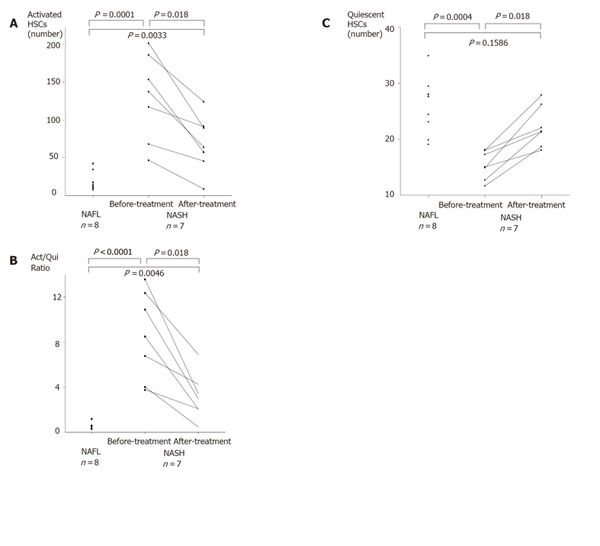
Average numbers of HSCs in five fields per slide at x200 magnification. A: number of activated HSCs; B: averaged ratio of activated HSCs to quiescent HSCs (Act-/Qui- Ratio: a ratio of activated HSCs to quiescent HSCs); C: averaged number of quiescent HSCs.
The number of activated HSCs and the ratio of activated/quiescent HSCs were significantly decreased after 48-wk losartan treatment (Figures 3A and 3B). On the other hand, quiescent HSCs slightly increased after losartan treatment (Figure 3C). There was no significant correlation between the decrease rate of activated HSCs and the improvement of hepatic pathologic findings after losartan treatment.
DISCUSSION
In the present study, we demonstrated that the activated HSCs and the ratio of activated/quiescent HSCs in NASH hepatic tissues were significantly higher as compared with those in NAFL, and activated HSCs in NASH livers were preferentially localized along fibrotic area. The number of activated HSCs tended to be correlated with the necroinflammatory grade and fibrotic stage. This result was very consistent with previous studies that demonstrated the activation of HSCs in NASH[5,6], and suggested that the hepatic necroinflammation activated and proliferated HSCs, and led to progress the hepatic fibrosis in NASH. In contrast, Wasington et al [6] reported no difference in the number of activated HSCs between NASH and NAFL. This discrepancy may be due to the difference of employed procedure for identifying activated HSCs. Although we judged activated HSCs by their distinctive form and disappearance of fat vesicle, together with double staining with anti-p75 and α-SMA antibodies, they identified activated HSCs only by positive staining for α-SMA. Since recent studies demonstrated that both activated and quiescent HSCs expressed α-SMA[10,11], activated HSCs identified by their procedure may include quiescent phenotypes.
We also investigated the changes of HSCs after 48-wk losartan treatment in patients with NASH. Both the number of activated HSCs and the ratio of activated/quiescent HSCs were significantly decreased, and quiescent HSCs were slightly increased by losartan treatment concurrently with the improvement of hepatic fibrosis. The depletion of activated HSCs might be due to inactivation of activated HSCs[12], or disappearance of activated HSCs by apoptosis[10]. An increase of quiescent HSCs after losartan treatment supported the former mechanism. However, an increase in quiescent HSCs was relatively small as compared with a decrease in activated HSCs after losartan treatment, suggesting that the latter mechanism may be dominant in a depletion of activated HSCs. Because it is difficult to recruit patients with both NASH and hypertension, and only seven patients were evaluated in this study, significant correlation between the decrease rate of activated HSCs and the improvement of hepatic pathologic findings after losartan treatment was not observed. However, our present data strongly suggested a causative role of HSCs in hepatic fibrosis of NASH.
Although detailed mechanisms for the suppressive effect of angiotensin II receptor antagonist on HSC activation could not be well defined in this study, we hypothesize that there are two different pathways. The first may be the direct inhibition to HSCs. Studies have revealed that an activated HSC expresses angiotensin II type 1 receptor[13], and synthesizes angiotensin II by itself[14]. Locally generated angiotensin II enhances the activation and proliferation of HSCs through autocrine and paracrine mechanisms[15]. Losartan inhibited activated HSCs possibly through the blocking of angiotensin II type 1 receptors expressed on the surface of HSCs. In accordance with this mechanism, angiotensin II receptor antagonist may have a therapeutic efficacy on other chronic liver diseases that HSCs are involved in the progression of hepatic fibrosis, including viral hepatitis, alcoholic liver injury, or autoimmune liver diseases[16-18]. Actually, Terui et al [19] reported that losartan improved the hepatic fibrosis in chronic type C hepatitis.
Another mechanism of the angiotensin II receptor antagonist may be the indirect inhibition of HSCs mediated through the improvement of hepatic necroinflammation. Chronic hepatic inflammation enhances the production of transforming growth factor-β1 (TGF-β1) from Kupffer cell and inflammatory cells[20]. TGF-β1 is a multifunctional and ubiquitous cytokine, and in chronic liver diseases, this peptide plays a causative role in hepatic fibrogenesis through the activation of HSCs. We demonstrated that the patients with NASH had elevated plasma TGF-β1 levels that were reversed by α-tocopherol[21]. Moreover, we have recently reported that losartan improves hepatic necroinflammation concurrently with a reduction of plasma TGF-β1 levels in the patients with NASH[8]. Angiotensin II is known to aggravate several pathogenic factors of NASH, such as insulin resistance, oxidative stress and hepatic iron overload[22-24]. In fact, our previous studies demonstrated that serum ferritin levels were significantly decreased and hepatic iron deposition disappeared in some patients with NASH after losartan treatment[8]. Angiotensin II receptor antagonist may also suppress the HSC activation through the decrease of TGF-β1.
In conclusion, inhibiting the action of angiotensin II by losartan results in a remarkable depletion of activated HSCs and a mild increase of quiescent HSCs, thereby suggesting the important involvement of HSCs in anti-fibrotic effect of angiotensin II receptor antagonist on the patients with NASH.
Footnotes
Supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science, No. 15590613, and a grant for Research on Intractable Disease from the Japanese Ministry of Public Welfare
S- Editor Kumar M and Guo SY L- Editor Elsevier HK E- Editor Cao L
References
- 1.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 2.Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology. 1994;107:1103–1109. doi: 10.1016/0016-5085(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 3.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 4.Shimada M, Hashimoto E, Taniai M, Hasegawa K, Okuda H, Hayashi N, Takasaki K, Ludwig J. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol. 2002;37:154–160. doi: 10.1016/s0168-8278(02)00099-5. [DOI] [PubMed] [Google Scholar]
- 5.Cortez-Pinto H, Baptista A, Camilo ME, de Moura MC. Hepatic stellate cell activation occurs in nonalcoholic steatohepatitis. Hepatogastroenterology. 2001;48:87–90. [PubMed] [Google Scholar]
- 6.Washington K, Wright K, Shyr Y, Hunter EB, Olson S, Raiford DS. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum Pathol. 2000;31:822–828. doi: 10.1053/hupa.2000.8440. [DOI] [PubMed] [Google Scholar]
- 7.Senoo H. Structure and function of hepatic stellate cells. Med Electron Microsc. 2004;37:3–15. doi: 10.1007/s00795-003-0230-3. [DOI] [PubMed] [Google Scholar]
- 8.Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, Hasegawa T, Tokusashi Y, Miyokawa N, Nakamura K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–1225. doi: 10.1002/hep.20420. [DOI] [PubMed] [Google Scholar]
- 9.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 10.Trim N, Morgan S, Evans M, Issa R, Fine D, Afford S, Wilkins B, Iredale J. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am J Pathol. 2000;156:1235–1243. doi: 10.1016/S0002-9440(10)64994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tokusashi Y, Asai K, Tamakawa S, Yamamoto M, Yoshie M, Yaginuma Y, Miyokawa N, Aoki T, Kino S, Kasai S, et al. Expression of NGF in hepatocellular carcinoma cells with its receptors in non-tumor cell components. Int J Cancer. 2005;114:39–45. doi: 10.1002/ijc.20685. [DOI] [PubMed] [Google Scholar]
- 12.Murata T, Arii S, Nakamura T, Mori A, Kaido T, Furuyama H, Furumoto K, Nakao T, Isobe N, Imamura M. Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of rat liver fibrosis in association with inactivation of hepatic stellate cells. J Hepatol. 2001;35:474–481. doi: 10.1016/s0168-8278(01)00169-6. [DOI] [PubMed] [Google Scholar]
- 13.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34:745–750. doi: 10.1053/jhep.2001.28231. [DOI] [PubMed] [Google Scholar]
- 14.Bataller R, Sancho-Bru P, Ginès P, Lora JM, Al-Garawi A, Solé M, Colmenero J, Nicolás JM, Jiménez W, Weich N, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125:117–125. doi: 10.1016/s0016-5085(03)00695-4. [DOI] [PubMed] [Google Scholar]
- 15.Bataller R, Ginès P, Nicolás JM, Görbig MN, Garcia-Ramallo E, Gasull X, Bosch J, Arroyo V, Rodés J. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118:1149–1156. doi: 10.1016/s0016-5085(00)70368-4. [DOI] [PubMed] [Google Scholar]
- 16.Martinelli AL, Ramalho LN, Zucoloto S. Hepatic stellate cells in hepatitis C patients: relationship with liver iron deposits and severity of liver disease. J Gastroenterol Hepatol. 2004;19:91–98. doi: 10.1111/j.1440-1746.2004.03255.x. [DOI] [PubMed] [Google Scholar]
- 17.Reeves HL, Burt AD, Wood S, Day CP. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis. J Hepatol. 1996;25:677–683. doi: 10.1016/s0168-8278(96)80238-8. [DOI] [PubMed] [Google Scholar]
- 18.Goddard CJ, Smith A, Hoyland JA, Baird P, McMahon RF, Freemont AJ, Shomaf M, Haboubi NY, Warnes TW. Localisation and semiquantitative assessment of hepatic procollagen mRNA in primary biliary cirrhosis. Gut. 1998;43:433–440. doi: 10.1136/gut.43.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terui Y, Saito T, Watanabe H, Togashi H, Kawata S, Kamada Y, Sakuta S. Effect of angiotensin receptor antagonist on liver fibrosis in early stages of chronic hepatitis C. Hepatology. 2002;36:1022. doi: 10.1053/jhep.2002.32679. [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka M, Tsukamoto H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor beta: implication for a pathogenetic role in alcoholic liver fibrogenesis. Hepatology. 1990;11:599–605. doi: 10.1002/hep.1840110412. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther. 2001;15:1667–1672. doi: 10.1046/j.1365-2036.2001.01083.x. [DOI] [PubMed] [Google Scholar]
- 22.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 23.Day CP, James OF. Steatohepatitis: a tale of two "hits". Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 24.George DK, Goldwurm S, MacDonald GA, Cowley LL, Walker NI, Ward PJ, Jazwinska EC, Powell LW. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311–318. doi: 10.1016/s0016-5085(98)70482-2. [DOI] [PubMed] [Google Scholar]