

Abstract
AIM: To investigate the expression of cyclooxygenase-2 (COX-2) and epithelial growth factor receptor (EGFR) throughout the progression of Barrett’s esophagus (BE).
METHODS: COX-2 and EGFR protein expressions were detected by using immunohistochemical method. A detailed cytomorphological changes were determined. Areas of COX-2 and EGFR expression were quantified by using computer Imaging System.
RESULTS: The expressions of both COX-2 and EGFR increased along with the progression from BE to esophagus adenocarcinoma (EAC). A positive correlation was found between COX-2 expression and EGFR expression.
CONCLUSION: COX-2 and EGFR may be cooperative in the stepwise progression from BE to EAC, thereby leading to carcinogenesis.
Keywords: Cycloxygenase -2 (COX-2), Epithelial growth factor receptor (EGFR), Barrett’s esophagus, Carcinogenesis
INTRODUCTION
Esophageal adenocarcinoma (EAC) is one of the most rapidly increasing cancers over the past two decades in Caucasian males in the United States[1,2]. After being diagnosed with adenocarcinoma, patients have only a 13-15% 5-year survival rate[3]. Most of the EAC are developed from Barrett’s esophagus (BE), a pre-malignant disease characterized by replacement of the normal esophageal squamous epithelium with a specialized intestinal metaplasia (SIM)[4]. The risk of EAC in BE patients has been estimated to be about 30 -125 times higher than the general population[2,5].
The mechanisms of the evolution from BE to EAC are largely unknown[6,7]. During the neoplastic transformation, aberrations in the cell cycle lead to uncontrolled cellular replication, a molecular mechanism is believed to be involved in all carcinogenesis[8]. Epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases lies at the head of a complex signal transduction cascade that modulates cell proliferation, differentiation, adhesion and migration[9]. EGFR is a transmembrane receptor composed of an extracellular ligand-binding domain and a cytoplasmic region with enzymatic activity. This structure enables signals to be transmitted across the plasma membrane activating gene expression and inducing cellular responses, such as proliferation and differentiation. Over-expression of EGFR has been shown to occur in various malignancies, including esophageal adenocarcinoma[10].
Another important enzyme involved in tumor growth is cycloxygenase-2 (COX-2)[11]. There are two isoforms of COX enzyme. COX-1 is constitutively expressed in most tissues; however, COX-2 has a markedly different expression pattern. Normally, COX-2 is undetectable in most tissues, but can be expressed at high levels after induction with a variety of substances, including inflammatory mediators and mitogens[12-14]. Epidemiological studies have shown that intake of aspirin, a nonsteroidal anti-inflammatory drug (NSAID) known as COX enzymes blocker, is associated with an up to 90% decreased risk of developing esophageal cancer[15,16]. Experimental studies also have indicated that over-expression of COX-2 is associated with decreased apoptosis and cell-cell adhesion, and with increased proliferation and differentiation[17,18].
Recently, experimental investigations have suggested a close relationship between COX-2 and EGFR. Expression of EGFR was markedly increased in the COX-2-transfected cells , and inhibition of COX-2 could suppress the induction of EGFR in these cells[19]. It has also been demonstrated that activation of EGFR can induce COX-2 expression and prostaglandin production[20]. Furthermore, a combination of an EGFR inhibitor with a nonselective COX-1/COX-2 inhibitor was shown to prevent the development of intestinal cancer in nude mice[21]. Taken together, these data link COX and EGFR, and provide a theoretical basis whereby the COX-2 and EGFR signaling pathway may have additive effects of tumor growth. Some studies on expression of COX-2[18] and EGFR[22] in the human Barrett’s esophagus specimens have been reported. However, to our knowledge, a detailed morphologic description of the esophageal epithelium expressing both COX-2 and EGFR in the BE metaplasia to dysplasia to adenocarcinoma sequence has not yet been performed.
In the present study, we therefore investigated the expressions of both COX-2 and EGFR in biopsy tissues from different histologic stages of BE by using immunohistochemical assay, and addressed the morphological features of Barrett’s epithelium with over-expressed protein levels of COX-2 and EGFR in the histologic stages of metaplasia and dysplasia. Furthermore, we set forth to determine the correlation between COX-2 and EGFR in progression from the metaplasia to dysplasia to adenocarcinoma sequence.
MATERIALS AND METHODS
Case selection and review
The study samples were collected retrospectively from 104 patients who had undergone esophageal biopsy between 1996 and 2004. All of these patients were recruited from the Barrett’s Esophagus registry at the University of Louisville with established diagnoses of SIM, indefinite/low-grade dysplasia (I/LGD), high-grade dysplasia (HGD), and EAC. No preoperative radio- or chemotherapy had been performed. None of the patients had received NSAIDs therapy. A microscope examination confirmed the diagnoses of SIM, LGD, HGD and EAC on these esophageal tissues reviewed by two pathologists independently, blinded to the subject’s clinical history. Alcian-blue staining was used to identify any presence of SIM. Hematoxylin-eosin staining was used to identify any presence of dysplasia and EAC. A control group consisted of 30 subjects with nondetectable Barrett’s (NB), defined by endoscopic findings of columnar-lined esophagus (CLE), but no pathologic evidence of SIM despite alcian-blue staining on all of the four-quadrant biopsies. This study was approved by the Institutional Review Board for Human Study at the University of Louisville.
Immunohistochemica assay for COX-2 and EGFR
COX-2 and EGFR protein expressions were determined by using an immunohistochemical assay. Staining was carried out on the paraffin-embedded tissues using the DAKO EnVision™+System Kit (DAKO Corporation, Carpinteria, CA) according to the manufacturer’s instructions. In brief, the sections were deparaffinized and hydrated. The slides were washed with TRIS-buffer, and peroxidase blocking was performed for 5 min. After rewashing, the slides were incubated separately with the monoclonal mouse COX-2 antibody (1:200) or EGFR antibody (1:200) (SantaCruz Biotechnology Inc, CA) for 30 min at room temperature. The chromogenic substrate diaminobenzidine was added as a visualization reagent. Finally, the slides were counterstained with haematoxylin. A negative control was included in each run.
Cytomorphology associated with localization of COX-2 and EGFR
Microscopic evaluation of the morphological features of epithelium was performed at high-power fields (X 400 magnification) on the COX-2 and EGFR staining sections. Two cytomorphological features were chosen as the criteria of assessment based on observed morphological differences among SIM, I/LGD, HGD and EAC along with NB columnar epithelium: (1) ordering of nuclei; and (2) the presence of basally located nuclei with an apical cytoplasmic layer. We considered the normal cell phenotype (NCP) when the epithelial cells were arranged in a single layer with their long axes orientated towards the center of the lumen, and had a basally located nucleus with a thick apical layer of cytoplasm, whereas the abnormal cell phenotype (ACP) included multiple cell layers, displayed and enlarged nucleus and less cytoplasm. Every slide was examined throughout to identify the NCP cell and ACP cell with COX-2 staining and EGFR staining. Then, the COX-2-stained and the EGFR-stained epithelial cells were categorized to NCP cells and ACP cells. The other cells were ignored during the evaluation.
Computer image analysis
A computer image analysis was performed to quantify the expressions of COX-2 and EGFR in totally 104 subjects diagnosed with SIM, I/LGD, HGD and EAC, and in 30 controls. The imaging fields were chosen randomly from various section levels to ensure objectivity of sampling. Five imaging fields were scanned for each specimen sample. All digital images were acquired with the microscope at 40x magnification using the Spot camera via the MetaMorph® Imaging System (Universal Imaging Corporation., Downingtown, PA) and stored as JPG data files (the resolutions were fixed as 200 pixels/inch). The acquired color images from the immunohistochemical staining were defined a standard threshold according to the software specification. The computer program then quantified the threshold area represented by color images. COX-2 and EGFR protein expressions were defined by the percentages of threshold area in acquired color images.
Statistical analysis
Spearman rank correlation coefficient (R) was used to analyze the correlation between COX-2 and EGFR expressions with the various histologic stages of Barrett’s esophagus. Multiple analysis of variance (MANOVA) was used to determine the differences between COX-2 and EGFR expressions, if any, between the different histologic stages of carcinogenesis. P values less than 0.05 were considered statistically significant.
RESULTS
One hundred and four subjects (92 males, 12 females) with Barrett’s esophagus were enrolled in this study. The study population consisted of 37 subjects with a diagnosis of SIM without dysplasia, 36 with I/LGD, 16 with HGD, and 15 with EAC. The control group consisted of 30 NB subjects. Patients identified as having no Barrett’s were mostly those with a short segment of CLE whereas those identified with SIM, I/LDG, HDG or EAC were mostly found to have a long segment of CLE (Table 1).
Table 1.
Demographics of 104 study subjects with Barrett’s esophagus and 30 control subjects
| NB | SIM | I/LDG | HDG | EAC | |
| Subjects | 30 | 37 | 36 | 16 | 15 |
| Gender (male:female) | 28:2 | 34:3 | 30:6 | 14:2 | 14:1 |
| Mean age (yr ) | 60 | 55 | 61 | 68 | 66 |
| Short-segment CLE | 27 | 21 | 7 | 1 | 0 |
| Long-segment CLE | 3 | 16 | 29 | 15 | 15 |
SIM: Specialized intestinal metaplasia; I/LGD: Indeterminate/low-grade dysplasia; HGD: High-grade dysplasia; EAC: Esophageal adenocarcinoma; NB: Nondetectable Barrett’s; CLE: Columnar-lined esophagus.
Cytomorphological features associated with localization of COX-2 and EGFR
A very strong COX-2 staining in all Barrett’s epithelium compared to a weak staining in normal squamous epithelium and CLE without Barrett’s epithelium was observed. In particular, in the regions with COX-2-positive staining, the epithelial cells presented cytomorphological features of both NCP cells and ACP cells in the histological stages of SIM, I/LGD, HGD and EAC (Figure 1). A positive EGFR staining was presented in all Barrett’s epithelium, but not as strong as that seen in the COX-2 staining. In contrast to COX-2 staining, the regions of positive EGFR expression exhibited ACP cells while those regions with weak or no EGFR staining exhibited NCP cells (Figure 2).
Figure 1.
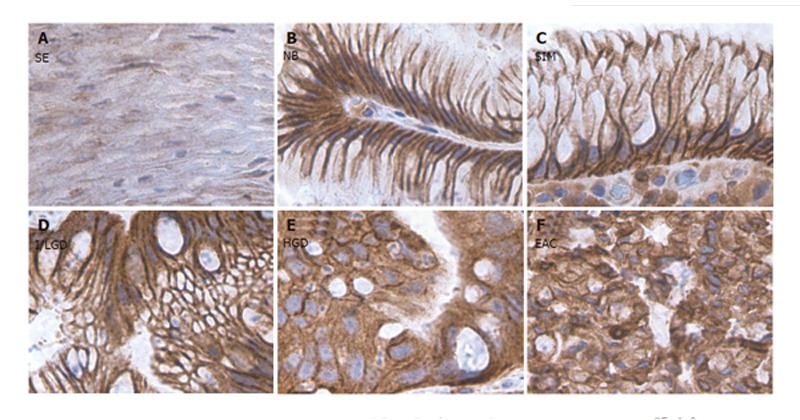
COX-2 staining in the sections from different stages of BE along with the sections from normal squamous and CLE nondetectable BE. A: Squamous epithelium (SE); B: Nondetectable BE in CLE (NB); C: Specialized intestinal metaplasia (SIM); D: Indeterminate/low-grade dysplasia (I/LGD); E: high-grade dysplasia (HGD); F: Esophageal adenocarcinoma (EAC). Strong COX-2 staining was detected in both NCP cells and ACP cells in the histological stages of SIM, I/LGD, HGD and EAC. A weak COX-2 staining was observed in the normal squamous epithelium.
Figure 2.
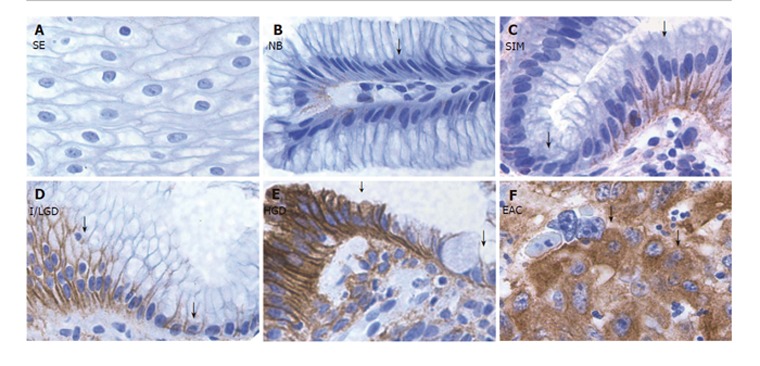
EGFR staining in the sections from different stages of BE along with the sections from normal squamous and CLE nondetectable BE. A: Squamous epithelium (SE); B: Nondetectable BE in CLE (NB); C: Specialized intestinal metaplasia (SIM); D: Indeterminate/low-grade dysplasia (I/LGD); E: high-grade dysplasia (HGD); F: Esophageal adenocarcinoma (EAC). Strong EGFR staining was detected in ACP cells (arrowhead) in the histological stages of SIM, I/LGD, HGD and EAC. A weak EGFR staining was found in NCP cells (arrow) and in the normal squamous epithelium.
Expression of COX-2 in the metaplasia to dysplasia to adenocarcinoma sequence
We observed significant increases in COX-2 expression in specimens with SIM (7.8 ± 0.8), I/LGD (10.0 ± 0.9), HGD (14.2 ± 1.8) and EAC (16.0 ± 2.2) as compared with the control specimens (6.9 ± 1) (P < 0.05). Furthermore, COX-2 expression appeared to increase with the histological severity of BE; there was a significant positive correlation between COX-2 expression and histopathologic changes from I/LGD to EAC (r = 0.97, P < 0.001) (Figure 3).
Figure 3.
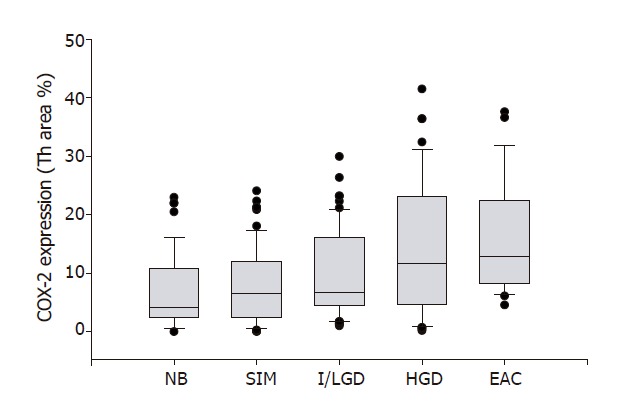
COX-2 expression and pathological severity of BE. COX-2 expression is plotted against the 4 stages of Barrett's esophagus. The line enclosed within the box represents the median value; the small circles indicate outlying data points. COX-2 expression in specimens with SIM, I/LGD, HGD and EAC was significantly increased in comparison with that in NB (P < 0.01). A significant positive correlation between COX-2 expression and histopathologic changes from SIM to EAC was observed (r = 0.97, P < 0.001).
Expression of EGFR in the metaplasia–dysplasia–adenocarcinoma sequence
We found significant increases in EGFR expression in specimens with SIM (2.1 ± 0.5), I/LGD (2.7 ± 0.6), HGD (3.9 ± 0.7) and EAC (5.2 ± 0.9) as compared with the control specimens (0.8 ± 0.4) (P < 0.05). Furthermore, EGFR expression appeared to increase with the histological severity of BE; there was a significant positive correlation between EGFR expression and histopathologic changes from I/LGD to EAC (r = 0.97, P < 0.001) (Figure 4).
Figure 4.
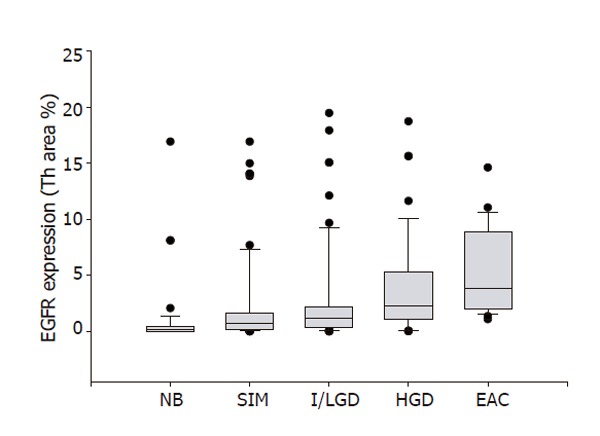
EGFR expression and pathological severity of BE. EGFR expression is plotted against the 4 stages of Barrett's esophagus. The line enclosed within the box represents the median value; the small circles indicate outlying data points. EGFR expression in specimens with SIM, I/LGD, HGD and EAC was significantly increased in comparison with that in NB (P < 0.01). A significant positive correlation between EGFR expression and histopathologic changes from SIM to EAC was found(r = 0.97, P < 0.001).
Correlation between COX-2 and EGFR in metaplasia–dysplasia–adenocarcinoma sequence
The data from computer quantification of COX-2 expression and EGFR expression in all immunohistochemically stained sections were used to determine the correlation between COX-2 and EGFR in the metaplasia-dysplasia- adenocarcinoma sequence. A significant positive correlation between COX-2 expression and EGFR expression was determined in the BE tissues (r = 0.94, P < 0.001) (Figure 5).
Figure 5.
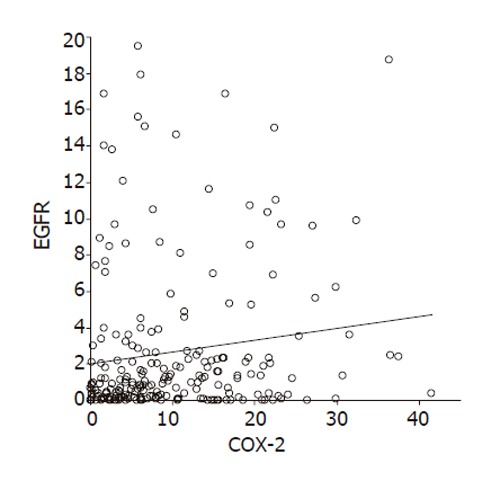
Relationship between COX-2 expression and EGFR expression in 104 patients. A significant positive correlation between COX-2 expression and EGFR expression was found (r = 0.94, P < 0.001).
DISCUSSION
Our results showed a detailed information on localization of COX-2 and EGFR protein within the metaplastic cells, dysplastic cells and cancer cells. We demonstrated a significant increase in both COX-2 and EGFR expressions in the metaplasia-dysplasia-adenocarcinoma sequence. In particular, weak COX-2 and EGFR stainings were found in the normal squamous epithelium and NB epithelium, while strong COX-2 and EGFR stainings were found in the region with histological changes of metaplasia, dysplasia and EAC. To our best of knowledge, this is the first study to examine the relationship between COX-2 and EGFR in respect to the histologic progression from BE to EAC. Since both COX-2 and EGFR expression levels increase dramatically in BE, each of these changes could enhance the tumorigenic potential of BE[10,11,18,22].
This study focused on the morphological features of Barrett’s epithelium with the expression of COX-2 and EGFR for the following reasons. First, evaluation from BE to EAC is primarily based on the cytomorphological characteristics[1,7,23,24]. Neoplasia develops in a series of steps through the development of a morphologically changed cell population. It is widely accepted that COX-2 and EGFR play important roles during carcinogenesis, and the over-expression of COX-2 and EGFR would be expected to predispose the epithelium to tumorigenic morphological change. Second, up-regulations of COX-2 and EGFR have been reported in BE and EAC, but no evidence has been provided for the joint signaling of COX-2 and EGFR in the induction of Barrett’s epithelium to adenocarcinoma transition. This induction remains an integral process occurring during critical phase of tumor progression. Third, although COX-2 and EGFR play different biological roles in tumor formation, targeting both COX-2 and EGFR to abrogate both pathways and their downstream targets achieved significant antitumor and antiangiogenic effects in the in vivo and in vitro studies[21,25]. Therefore, the two proteins might be cooperatively carcinogenic, which intensify the transformation effects on Barrett’s metaplasia to dysplasia to adenocarcinoma sequence.
The exact function of COX-2 in tumor development and progression in vivo is not known as yet. One possibility follows that over-expression of COX-2 leads to high levels of prostaglandins synthesis in the tissues. Prostaglandins produced by COX-2 may subsequently facilitate tumor progression by acting as growth factors, differentiation factors, immunosuppressors and angiogenic agents[26,27]. Other possibilities from recent studies indicate that reactive oxygen species (ROS) are generated during the conversion from prostaglandins G2 to prostaglandins H2[28,29], this enhanced level of ROS parallels the levels of COX-2 mRNA. The COX-2-derived oxidative DNA damage may contribute to the accumulation of genetic damage, and the accumulation of genetic and epigenetic aberrations produces one or more clones with tumorigenic potential. In addition, the COX-2 enzyme itself may promote tumor development and progression by a prostaglandins-independent pathway. This is supported by the evidences that NSAIDs suppressed proliferative activity and inhibited the growth in colon cancer cells without repressing prostaglandins synthesis[30]. Over-expression of COX-2 can also up-regulate anti-apoptotic oncoprotein such as BCL-2 and multiple proliferation-promoting cyclins and cyclin-dependent kinases, and down-regulate some tumor suppressor proteins[17,31]. The postulation for the involvement of COX-2 over-expression related to the progression in metaplasia to dysplasia to adenocarcinoma sequence is based on extensive studies of the relationship between COX-2 and tumor development and progression in multiple organ system.
Our data demonstrated a strong immunoreactivity for COX-2 protein within metaplastic cells, dysplastic, and malignant cells when compared to normal squamous cells and NB columnar cells. The cytomorphological features of COX-2 distribution in all epithelium may reflect two aspects of the effect of COX in contribution to the development of tumor. COX-2 is an important factor to maintain the microenvironment for tumor growth, and COX-2 may not be the critical factor in contribution to the phenotype changes of pre-malignant cells. COX-2 is undetectable in most tissues in the physiological condition[13,14]; the animals lacking COX-2 and prostaglandins synthesis show no innate gastrointestinal pathology[32]. These features of COX-2 do not confirm COX-2 expression as a central event in carcinogenesis. On the other hand, it is also possible that duodenal-gastro reflux and/or rapid growth and invasion of cancer cells in BE would stimulate local inflammation to inflammatory mediators and mitogens[12,13], which, in turn, would induce COX-2 synthesis. Nevertheless, the high level of COX-2 synthesis would prolong the survival of these abnormal cells and increase the chance for tumorigenesis.
EGFR signaling module plays a fundamental role in the morphogenesis of a diverse spectrum of organisms, and has been highly conserved through the course of evolution. In humans, the EGFR family, which consists of the EGFR itself and the receptors known as HER 2-4, and more than 30 ligands lie at the head of a complex, multi-layered signal-transduction network[33]. The aberrant activity of the members in this family has been shown to play a key role in the development and growth of tumor cells[9]. The EGFR family is over-expressed in various types of human neoplasms, such as esophagus, thyroid, breast, testis, bladder, melanoma, pancreas, colon, cervical carcinomas, renal carcinoma, prostate and liver cancer[10]. The underlying mechanism for EGFR involvement in carcinogenesis is likely to reduce the requirement for exogenously supplied growth factors to maintain tumorigenic cells proliferation. Over-expression of EGFR can hypersensitize the cells to low concentration of growth factors to maintain their viability, and to facilitate their phenotype transformation mediated by these growth factors[34,35]. The aberrant activity of the EGFR might regulate the expression of specific oncogenes or tumor suppressor genes involved in the intracellular signal transduction pathway to synthesize endogenously produced growth factors[36]. Several studies have also supported the notion that the endogenous growth factors might function via an intracrine, juxtacrine, autocrine or paracrine mechanisms to control cell proliferation[34-36].
The results obtained from this study for EGFR staining showed a significant effect of EGFR in contribution to the cytomorphological changes. A large amount of EGFR protein was expressed in the epithelial cells displaying multiple cell layers, enlarged nucleus and less cytoplasm in SIM, I/LGD, HGD and EAC. This over-expression was consistent across the BE progression, thereby indicating an important role of EGFR in the initiation of Barrett’s cells transition to adenocarcinoma. The phenotypic effects of abnormal function of EGFR protein are far more dramatic. Recent studies have indicated that animals lacking EGFR have abnormal eyes and epidermal tissues and die due to defects in the development of epithelial organ[37,38]. Therefore, EGFR may play a key role in the morphogenesis of metaplasia, dysplasia and adenocarcinoma. In our study, the immunoreactivity of EGFR was observed in the same increasing trend with the metaplasia to dysplasia to adenocarcinoma sequence as that seen in COX-2 immunoreactivity, thus suggesting a cooperative effect of these two proteins.
In summary, our study demonstrates a positive relationship between expression of COX-2 and EGFR in the progression from metaplasia to dysplasia to adenocarcinoma in subjects with BE. This suggests that both COX-2 and EGFR may increase the likelihood of Barrett’s cells to undergo abnormal cell cycling or gene expression. Further studies are needed to correlate the COX-2 and EGFR expression pattern with clinical outcome. Animal models and tissue culture are also required to determine the exact mechanism of COX-2 and EGFR action in the metaplasia to dysplasia to adenocarcinoma sequence.
Footnotes
Supported by the University of Louisville Hospital and the Society for Surgery of the Alimentary Tract Career Development Award
S- Editor Kumar M and Guo SY L- Editor Elsevier HK E- Editor Kong LH
References
- 1.Blot WJ, Devesa SS, Kneller RW, Fraumeni JF Jr. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA. 1991;265:1287–1289. [PubMed] [Google Scholar]
- 2.Pera M, Cameron AJ, Trastek VF, Carpenter HA, Zinsmeister AR. Increasing incidence of adenocarcinoma of the esophagus and esophagogastric junction. Gastroenterology. 1993;104:510–513. doi: 10.1016/0016-5085(93)90420-h. [DOI] [PubMed] [Google Scholar]
- 3.Farrow DC, Vaughan TL. Determinants of survival following the diagnosis of esophageal adenocarcinoma (United States) Cancer Causes Control. 1996;7:322–327. doi: 10.1007/BF00052937. [DOI] [PubMed] [Google Scholar]
- 4.Isolauri J, Luostarinen M, Isolauri E, Reinikainen P, Viljakka M, Keyriläinen O. Natural course of gastroesophageal reflux disease: 17-22 year follow-up of 60 patients. Am J Gastroenterol. 1997;92:37–41. [PubMed] [Google Scholar]
- 5.Altorki NK, Oliveria S, Schrump DS. Epidemiology and molecular biology of Barrett's adenocarcinoma. Semin Surg Oncol. 1997;13:270–280. doi: 10.1002/(sici)1098-2388(199707/08)13:4<270::aid-ssu9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Geboes K. Barrett's esophagus: the metaplasia-dysplasia-carcinoma sequence: morphological aspects. Acta Gastroenterol Belg. 2000;63:13–17. [PubMed] [Google Scholar]
- 7.Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, Kerr D, Young LS. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol. 1999;154:965–973. doi: 10.1016/S0002-9440(10)65346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 9.Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37 Suppl 4:S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 10.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 11.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 12.Evett GE, Xie W, Chipman JG, Robertson DL, Simmons DL. Prostaglandin G/H synthase isoenzyme 2 expression in fibroblasts: regulation by dexamethasone, mitogens, and oncogenes. Arch Biochem Biophys. 1993;306:169–177. doi: 10.1006/abbi.1993.1496. [DOI] [PubMed] [Google Scholar]
- 13.Lee SH, Soyoola E, Chanmugam P, Hart S, Sun W, Zhong H, Liou S, Simmons D, Hwang D. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem. 1992;267:25934–25938. [PubMed] [Google Scholar]
- 14.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci U S A. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funkhouser EM, Sharp GB. Aspirin and reduced risk of esophageal carcinoma. Cancer. 1995;76:1116–1119. doi: 10.1002/1097-0142(19951001)76:7<1116::aid-cncr2820760703>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 16.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW Jr. Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- 17.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 18.Zimmermann KC, Sarbia M, Weber AA, Borchard F, Gabbert HE, Schrör K. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res. 1999;59:198–204. [PubMed] [Google Scholar]
- 19.Yoshimoto T, Takahashi Y, Kinoshita T, Sakashita T, Inoue H, Tanabe T. Growth stimulation and epidermal growth factor receptor induction in cyclooxygenase-overexpressing human colon carcinoma cells. Adv Exp Med Biol. 2002;507:403–407. doi: 10.1007/978-1-4615-0193-0_62. [DOI] [PubMed] [Google Scholar]
- 20.Ranelletti FO, Almadori G, Rocca B, Ferrandina G, Ciabattoni G, Habib A, Galli J, Maggiano N, Gessi M, Lauriola L. Prognostic significance of cyclooxygenase-2 in laryngeal squamous cell carcinoma. Int J Cancer. 2001;95:343–349. doi: 10.1002/1097-0215(20011120)95:6<343::aid-ijc1060>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 21.Tortora G, Caputo R, Damiano V, Melisi D, Bianco R, Fontanini G, Veneziani BM, De Placido S, Bianco AR, Ciardiello F. Combination of a selective cyclooxygenase-2 inhibitor with epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 and protein kinase A antisense causes cooperative antitumor and antiangiogenic effect. Clin Cancer Res. 2003;9:1566–1572. [PubMed] [Google Scholar]
- 22.Jankowski J, Hopwood D, Pringle R, Wormsley KG. Increased expression of epidermal growth factor receptors in Barrett's esophagus associated with alkaline reflux: a putative model for carcinogenesis. Am J Gastroenterol. 1993;88:402–408. [PubMed] [Google Scholar]
- 23.Reid BJ, Haggitt RC, Rubin CE, Roth G, Surawicz CM, Van Belle G, Lewin K, Weinstein WM, Antonioli DA, Goldman H. Observer variation in the diagnosis of dysplasia in Barrett's esophagus. Hum Pathol. 1988;19:166–178. doi: 10.1016/s0046-8177(88)80344-7. [DOI] [PubMed] [Google Scholar]
- 24.Haggitt RC. Barrett's esophagus, dysplasia, and adenocarcinoma. Hum Pathol. 1994;25:982–993. doi: 10.1016/0046-8177(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Zhang X, Li M, Wang Z, Wieand HS, Grandis JR, Shin DM. Simultaneously targeting epidermal growth factor receptor tyrosine kinase and cyclooxygenase-2, an efficient approach to inhibition of squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004;10:5930–5939. doi: 10.1158/1078-0432.CCR-03-0677. [DOI] [PubMed] [Google Scholar]
- 26.Marnett LJ. Aspirin and the potential role of prostaglandins in colon cancer. Cancer Res. 1992;52:5575–5589. [PubMed] [Google Scholar]
- 27.Hla T, Ristimäki A, Appleby S, Barriocanal JG. Cyclooxygenase gene expression in inflammation and angiogenesis. Ann N Y Acad Sci. 1993;696:197–204. doi: 10.1111/j.1749-6632.1993.tb17152.x. [DOI] [PubMed] [Google Scholar]
- 28.Chung HY, Kim HJ, Kim JW, Yu BP. The inflammation hypothesis of aging: molecular modulation by calorie restriction. Ann N Y Acad Sci. 2001;928:327–335. [PubMed] [Google Scholar]
- 29.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 30.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 31.Goldberg Y, Nassif II, Pittas A, Tsai LL, Dynlacht BD, Rigas B, Shiff SJ. The anti-proliferative effect of sulindac and sulindac sulfide on HT-29 colon cancer cells: alterations in tumor suppressor and cell cycle-regulatory proteins. Oncogene. 1996;12:893–901. [PubMed] [Google Scholar]
- 32.Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 33.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 34.Logan A. Intracrine regulation at the nucleus--a further mechanism of growth factor activity. J Endocrinol. 1990;125:339–343. doi: 10.1677/joe.0.1250339. [DOI] [PubMed] [Google Scholar]
- 35.Sporn MB, Roberts AB. Autocrine secretion--10 years later. Ann Intern Med. 1992;117:408–414. doi: 10.7326/0003-4819-117-5-408. [DOI] [PubMed] [Google Scholar]
- 36.Aaronson SA. Growth factors and cancer. Science. 1991;254:1146–1153. doi: 10.1126/science.1659742. [DOI] [PubMed] [Google Scholar]
- 37.Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science. 1995;269:234–238. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- 38.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]