

Abstract
Ultrastructure of Kupffer cells and hepatocytes in liver bioptate was evaluated in a 17-year-old boy with Dubin–Johnson syndrome (DJS). The liver tissue obtained by needle biopsy was fixed in glutaraldehyde and paraformaldehyde and routinely processed for electron microscopic analysis. The ultrastructural examinations of liver bioptate revealed the accumulation of membrane-bound, electron-dense lysosomal granules within the cytoplasm of hepatocytes, characteristic of DJS. They were located mainly in the vicinity of the biliary pole, and preferentially in the centrilobular region that corresponded to the pigment deposits seen under light microscope. The presence of the granules was accompanied by dilated elements of the granular endoplasmic reticulum and paracrystalline mitochondrial inclusions as well as dilation of the bile canaliculi. The changes in hepatocytes co-existed with marked stimulation and enhanced phagocytic activity of Kupffer cells. This was manifested in the accumulation of pigment deposits within their cytoplasm that corresponded to those observed in hepatocytes. Hyperactive pericentral Kupffer cells which are involved in the response to pigmentary material originating from disintegrated hepatocytes may play an essential role in the development of DJS.
Keywords: Liver biopsy, Electron microscopic study, Lysosomal granules, Functional hyperbilirubinemia, Dubin–Johnson syndrome
INTRODUCTION
A rise in serum bilirubin concentration above the norm may be caused by its increased production (hemolytic processes, shunt-bilirubinemia), impairment of its uptake, conjugation and discharge from hepatocytes (congenital non-hemolytic functional hyperbilirubinemia) or excretion of unconjugated and conjugated bilirubin from damaged hepatocytes or biliary canaliculi to the blood. Clinical picture can be similar, as jaundice is the predominant and often isolated symptom in the above pathologies. In doubtful cases, morphological examination of a liver bioptate is a decisive step[1-3].
The main aim of the paper was to demonstrate the ultrastructure of Kupffer cells and hepatocytes in liver biopsy from a patient with Dubin-Johnson syndrome (DJS). To the best of our knowledge, it is the first report on the ultrastructure of Kupffer cells in this disorder.
CASE REPORT
Our report presents data concerning a 17-year-old boy suffering from hyperbilirubinemia (up to 119.7 μmol/L) with concomitant normal activity of aminotransferases from 6 years of age. He was suspected of congenital functional hyperbilirubinemia. However, since abdominal complaints and hepatomegaly were found to coexist with scleral xanthosis, the boy was sent for further diagnosis to the 3rd Department of Pediatrics, Medical University of Białystok. On admission, laboratory tests revealed high serum bilirubin concentration (120.55 μmol/L) with the predominance of unconjugated fraction (82.94 μmol/L).
However, the activities of aminotransferases (ALT, AST), alkaline phosphatase and GGTP were not increased. Differential diagnosis excluded: hemolytic disorders, infections with hepatotropic viruses (HBV, HCV,
CMV, EBV, HSV), metabolic disorders (cystic fibrosis, Wilson’s disease, α-1 antitrypsin deficiency, hemochromatosis), hypothyroidism, autoimmune liver diseases, parasitic and epizootic diseases. The ultrasonographic examination of the abdominal cavity confirmed hepatomegaly with no changes in parenchyma echogenicity. Liver biopsy was performed to elucidate the still unclear cause of jaundice.
For ultrastructural analysis, small, fresh tissue blocks (1 mm3) were cut off from the cylinder of liver tissue obtained by needle biopsy. The remaining considerable part of the collected tissue was submitted for examination by the light microscopy.
The material for electron microscopic analysis was fixed in 10 g/L glutaraldehyde and 8 g/L paraformaldehyde in 0.1 mol/L cacodylate buffer (pH 7.4). Subsequently, fixation was carried out in osmium tetroxide; then the material was routinely processed for embedding in Epon 812. Ultrathin sections were contrasted with uranyl acetate and lead citrate, and examined using an Opton 900 PC transmission electron microscope (Zeiss, Oberkochen, Germany).
The liver specimen obtained by needle biopsy was grossly dark and greenish, the striking characteristics for DJS. Liver histology showed numerous distinct coarse brownish pigment deposits in the cytoplasm of the hepatocytes.
Ultrastructural examination of these cells showed electron-dense, lysosomal granules, characteristic of DJS, that corresponded to the intracytoplasmic pigment deposits seen in the light microscopic sections. This pigmentary material was frequently surrounded by a single, fine continuous or discontinuous membrane; sometimes no such membrane could be found. The lysosomal pigment granules demonstrated great variability in shape, size, content, and electron density and were located mainly in the vicinity of the biliary pole (Figure 1), and preferentially found in the centrilobular region. Moderately electron-dense finely granular background alternating with denser irregular areas and pleomorphic dark bodies (clearly seen against a finely homogenous granular matrix) characterized these granules. The pleomorphic very dense bodies frequently occupied a considerable part or almost whole lysosomes. Some hepatocytes filled with coarse electron-dense granules were disintegrated. Dilation of bile canaliculi with loss of microvilli typical for DJS was observed (Figure 1). Dilated elements of the granular endoplasmic reticulum (Figure 1), the presence of paracrystalline mitochondrial inclusions, enlarged Golgi complex and dispersed lipid droplets within the cytoplasm of hepatocytes were also noted. The vascular pole of the hepatocytes showed alteration of microvilli including their disintegration; a fine granular material could be occasionally seen in Disse’s space. Sometimes this space was discontinued. The basement membrane of some hepatocytes was distinctly outlined, and even thickened.
Figure 1.
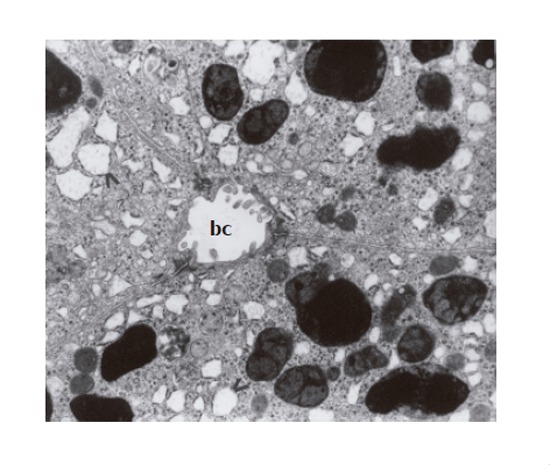
Several membrane-bound granules – varying in size, content, and electron density – located near the biliary pole of hepatocytes; broadening of granular endoplasmic reticulum (>); dilation of bile canaliculus (cb) with the loss of microvilli (×7 000).
The ultrastructural examinations of the liver bioptate also showed, especially in the centrilobular region, an increase in hepatic macrophages. Kupffer cells in the lumen of hepatic sinusoidal vessels were frequently hypertrophied and markedly activated, blocking the vascular lumen and demonstrating features of hyperactivity (Figure 3). They contained electron-dense dark pigment granules, varying in shape and size (Figures 2 and 3), often occupying almost whole lysosomes. This material corresponded to the pigment deposits present in hepatocytes (Figure 2). Bile deposits were sometimes observed near the plentiful phagocytized pigment granules (Figure 3).
Figure 3.
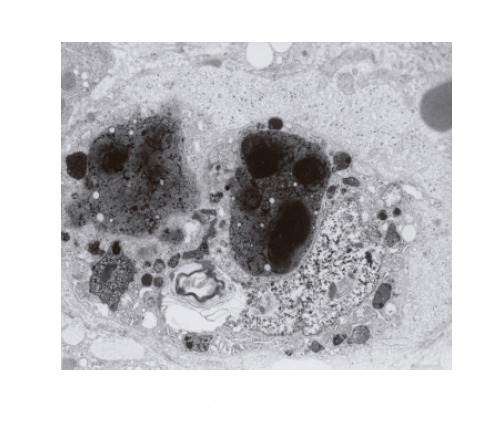
A hypertrophic, evidently activated Kupffer cell with phagocytized, very thick, black pigment deposits and biliary lamellar material. The cell blocks the sinusoidal vascular lumen. Distinctly dilated Disse’s space contains a fine granular material (×4 400).
Figure 2.
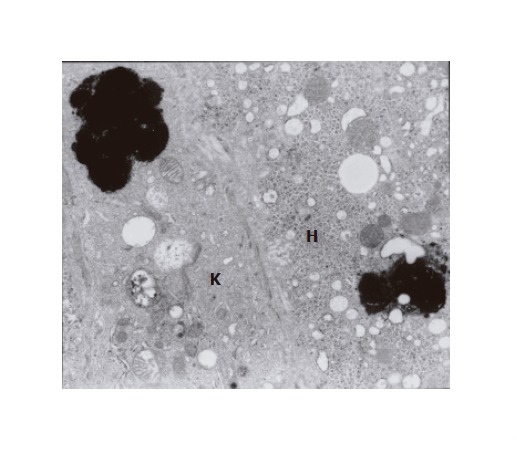
Fragment of Kupffer cell with phagocytized thick black pigment granules; the cell adheres to a hepatocyte (H) containing a similar pigment deposit (×7 000).
Since the morphological picture of the liver bioptate was typical of DJS, leaving no doubt, we did not perform very expensive genetic tests (mutation in the ABCC2/MRP2 gene[4]).
DISCUSSION
Macro- and microscopic, especially ultrastructural, changes observed in the liver bioptate in our patient were characteristic of DJS. The hepatocytes contained numerous electron-dense lysosomal, membrane-bound granules located mainly near the biliary pole that corresponded to the coarse brownish pigment deposits seen under light microscope. The granules were accompanied by dilated endoplasmic reticulum and paracrystalline inclusions as well as alterations in bile canaliculi of hepatocytes.
However, Kupffer cells demonstrated features of hyper-reactivity. They accumulated pigment granules that corresponded to those observed in hepatocytes.
Morphological abnormalities of hepatocytes observed in the course of our studies were similar to those found by other authors in this syndrome[3,5-7]. It is worth noting that although the pigment granules showed characteristic microscopic features, their exact composition has not been clarified[1,6]. The results of ultrastructural and histochemical studies by Luo et al[2] suggest that this deposit material is a lipofuscin–melanin complex.
Up to now, there have been no reports describing the electron microscope picture of Kupffer cells in DJS.
It has been known that Kupffer cells are the largest population of tissue macrophages, predominantly distributed in the lumen of hepatic sinusoids. According to Naito et al[8], these cells demonstrate unique differentiation mechanisms, metabolic functions and responsiveness to inflammatory agents. They exhibit endocytic activity against blood-borne material entering the liver as well as phagocytic function in relation to cellular debris[8,9]. Kupffer cells also play an important role in the modulation of drug metabolic enzymes[10], in the carcinogenesis of hepatocellular carcinoma[11] and liver fibrosis[12].
We assume that pigment deposits absorbed by Kupffer cells in the course of DJS may have originated from disintegrating, pigment-load hepatocytes. The present study indicates that it is the process of phagocytosis that plays a significant role in the accumulation of thick black granules within the cytoplasm of these macrophages. This process is strictly connected with the morphological status and function of lysosomes of Kupffer cells. This is confirmed by the fact that the number of lysosomes, both primary and secondary, within the cytoplasm of hyperactivated macrophages was markedly increased in our observations. It can be suggested that in the course of DJS, due to phagocytosis, remnants of pigment-load hepatocytes trapped by the hyperactivated Kupffer cells with the involved macrophage scavenger receptors are shifted to primary lysosomes, i.e. intracytoplasmic reservoirs containing lytic enzymes, which undergo degradation. Then, the lysosomes are transformed into the secondary ones[8].
Unlike in the Gilbert’s syndrome, in DJS clinical manifestation of hepatomegaly can be found even in the neonatal-infantile period[3,7,13-14]. In our patient, the cause of hyperbilirubinemia was unknown for many years. DJS was not taken into account because of atypical course of the disease (predominance of unconjugated bilirubin in the blood serum). However, the predominance of non-conjugated bilirubin in the blood serum has been noted in some diagnosed DJS cases[1].
We emphasize that hyperactive pericentral Kupffer cells may play an essential role in the development of the syndrome. They are involved in the response to pigmentary material originating from disintegrated hepatocytes.
Footnotes
S- Editor Guo SY L- Editor Elsevier HK E- Editor Bai SH
References
- 1.Berk PD. Bilirubin metabolism and the hereditary hyperbilirubinemias. Semin Liver Dis. 1994;14:321–322. doi: 10.1055/s-2007-1007321. [DOI] [PubMed] [Google Scholar]
- 2.Luo Z, Zhang L, Li Y. [Clinical pathology of Dubin-Johnson syndrome] Zhonghua Gan Zang Bing Za Zhi. 2000;8:45–47. [PubMed] [Google Scholar]
- 3.Shimizu T, Tawa T, Maruyama T, Oguchi S, Yamashiro Y, Yabuta K. A case of infantile Dubin-Johnson syndrome with high CT attenuation in the liver. Pediatr Radiol. 1997;27:345–347. doi: 10.1007/s002470050147. [DOI] [PubMed] [Google Scholar]
- 4.Keitel V, Kartenbeck J, Nies AT, Spring H, Brom M, Keppler D. Impaired protein maturation of the conjugate export pump multidrug resistance protein 2 as a consequence of a deletion mutation in Dubin-Johnson syndrome. Hepatology. 2000;32:1317–1328. doi: 10.1053/jhep.2000.19791. [DOI] [PubMed] [Google Scholar]
- 5.Baba N, Ruppert RD. The Dubin-Johnson syndrome: electron microscopic observation of hepatic pigment--a case study. Am J Clin Pathol. 1972;57:306–310. doi: 10.1093/ajcp/57.3.306. [DOI] [PubMed] [Google Scholar]
- 6.Cotutiu C, Moraru I, Ionescu V. Ultrastructural data in a case of Dubin-Johnson syndrome. Acta Morphol Acad Sci Hung. 1971;19:313–322. [PubMed] [Google Scholar]
- 7.Tsai WH, Teng RJ, Chu JS, Chang MH, Ho MM. Neonatal Dubin-Johnson syndrome. J Pediatr Gastroenterol Nutr. 1994;18:253–254. doi: 10.1097/00005176-199402000-00023. [DOI] [PubMed] [Google Scholar]
- 8.Naito M, Hasegawa G, Ebe Y, Yamamoto T. Differentiation and function of Kupffer cells. Med Electron Microsc. 2004;37:16–28. doi: 10.1007/s00795-003-0228-x. [DOI] [PubMed] [Google Scholar]
- 9.Yano H, Kinoshita S, Kira S. Effects of acute moderate exercise on the phagocytosis of Kupffer cells in rats. Acta Physiol Scand. 2004;182:151–160. doi: 10.1111/j.1365-201X.2004.01343.x. [DOI] [PubMed] [Google Scholar]
- 10.Ding H, Tong J, Wu SC, Yin DK, Yuan XF, Wu JY, Chen J, Shi GG. Modulation of Kupffer cells on hepatic drug metabolism. World J Gastroenterol. 2004;10:1325–1328. doi: 10.3748/wjg.v10.i9.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K, He X, Lei XZ, Zhao LS, Tang H, Liu L, Lei BJ. Pathomorphological study on location and distribution of Kupffer cells in hepatocellular carcinoma. World J Gastroenterol. 2003;9:1946–1949. doi: 10.3748/wjg.v9.i9.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Yu WP, Gao L, Wei KB, Ju JL, Xu JZ. Effects of lipopolysaccharides stimulated Kupffer cells on activation of rat hepatic stellate cells. World J Gastroenterol. 2004;10:610–613. doi: 10.3748/wjg.v10.i4.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haimi-Cohen Y, Amir J, Merlob P. Neonatal and infantile Dubin-Johnson syndrome. Pediatr Radiol. 1998;28:900. doi: 10.1007/s002470050494. [DOI] [PubMed] [Google Scholar]
- 14.Kimura A, Ushijima K, Kage M, Mahara R, Tohma M, Inokuchi T, Shibao K, Tanaka N, Fujisawa T, Ono E. Neonatal Dubin-Johnson syndrome with severe cholestasis: effective phenobarbital therapy. Acta Paediatr Scand. 1991;80:381–385. doi: 10.1111/j.1651-2227.1991.tb11867.x. [DOI] [PubMed] [Google Scholar]