

Abstract
Hepatic fibrosis is a wound healing response, involving pathways of inflammation and fibrogenesis. In response to various insults, such as alcohol, ischemia, viral agents, and medications or hepatotoxins, hepatocyte damage will cause the release of cytokines and other soluble factors by Kupffer cells and other cell types in the liver. These factors lead to activation of hepatic stellate cells, which synthesize large amounts of extracellular matrix components. With chronic injury and fibrosis, liver architecture and metabolism are disrupted, eventually manifesting as cirrhosis and its complications. In addition to eliminating etiology, such as antiviral therapy and pharmacological intervention, it is encouraging that novel strategies are being developed to directly address hepatic injury and fibrosis at the subcellular and molecular levels. With improvement in understanding these mechanisms and pathways, key steps in injury, signaling, activation, and gene expression are being targeted by molecular modalities and other molecular or gene therapy approaches. This article intends to provide an update in terms of the current status of molecular therapy for hepatic injury and fibrosis and how far we are from clinical utilization of these new therapeutic modalities.
Keywords: Fibrosis, Gene therapy, Hepatic stellate cell, Hepatocyte, Injury
INTRODUCTION
Hepatic fibrosis is a wound-healing process after chronic liver injury, and is characterized by the activation of hepatic stellate cells (HSC) and excess production of extracellular matrix (ECM) components. The activation of HSC involves the transdifferentiation from a quiescent state into myofibroblast-like cells with the appearance of smooth muscle α-actin (SMA) and loss of cellular vitamin A storage[1]. The activated HSC are distinguished by accelerated proliferation and enhanced production of ECM components. Cross-talks between parenchymal and non-parenchymal cells constitute the major interactions in the development of hepatic injury and fibrosis. Soluble factors, such as cytokines, chemokines or reactive oxygen species (ROS) are the intermediates in these cross-talks, and are possible targets for therapeutic consideration. A schematic illustration shown in Figure 1 represents the major cascades of the injury process, including the interactions between damaged hepatocytes and Kupffer cell activation, proliferation and activation of HSC, and the excess production of ECM components during hepatic fibrosis. Along with these cascades, possible molecular therapeutic targets are provided in italics. From our improved understanding of the mechanisms underlying liver injury and fibrosis, key steps, such as signaling, activation, and gene expression in specific cell types of the liver, have been targeted by molecular modalities[2,3]. However, various molecular therapeutic approaches carry advantages and disadvantages and patient safety is the primary concern in considering the use of novel modalities[4]. Thus, people may ask: “What is the current status of molecular therapy for hepatic injury and fibrosis” “How far from now these novel modalities will be possibly utilized in patients” This concise review intends to answer these questions.
Figure 1.
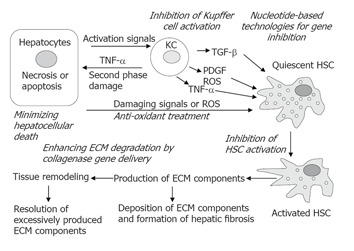
Strategies for the prevention and treatment of hepatic fibrosis. A simplified version of the pathogenesis of hepatic fibrosis is illustrated schematically. At various stages, possible therapeutic strategies are provided in italics. See the text for details. KC = Kupffer cells; ECM = extracellular matrix; HSC = hepatic stellate cells; PDGF = platelet-derived growth factor; ROS = reactive oxygen species; TNF-α = tumor necrosis factor-α; TGF-β = transforming growth factor-β.
TARGETING SPECIFIC MOLECULES OR PATHWAYS
Inhibition of hepatocellular apoptosis
Chronic hepatocellular death via necrosis and/or apoptosis initiates inflammatory responses and hepatic fibrogenesis. Ideally, hepatocytes regenerate and replace dying cells; however, hepatocellular regeneration in chronic liver injury is often inhibited due to the imbalance of growth factors and distortion of liver architecture and circulation. Cellular debris and apoptotic bodies accumulate and initiate inflammatory responses, which may form a self-amplifying loop that further compromises recovery from injury, and facilitates the fibrogenic process[5]. HSC may engulf the apoptotic bodies, which has been demonstrated in vitro, and phagocytosis of apoptotic bodies by quiescent HSC facilitates the phenotypic transdifferentiation to myofibroblasts[6]. Activated HSC with engulfed apoptotic bodies were identified in a rat model of bile-duct ligation and liver biopsy specimens from HCV infection[7].
Tumor necrosis factor-α (TNF-α) (via TNF-receptor-1) can initiate apoptosis[8,9]. This extrinsic pathway is signaled through cell surface death receptors[9], including Fas (also known as CD95), TNF-α-receptor-1, and TNF-α-related-apoptosis-inducing-ligand receptors 1 & 2 (TRAIL-R1 and R2). Examples of this extrinsic pathway of programmed cell death include autoimmune hepatitis, viral hepatitis[10,11], chronic alcohol consumption, D-galactosamine (GalN) plus lipopolysaccharide (LPS)-induced acute liver injury, and ischemia/reperfusion-associated liver injury. In these forms of injury, cytotoxic cytokines or chemokines play a crucial role in the mediation of the injury process[12-18]. In contrast to the extrinsic apoptotic pathway, the intrinsic pathway is based on damage or dysfunction of intracellular organelles, such as lysosomes, endoplasmic reticulum, nucleus and mitochondria[19]. This mechanism involves changes in membrane permeability and integrity of the subcellular organelles. Examples are that damage to nuclear or mitochondrial DNA can initiate apoptosis, and that the release of cytochrome C from miochondria triggers apoptotic cascades. This intrinsic pathway of programmed cell death is often seen in drug toxicity or hepatotoxin-induced liver injury, such as acetaminophen overdose, alcohol toxicity, etc[18,20].
Inhibiting apoptosis will alleviate liver injury, and in turn, delay or stop the progression of hepatic fibrosis. Molecular therapy has specifically targeted various steps in the apoptosis pathway including inhibition of cell death receptors, mediators of the apoptosis cascade, and regulation of the apoptosis cascade. One target is to inhibit Fas receptor expression, which signals the initiation of the apoptosis cascade. Another possible target includes inhibition of caspase expression. Caspases are proteolytic enzymes classified according to their function into ICE-like caspases (caspase 1, 4, 5, 13), death signaling or initiator caspases (caspase 2, 8, 9, 10), and death effector or executioner caspases (caspase 3, 6, 7).
Zhang et al[21] employed antisense oligodeoxynucleotide (ODN) ISIS 22023, specific for mouse Fas to prevent Fas ligand (Jo2 or CD95)-induced fulminant liver failure and mortality. Antisense ODN treatment reduced Fas mRNA and protein expression in the liver by 90%, and completely prevented mortality which was 100% in the control group with extensive hepatocyte apoptosis and liver hemorrhage. In a separate study, Song et al[22] evaluated small interfering RNA (siRNA) against Fas for its ability to inhibit apoptosis and protect the liver in the same animal model. siRNA against the Fas receptor was administered via tail vein injection, which led to 88% of uptake by hepatocytes. The injection decreased Fas mRNA expression by 8 to 10-fold when compared to controls, and the decline in Fas mRNA and Fas protein was sustained for up to 10 d. Control mice displayed bridging fibrosis, whereas mice receiving anti-Fas siRNA injections had no evidence of necrosis or fibrosis. Mice treated with Fas-specific siRNA also had an improved mortality. Eighty-two percent of mice survived over 10 d of observation, whereas all control mice died within 3 d of lethal challenge with Jo2. Thus, both studies demonstrate that the inhibition of Fas receptor synthesis by either specific antisense ODN or siRNA protects animals from Jo2-induced fulminant liver failure and mortality[21-22].
Regulators of apoptosis in the Bcl-2 family have also been targeted with molecular approaches. Bcl-2 is a member of the proto-oncogene family, which blocks cytochrome C release during apoptosis signaling. Several gene products in the Bcl-2 family inhibit apoptosis, including Bcl-xL, Bcl-w, A1, Mcl-1 and Boo, whereas others, such as Bax, Bak, Bok, Bik, Bad and Bid, promote apoptosis[23]. Zhang et al[24] evaluated effects of the antisense against Bcl-xL (ISIS16009) and Bid on Jo2-induced fulminant liver failure in mice. Pre-administration of ISIS16009 reduced liver Bcl-xL expression, potentiated liver apoptosis and increased lethality in this mouse model of Fas-mediated fulminant hepatitis. Antisense ODN against Bid caused an 80% decrease in Bid expression. The reduction of Bid expression by the antisense treatment protected mice from Fas-mediated fulminant hepatitis and death with 100% survival in treated mice[24]. Therefore, Bid seems to be a reasonable target for reducing liver injury in which apoptosis is a predominant pathway of cell death, such as Fas ligand-induced acute liver injury.
Caspase 8 is a downstream target of death receptors and is important in the mediation of apoptosis by Fas and TNF-α. Zender et al[25] evaluated the effects of siRNA against caspase 8 in acute liver damage induced by Jo2 and adenovirus expressing Fas ligand (AdFasL). siRNA against specific caspase 8 target sequences was transfected into Hep G2 cells, and the transfected cells were further infected with AdFasL. Their results confirmed that the inhibition of caspase 8 by siRNA prevented Fas-mediated apoptosis in Hep G2 cells. The animal experiments involved siRNA delivery and administration of Fas antibody or AdFasL. The siRNA delivery resulted in a greater than 3-fold reduction in caspase 8 mRNA expression. Improved survival was significant even when caspase 8 siRNA was administered during ongoing acute liver failure. In addition, the study is of particular interest in that caspase 8 siRNA treatment was successful not only in acute liver failure mediated by specific Fas agonistic agents (Jo2 and AdFasL), but also in acute liver failure caused by an injection of a large amount of wild-type adenoviruses.
Transforming growth factor-β (TGF-β)
TGF-β is the most potent cytokine for enhancing hepatic fibrogenesis. It suppresses hepatocyte proliferation, stimulates the activation of HSC, promotes ECM production, and mediates hepatocyte apoptosis[26]. Smad3 (a transcriptional factor in TGF-β receptor downstream signal transduction) knock-out mice did not develop hepatic fibrosis[27]; while, transgenic mice overexpressing TGF-β1 developed hepatic fibrosis faster than wild type mice, and the fibrosis regressed slower after the withdrawal of the fibrogenic agent[28-30]. Many different strategies of molecular therapy have focused on the inhibition of TGF-β effects by blocking its synthesis, using TGF-β binding proteins, soluble receptors, or targeting its downstream signal transduction pathways.
Qi et al[31] evaluated adenoviral expression of truncated TGF-β receptor type II to abolish TGF-β signaling in liver fibrosis mediated by dimethylnitrosamine (DMN). DMN induces fatty degeneration of hepatocytes, activation of HSC, macrophage infiltration and secretion of TGF-β. Sprague-Dawley rats were given a single infusion of adenoviral vectors (AdCATβ-TR) encoding the TGF-β receptor II gene (TGF-βRII) via portal vein injection. A greater than 20-fold increase in truncated receptor mRNA expression with the adenoviral gene delivery was detected in animals after the TGF-β receptor type II gene delivery with adenoviral vectors. The gene delivery improved the animal mortality, decreased liver hydroxyproline content by 3.4-fold, hyaluronate levels by nearly 20-fold, AST by 100-fold, and ALT by 63-fold. Less hepatocyte injury and fibrosis with a decreased infiltration of monocytes/macrophages and reduced semiquantitative score of fibrosis in histopathology were reported, which was consistent with decreased gene expression of collagen type I, fibronectin, smooth muscle α-actin (SMA), and TGF-β1 in the treated animals. Yata et al[32] administered a soluble TGF-β type II receptor (STR) protein in CCl4-induced liver fibrogenesis in BALB/c mice. Elevated STR levels were associated with a reduction in procollagen type I mRNA expression in the mouse liver. In the 8-wk study, lower concentrations of STR had the greatest effect on procollagen type I mRNA expression. Quantitative morphometrics showed that lower concentrations of STR were the most anti-fibrogenic. These findings verified the antifibrotic effect of inhibiting TGF-β in chronic hepatic injury.
Tumor necrosis factor-α (TNF-α)
TNF-α is a key cytokine involved in many forms of liver injury, and may play a crucial role in HSC activation and hepatocyte regeneration[33-34]. The major cell type for TNF-α production in the liver is Kupffer cells which release TNF-α when activated by factors released by damaged hepatocytes, and by ROS occurrence. TNF-α participates in the second phase of hepatocellular damage via apoptosis and TRAIL receptor activation in alcoholic or other hepatotoxin-induced liver injury[35]. It acts on HSC, and may contribute to the activation process[36]. Accordingly, reducing TNF-α production, or blocking its action will significantly minimize liver injury caused by alcohol toxicity, acetaminophen overdose, or ischemia/reperfusion-associated liver injury in animal models[35,37].
Ponnappa et al[38] has developed a pH-sensitive liposomal formulation consisting of phosphatidyl ethanolamine, cholesteryl hemisuccinate, and cholesterol in a specific ratio (7:4:2), to deliver antisense ODN against TNF-α. This liposomal formulation, which is characterized by its feature of lysosomal resistance, helps release encapsulated-antisense ODN (TJU-2755) into the cytoplasm of Kupffer cells. Rats that received pretreatment with liposome-encapsulated TJU-2755 had a decreased expression of TNF-α mRNA and TNF-α secretion in response to LPS challenge. In subsequent studies, the in vivo efficacy of TJU-2755 was assessed against LPS-induced liver damage in ethanol-fed rats[39]. Liver damage was induced in male Lewis rats fed an ethanol-containing liquid diet for 8-10 wk followed by an intravenous injection of LPS. Pretreatment of the animals with TJU-2755 encapsulated in pH-sensitive anionic liposomes prevented liver damage by 60-70%, as assessed by the release of liver enzymes and histology. These results indicate that pH-sensitive, anionic liposomes can be used to effectively deliver TNF-α antisense ODN against TNF-α-mediated liver injury[39].
Platelet-derived growth factor (PDGF)
PDGF is the most potent mitogen for HSC with effects in growth stimulation, chemotaxis, and intracellular signaling. PDGF expression is upregulated in hepatic injury, as are PDGF receptors in activated HSC. The tyrosine kinase activity of PDGF receptors signals through PI-3K, Ras, Raf-1, and ERK, leading to nuclear translocation and activation of nuclear transcription factors[40]. Interrupting PDGF signal effects with specific agents inhibiting tyrosine phosphorylation, or interrupting down-stream signal transduction pathways attenuates hepatic fibrosis by preventing proliferation and chemotaxis of HSC in vitro and in vivo[40]. Selectively targeting the PDGF receptor by specific antibodies or agents has been considered to be a valuable strategy to block the progression of hepatic fibrogenesis[41-42].
Borkham-Kamphorst et al[43] cloned a chimera of PDGF receptor type β gene, fused to the IgG-Fc domain of human immunoglobulin, into an adenoviral vector. The gene product, soluble PDGF receptor, has a high ligand binding capacity, 100 to 1000-fold higher than that of extracellular monomeric PDGF receptors. The effects of soluble PDGF receptor gene therapy on PDGF-induced DNA synthesis, tyrosine autophosphorylation, and activation of ERK, P13K, and protein kinase B in HSC were evaluated in vitro. The in vitro studies with rat HSC transfected with AdsPDGFR showed decreased levels of PDGF-BB mRNA and thrombospondin-1 with lowered expression of collagen type I mRNA. The receptor gene delivery, however, did not affect levels of SMA or desmin levels in HSC, indicating that it did not significantly affect the HSC activation[43]. In their subsequent study, antisense against PDGF β-chain incorporated into adenoviral vector serotype 5 (Ad5-CMV-PDGF) was transduced in cultured rat HSC[44]. The antisense clearly exhibited the ability to down-regulate endogenous PDGF β-chain and PDGF R-β mRNA as well as procollagen type I gene expression in culture-activated HSC and rat livers[44].
Kupffer cells
Due to the critical role of Kupffer cells in the mediation of liver injury and fibrogenesis, inhibiting Kupffer cell activity has been proposed as a means to reduce liver injury. Ogushi et al[45] modulated Kupffer cell function by administering double stranded antisense ODN targeting NF-κB, the p65 subunit. The mice were first sensitized with intraperitoneal injection of heat killed Propionibacterium acnes. Phosphorothioate modified antisense ODN was delivered with hemagglutinating virus of Japan (HVJ)-liposomal complexes via portal vein injection on d 4 and 7 after P acnes priming challenge. The treated mice also had decreased levels of activated NF-κB translocation as shown by p65 staining. Then, the mice were injected with LPS and sacrificed later. The HVJ-liposome-mediated antisense ODN against NF-κB significantly improved animal survival, and the mortality was decreased to 15% in mice treated with NF-κB decoy ODN as compared to 90% in control mice that died within 24 hours. The treatment with decoy antisense ODN against NF-κB also suppressed the production of proinflammatory cytokines (IL-1β, TNF-α, IL-18, and IL-12) by Kupffer cells and decreased mRNA expression of IFN-γ and Fas-L. Less infiltration of inflammatory cells and destruction of sinusoidal architecture were documented by histopathology in the animals treated with antisense ODN against NF-κB[45].
REACTIVE OXYGEN SPECIES AND FREE RADICAL SCAVENGERS
Oxidative stress is an important mechanism in liver injury. ROS include superoxide anions, hydroxyl radicals, hydrogen peroxide, and hydroxyethyl radicals (HER), and are generated from a variety of insults such as drug/toxin metabolites, ischemia/reperfusion, and alcohol metabolism. ROS are involved in necrosis and apoptosis of hepatocytes, and contribute to HSC activation[2,46]. Several major classes of free radical scavengers, such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GSH-P), as well as SOD mimics, were investigated in various forms of liver injury, and they afforded effective protection against the oxidative insults to the tissue[47-48].
Three isoforms of SOD exist. Copper/Zinc (Cu/Zn) SOD is localized to the cytosol and nucleus, manganese (Mn) SOD is localized to the mitochondria and, extracellular (EC) SOD is primarily localized to the interstitial matrix. We employed polycationic liposomes previously formulated by our group with polycationic lipids (PCL)[49] to deliver the EC-SOD gene for the prevention of acute liver injury[50]. Our in vitro experiments showed that transfection of the EC-SOD plasmid in Hep G2 and Hep3B cells led to an increase in SOD activity in cell culture medium. Transfected cells were more resistant to the exposure of superoxide anions generated by hypoxanthine and xanthine oxidase, and to HER by an HER-generating system. We employed polycationic liposomes to deliver the human EC-SOD gene via portal vein injection in mice[50]. Two days after the gene delivery, a 55-fold increase in human EC-SOD gene expression was detected. When mice receiving the EC-SOD gene delivery were challenged with GalN and LPS, their serum ALT levels were much lower than those receiving control vector delivery, whereas their serum SOD levels were higher. Moreover, liver levels of the reduced form of glutathione (GSH) were preserved, and lipid peroxidation products, malondialdehyde (MDA) and 4-hydroxyalkenal (HAE) were reduced. Liver histology confirmed the effectiveness of the gene delivery. Thus, the overexpression of the human EC-SOD gene protected the mice from hepatotoxin-induced liver injury[50]. Laukkaen et al[51] achieved similar results when adenovirus-derived EC-SOD was delivered to prevent acetaminophen-induced acute liver injury in mice. Delivery of the Cu/Zn-SOD gene by adenoviral vector was also shown to protect rats from alcohol-induced liver injury[52]. Cu/Zn-SOD was expressed in approximately 60% of liver cells with 3-5 fold higher levels of SOD compared to controls. The treatment of alcohol-fed rats with Cu/Zn-SOD gene delivery attenuated alcohol-induced elevation of serum aminotransferase levels with a 60% decrease of ALT levels and a 39% decrease in AST levels. Liver histopathology demonstrated less inflammation, reduced steatosis and no detectable necrosis. Free radical adduct formation measured by electron spin resonance (ESR) spectroscopy was reduced in mice treated with Ad.Cu/Zn-SOD. Nuclear extracts examined by electrophoretic mobility shift assays demonstrated a decrease in activation and translocation of NF-κB. Thus, these studies provide substantial evidence that the delivery of SOD isoforms will protect the liver from acute and chronic liver injury in which the generation of ROS is a major component in the pathogenesis of the injury process.
Zhong et al[53] evaluated adenoviral delivery of either Mn-SOD or Cu/Zn-SOD in a cholestatic model of liver injury. Rats were treated with AdMnSOD or AdCu/ZnSOD 3 d prior to bile duct ligation (BDL). Over 90% of hepatocytes were infected by virus. Mn-SOD and Cu/Zn-SOD activity increased 4-fold at d 3 and remained elevated up to 2-fold for 3 wk after the injection of adenoviruses. Elevated serum aminotransferase levels in rats with BDL were abrogated by 70% with AdMnSOD and 40% with AdCu/ZnSOD. Alkaline phosphatase (ALP) release was reduced by 50% with AdMnSOD and by 25% with AdCu/ZnSOD. AdMnSOD was also able to abrogate the increase in 4-hydroxynonenal (lipid peroxidation product). Enhanced NF-κB activation, TGF-β and TNF-α expression in the cholestatic liver were reversed by AdMnSOD delivery. Furthermore, BDL caused enhanced fibrosis and a 20-fold increased level of procollagen type I (α1) mRNA in the liver. These changes were reversed significantly by prior Ad-Mn-SOD gene delivery. Thus, it appears that antioxidant gene delivery is a promising approach to prevent a variety of forms of liver injury, including drug-induced toxicity, ethanol consumption, and cholestasis or ischemia/reperfusion-associated liver injury[54], as well as hepatic fibrogenesis[53].
EXTRACELLULAR MATRIX REMODELING
ECM is composed of collagens, glycoproteins, proteoglycans, glycosaminoglycans, and other proteins[55-56]. In the normal liver, collagens types I, III, V and XI are principally found in the capsule, around large vessels, and in the portal triad. Of note, the ECM components of the subendothelial space are typically low in collagen type I. Changes in the proportion of ECM components also contribute to the activation of HSC. Matrix metalloproteinases (MMPs) are important in the modeling and degradation of ECM, and they are classified according to their target substrates as interstitial collagenases, gelatinases, stromelysins, and metalloelastases[57-58]. Stromelysin-1 degrades proteoglycans and glycoproteins. MMP-1 degrades collagen type I, while MMP-2 and MMP-9 degrade collagen type IV. These MMPs require activation by proteolytic cleavage and are inactivated by tissue inhibitors of metalloproteinases (TIMPs). The balance of production and degradation of ECM components maintains normal liver structure. The increased and disordered deposition of ECM components that occurs in fibrotic liver disease, especially fibrillar collagen types I and III, is due to unbalanced excessive production and reduced degradation. Enhancing the production of collagenase by gene delivery is a possible strategy to promote tissue remodeling and minimize the net accumulation of ECM components in chronic liver injury. Various collagenase genes have been delivered in attempts to decrease ECM deposition in animal models of hepatic fibrosis. Iimuro et al[59] evaluated the effectiveness of adenovirus-mediated gene delivery of matrix metalloproteinase-1 on the attenuation of liver fibrosis induced by thioacetamide or BDL in rats. The administration of Ad5MMP-1 significantly attenuated hepatic fibrosis with resolution within 2 wk, a decrease in cross-linked collagen type I, hydroxyproline content, and SMA-positive HSC, which corresponded to a decreased number of activated HSC in the liver sections. The treatment with Ad5MMP1 induced a transient increase in serum ALT levels that resolved by wk 4. This may reflect hepatocyte damage induced by adenoviral gene delivery. Subsequently, enhanced hepatocyte proliferation was found in the animals treated with MMP-1 gene therapy.
Siller-López et al[60] evaluated the efficacy of adenoviral delivery of MMP8 in ameliorating liver cirrhosis induced by either BDL or CCl4 intoxication in rats. Marked expression of MMP-8 was found to reach a level of 550 pg/mL in AdMMP8-treated cirrhotic animals. This was accompanied with a significant decrease of a fibrotic score by 45% in BDL rats, as indicated by decreased liver hydroxyproline content. Moreover, TIMP-1, MMP-9, and HGF were found to be increased, whereas, TGF-β was decreased in animals receiving the MMP-8 gene delivery. The gene delivery also improved ascites, hepatic function tests, and gastric varices in these cirrhotic rats. Thus, MMP-8 gene delivery seems to be highly effective in accelerating the resolution of the deposited ECM components in experimental animal models of hepatic fibrosis. However, immunoreaction to the adenoviral vectors or a high viral titer used, caused marked hepatocyte damage in those animals receiving adenoviral gene delivery[60].
Urokinase-type plasminogen activator (uPA) is known to be an initiator of matrix proteolysis and promotes degradation of the extracellular matrix. Salgado et al[61] employed adenoviral vector to deliver human uPA in a rat model of hepatic fibrosis induced by chronic CCl4 intoxication. At the end of 6 wk of repeated CCl4 injections, rats were given Ad-ΔhuPA via iliac vein injection. Animals were sacrificed 2 d after Ad-ΔhuPA treatment for evaluating the gene delivery efficacy. Human uPA gene expression was detected in 46-80% of hepatocytes with marked elevation of human uPA in serum. The successful human uPA gene delivery resulted in an almost complete resolution of periportal and centrilobular fibrosis when compared to the animals with a control vector injection (Ad-GFP) as evidenced by histochemical staining for ECM components. The gene delivery also led to an elevated MMP-2 activity, and induced hepatocyte proliferation with increased levels of HGF. Thus, the study indicated that enhanced uPA expression by a gene delivery approach may activate MMP activity and accelerate ECM degradation, as well as facilitate a resolution of hepatic fibrosis. The same group performed another experiment in BDL rats and found that combination of adenoviral delivery of human uPA gene with biliodigestive anastomosis achieved a synergistic effect in reducing hepatic fibrosis[62]. These studies provide substantial evidence that hepatic fibrosis is reversible in animal models when the underlying injury process is attenuated or stopped and an accelerated tissue remodeling occurs.
CONCLUSIONS AND PROSPECTS
New understanding and insights into the pathogenesis of hepatocellular damage and hepatic fibrogenesis, and the development of molecular techniques, such as new viral or non-viral vector systems, RNAi, ribozyme and antisense technologies have made it possible to modulate the expression of specific genes involved in the key pathways of liver injury and fibrosis. The fact that blocking Fas-ligand activity by siRNA or antisense against Fas receptor almost completely prevented Fas ligand-induced fulminant liver failure and animal mortality is noteworthy. It is also striking that silencing of a key enzyme, caspase 3 or 7, in an apoptotic cascade by siRNA blocks the cell damage. The transfer of anti-oxidant genes, such as isoforms of SOD, by viral or non-viral vectors prevents a variety of forms of liver injury in which ROS generation is involved in the pathogenic pathways. Alternatively, when the overexpression of fibrogenic cytokines, such as TGF-β and PDGF and their receptors are inhibited at the mRNA level with antisense, ribozymes or siRNA, or interrupted by specific antibodies or protein molecules, the interventions result in marked amelioration of hepatic fibrosis. Moreover, delivery of collagenase genes by adenoviral vectors promotes the resolution of excessive deposition of ECM components and reverses hepatic fibrosis. Although these progresses in treatment of liver injury and fibrogenesis are encouraging, one issue that requires additional attention remains that targeting these molecular therapeutics to specific cell types (hepatocytes, Kupffer cells or HSC) is critical in avoiding undesired effects on other organs or cell types[39,41,63].
Gene therapy itself is still in its infancy, and ideal gene delivery systems for selective gene transfer with high and prolonged gene expression, as well as less cytotoxicity or immunogenicity remain to be developed. Nevertheless, molecular therapeutics has already proven to be powerful research tools and explorative experimental medicine. They have demonstrated some promise as novel modalities in reducing liver injury, inhibiting HSC activation, and promoting the resolution of fibrotic deposition. However, the transition of promising molecular therapeutics from animal models to clinical trials for the validation of their clinical efficacy, adverse effects, target patient population will usually take a quite long period of time. One may anticipate that in combination with other more standard therapeutic modalities, these molecular approaches may yield effective treatments for hepatic injury and fibrogenesis in man in the foreseeable future.
Footnotes
Supported by NIH grant (DK069939), and the Liver Scholar Award by the American Liver Foundation to J.W.
S- Editor Guo SY and Pan BR L- Editor Elsevier HK E- Editor Kong LH
References
- 1.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 2.Wu J, Zern MA. Hepatic stellate cells: a target for the treatment of liver fibrosis. J Gastroenterol. 2000;35:665–672. doi: 10.1007/s005350070045. [DOI] [PubMed] [Google Scholar]
- 3.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen TH, Ferry N. Liver gene therapy: advances and hurdles. Gene Ther. 2004;11 Suppl 1:S76–S84. doi: 10.1038/sj.gt.3302373. [DOI] [PubMed] [Google Scholar]
- 5.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 6.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 7.Torok N, Wu J, Zern MA, Halsted C, French S, Friedman SL, Zhan SS. Phagocytosis of apoptotic bodies by hepatic stellate cells occurs in vivo and is an important mechanism in liver fibrogenesis. Digestive Disease Week (DDW), May 14-19, 2005, Chicago, IL (oral presentation, Abstract No. 80) Gastroenterology. 2005;125:4(S2): A15. [Google Scholar]
- 8.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 9.Gores GJ, Kaufmann SH. Is TRAIL hepatotoxic. Hepatology. 2001;34:3–6. doi: 10.1053/jhep.2001.25173. [DOI] [PubMed] [Google Scholar]
- 10.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 11.Kountouras J, Zavos C, Chatzopoulos D. Apoptosis in hepatitis C. J Viral Hepat. 2003;10:335–342. doi: 10.1046/j.1365-2893.2003.00452.x. [DOI] [PubMed] [Google Scholar]
- 12.Rivero M, Crespo J, Fábrega E, Casafont F, Mayorga M, Gomez-Fleitas M, Pons-Romero F. Apoptosis mediated by the Fas system in the fulminant hepatitis by hepatitis B virus. J Viral Hepat. 2002;9:107–113. doi: 10.1046/j.1365-2893.2002.00338.x. [DOI] [PubMed] [Google Scholar]
- 13.Fox CK, Furtwaengler A, Nepomuceno RR, Martinez OM, Krams SM. Apoptotic pathways in primary biliary cirrhosis and autoimmune hepatitis. Liver. 2001;21:272–279. doi: 10.1034/j.1600-0676.2001.021004272.x. [DOI] [PubMed] [Google Scholar]
- 14.Nakama T, Hirono S, Moriuchi A, Hasuike S, Nagata K, Hori T, Ido A, Hayashi K, Tsubouchi H. Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 2001;33:1441–1450. doi: 10.1053/jhep.2001.24561. [DOI] [PubMed] [Google Scholar]
- 15.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 16.Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology. 2004;126:1387–1399. doi: 10.1053/j.gastro.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Realdon S, Gerotto M, Dal Pero F, Marin O, Granato A, Basso G, Muraca M, Alberti A. Proapoptotic effect of hepatitis C virus CORE protein in transiently transfected cells is enhanced by nuclear localization and is dependent on PKR activation. J Hepatol. 2004;40:77–85. doi: 10.1016/j.jhep.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Jaeschke H, Gujral JS, Bajt ML. Apoptosis and necrosis in liver disease. Liver Int. 2004;24:85–89. doi: 10.1111/j.1478-3231.2004.0906.x. [DOI] [PubMed] [Google Scholar]
- 19.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Danielsson A, Zern MA. Toxicity of hepatotoxins: new insights into mechanisms and therapy. Expert Opin Investig Drugs. 1999;8:585–607. doi: 10.1517/13543784.8.5.585. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Cook J, Nickel J, Yu R, Stecker K, Myers K, Dean NM. Reduction of liver Fas expression by an antisense oligonucleotide protects mice from fulminant hepatitis. Nat Biotechnol. 2000;18:862–867. doi: 10.1038/78475. [DOI] [PubMed] [Google Scholar]
- 22.Song E, Lee SK, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9:347–351. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 23.Pellegrini M, Strasser A. A portrait of the Bcl-2 protein family: life, death, and the whole picture. J Clin Immunol. 1999;19:365–377. doi: 10.1023/a:1020598632068. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Taylor J, Luther D, Johnston J, Murray S, Wyatt JR, Watt AT, Koo S, York-DeFalco C, Stecker K, et al. Antisense oligonucleotide inhibition of Bcl-xL and Bid expression in liver regulates responses in a mouse model of Fas-induced fulminant hepatitis. J Pharmacol Exp Ther. 2003;307:24–33. doi: 10.1124/jpet.103.050435. [DOI] [PubMed] [Google Scholar]
- 25.Zender L, Hutker S, Liedtke C, Tillmann HL, Zender S, Mundt B, Waltemathe M, Gosling T, Flemming P, Malek NP, et al. Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci USA. 2003;100:7797–7802. doi: 10.1073/pnas.1330920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 27.Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 28.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, Rose-John S, zum Büschenfelde KH, Blessing M. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol. 1999;276:G1059–G1068. doi: 10.1152/ajpgi.1999.276.4.G1059. [DOI] [PubMed] [Google Scholar]
- 30.Schnur J, Oláh J, Szepesi A, Nagy P, Thorgeirsson SS. Thioacetamide-induced hepatic fibrosis in transforming growth factor beta-1 transgenic mice. Eur J Gastroenterol Hepatol. 2004;16:127–133. doi: 10.1097/00042737-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H. Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci USA. 1999;96:2345–2349. doi: 10.1073/pnas.96.5.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yata Y, Gotwals P, Koteliansky V, Rockey DC. Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: implications for antifibrotic therapy. Hepatology. 2002;35:1022–1030. doi: 10.1053/jhep.2002.32673. [DOI] [PubMed] [Google Scholar]
- 33.Marra F. Chemokines in liver inflammation and fibrosis. Front Biosci. 2002;7:d1899–d1914. doi: 10.2741/A887. [DOI] [PubMed] [Google Scholar]
- 34.Diehl AM. Liver regeneration. Front Biosci. 2002;7:e301–e314. doi: 10.2741/A925. [DOI] [PubMed] [Google Scholar]
- 35.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 36.Reeves HL, Friedman SL. Activation of hepatic stellate cells--a key issue in liver fibrosis. Front Biosci. 2002;7:d808–d826. doi: 10.2741/reeves. [DOI] [PubMed] [Google Scholar]
- 37.Diehl AM. Tumor necrosis factor and its potential role in insulin resistance and nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:619–38, x. doi: 10.1016/j.cld.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Ponnappa BC, Dey I, Tu GC, Zhou F, Aini M, Cao QN, Israel Y. In vivo delivery of antisense oligonucleotides in pH-sensitive liposomes inhibits lipopolysaccharide-induced production of tumor necrosis factor-alpha in rats. J Pharmacol Exp Ther. 2001;297:1129–1136. [PubMed] [Google Scholar]
- 39.Ponnappa BC, Israel Y. Targeting Kupffer cells with antisense oligonucleotides. Front Biosci. 2002;7:e223–e233. doi: 10.2741/A918. [DOI] [PubMed] [Google Scholar]
- 40.Pinzani M. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 2002;7:d1720–d1726. doi: 10.2741/A875. [DOI] [PubMed] [Google Scholar]
- 41.Beljaars L, Meijer DK, Poelstra K. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front Biosci. 2002;7:e214–e222. doi: 10.2741/A917. [DOI] [PubMed] [Google Scholar]
- 42.Beljaars L, Weert B, Geerts A, Meijer DK, Poelstra K. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue. Biochem Pharmacol. 2003;66:1307–1317. doi: 10.1016/s0006-2952(03)00445-3. [DOI] [PubMed] [Google Scholar]
- 43.Borkham-Kamphorst E, Stoll D, Gressner AM, Weiskirchen R. Inhibitory effect of soluble PDGF-beta receptor in culture-activated hepatic stellate cells. Biochem Biophys Res Commun. 2004;317:451–462. doi: 10.1016/j.bbrc.2004.03.064. [DOI] [PubMed] [Google Scholar]
- 44.Borkham-Kamphorst E, Stoll D, Gressner AM, Weiskirchen R. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem Biophys Res Commun. 2004;321:413–423. doi: 10.1016/j.bbrc.2004.06.153. [DOI] [PubMed] [Google Scholar]
- 45.Ogushi I, Iimuro Y, Seki E, Son G, Hirano T, Hada T, Tsutsui H, Nakanishi K, Morishita R, Kaneda Y, et al. Nuclear factor kappa B decoy oligodeoxynucleotides prevent endotoxin-induced fatal liver failure in a murine model. Hepatology. 2003;38:335–344. doi: 10.1053/jhep.2003.50298. [DOI] [PubMed] [Google Scholar]
- 46.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen WD, Kim DH, Alam HB, Provido HS, Kirkpatrick JR. Polyethylene glycol-superoxide dismutase inhibits lipid peroxidation in hepatic ischemia/reperfusion injury. Crit Care. 1999;3:127–130. doi: 10.1186/cc358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferret PJ, Hammoud R, Tulliez M, Tran A, Trébéden H, Jaffray P, Malassagne B, Calmus Y, Weill B, Batteux F. Detoxification of reactive oxygen species by a nonpeptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology. 2001;33:1173–1180. doi: 10.1053/jhep.2001.24267. [DOI] [PubMed] [Google Scholar]
- 49.Liu L, Zern MA, Lizarzaburu ME, Nantz MH, Wu J. Poly(cationic lipid)-mediated in vivo gene delivery to mouse liver. Gene Ther. 2003;10:180–187. doi: 10.1038/sj.gt.3301861. [DOI] [PubMed] [Google Scholar]
- 50.Wu J, Liu L, Yen RD, Catana A, Nantz MH, Zern MA. Liposome-mediated extracellular superoxide dismutase gene delivery protects against acute liver injury in mice. Hepatology. 2004;40:195–204. doi: 10.1002/hep.20288. [DOI] [PubMed] [Google Scholar]
- 51.Laukkanen MO, Leppanen P, Turunen P, Tuomisto T, Naarala J, Yla-Herttuala S. EC-SOD gene therapy reduces paracetamol-induced liver damage in mice. J Gene Med. 2001;3:321–325. doi: 10.1002/jgm.194. [DOI] [PubMed] [Google Scholar]
- 52.Wheeler MD, Kono H, Yin M, Rusyn I, Froh M, Connor HD, Mason RP, Samulski RJ, Thurman RG. Delivery of the Cu/Zn-superoxide dismutase gene with adenovirus reduces early alcohol-induced liver injury in rats. Gastroenterology. 2001;120:1241–1250. doi: 10.1053/gast.2001.23253. [DOI] [PubMed] [Google Scholar]
- 53.Zhong Z, Froh M, Wheeler MD, Smutney O, Lehmann TG, Thurman RG. Viral gene delivery of superoxide dismutase attenuates experimental cholestasis-induced liver fibrosis in the rat. Gene Ther. 2002;9:183–191. doi: 10.1038/sj.gt.3301638. [DOI] [PubMed] [Google Scholar]
- 54.Wheeler MD, Katuna M, Smutney OM, Froh M, Dikalova A, Mason RP, Samulski RJ, Thurman RG. Comparison of the effect of adenoviral delivery of three superoxide dismutase genes against hepatic ischemia-reperfusion injury. Hum Gene Ther. 2001;12:2167–2177. doi: 10.1089/10430340152710513. [DOI] [PubMed] [Google Scholar]
- 55.Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–372. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 56.Rojkind M, Giambrone MA, Biempica L. Collagen types in normal and cirrhotic liver. Gastroenterology. 1979;76:710–719. [PubMed] [Google Scholar]
- 57.Gressner AM. The cell biology of liver fibrogenesis - an imbalance of proliferation, growth arrest and apoptosis of myofibroblasts. Cell Tissue Res. 1998;292:447–452. doi: 10.1007/s004410051073. [DOI] [PubMed] [Google Scholar]
- 58.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 59.Iimuro Y, Nishio T, Morimoto T, Nitta T, Stefanovic B, Choi SK, Brenner DA, Yamaoka Y. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124:445–458. doi: 10.1053/gast.2003.50063. [DOI] [PubMed] [Google Scholar]
- 60.Siller-López F, Sandoval A, Salgado S, Salazar A, Bueno M, Garcia J, Vera J, Gálvez J, Hernández I, Ramos M, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126:1122–133; discussion 949. doi: 10.1053/j.gastro.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 61.Salgado S, Garcia J, Vera J, Siller F, Bueno M, Miranda A, Segura A, Grijalva G, Segura J, Orozco H, et al. Liver cirrhosis is reverted by urokinase-type plasminogen activator gene therapy. Mol Ther. 2000;2:545–551. doi: 10.1006/mthe.2000.0210. [DOI] [PubMed] [Google Scholar]
- 62.Miranda-Díaz A, Rincón AR, Salgado S, Vera-Cruz J, Gálvez J, Islas MC, Berumen J, Aguilar-Cordova E, Armendáriz-Borunda J. Improved effects of viral gene delivery of human uPA plus biliodigestive anastomosis induce recovery from experimental biliary cirrhosis. Mol Ther. 2004;9:30–37. doi: 10.1016/j.ymthe.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Wu J, Nantz MH, Zern MA. Targeting hepatocytes for drug and gene delivery: emerging novel approaches and applications. Front Biosci. 2002;7:d717–d725. doi: 10.2741/A806. [DOI] [PubMed] [Google Scholar]