

Abstract
Objective
The aim of this study is to evaluate the auditory phenotype in subjects with OTOF gene mutations to describe genotype-phenotype correlations.
Material-methods
Twenty-two affected members from three families with homozygous OTOF mutations were included. Nine subjects were evaluated audiologically with otoscopic examination, pure-tone audiometry, tympanometry with acoustic reflex testing, auditory brain stem responses, and otoacoustic emission tests.
Results
Homozygous c.4718T>C (p.Ile1573Thr) mutation was associated with the auditory neuropathy/auditory dys-synchrony (AN/AD) phenotype and with progressive sensorineural hearing loss in four siblings in one family, while homozygous c.4467dupC (p.I1490HfsX19) was associated with severe to profound sensorineural hearing loss without AN/AD in four relatives in another family. Homozygous c.1958delC (p.Pro653LeufsX13) mutation was associated with moderate sensorineural hearing loss without AN/AD in one affected person in an additional family.
Conclusions
The audiological phenotype associated with different OTOF mutations appears to be consistently different suggesting the presence of a genotype-phenotype correlation.
Keywords: Auditory neuropathy, autosomal recessive, hearing loss, OTOF
1. Introduction
Congenital (or prelingual) inherited hearing impairment affects approximately one in 1,000 newborns. Genetic etiologies are responsible for 60-80% of cases with congenital hearing loss. Hereditary hearing loss is classified into two main categories: non-syndromic (90%), of which majority are autosomal recessive (DFNB), and syndromic (10%). The inheritance pattern of non-syndromic hearing loss can be autosomal recessive, autosomal dominant, X-linked and mitochondrial [1]. Mutations in 43 genes, including OTOF (MIM 603681), have been implicated in non-syndromic autosomal recessive sensorineural hearing loss (SNHL) [2].
Yasunaga et al. [3] first identified mutations in OTOF, which is located on the short arm of human chromosome 2 and codes for otoferlin, as a cause of non-syndromic autosomal recessive SNHL at the DFNB9 locus. To date, 93 mutations in OTOF have been reported [4]. Otoferlin was found to be preferentially expressed in sensory hair cells of the cochleathe vestibule, and also expressed in the brain [3,5]. In the murine mature cochlea, otoferlin is present in the inner hair cells and plays an essential role in a late step of synaptic vesicle exocytosis and likely acts as a sensor triggering membrane fusion [6,7]. OTOF-related hearing loss is frequently associated with auditory neuropathy/dys-synchrony (AN/AD) [8-11]. The auditory neuropathies are characterized by disturbed pure tone audiometry, auditory brain stem responses (ABR) and preservation of otoacoustic emissions (OAEs). In this disease, auditory nerve and/or inner hair cells are damaged, however, the outer hair cells are intact [12].
Most individuals previously reported with OTOF mutations exhibited severe-to-profound SNHL [6, 8, 9, 11, 13-18]. However, available data on genotype-phenotype correlation remain limited. In this study, we analyzed auditory phenotype of three families that we determined as having OTOF mutations.
2. Material and methods
This study was approved by the Ankara University Ethics Committee (Turkey) and by the IRB at the University of Miami (USA). All families were Turkish and after careful evaluation, they were negative for the presence of syndromic findings or environmental exposure. Otoscopic examination, pure-tone audiometry, tympanometry measurement with acoustic reflex, and transient otoacoustic emission (TEOAE) test were performed. ABR test was performed on one member in one family. The degree of hearing loss was determined by pure tone audiometry. Degree of hearing impairment was defined by pure tone average (PTA) threshold levels at 0.5, 1, 2 and 4 kHz. Hearing loss was classified as mild (PTA 21-40 dB HL), moderate (PTA 41-70 dB HL), severe (PTA 71-95 dB HL) and profound (PTA > 95 dB HL) (European Concerted Action Project on Genetics of Hearing Impairment 1996). Computed tomography of temporal bone was performed on one affected member in each family.
3. Results
In this study three families with homozygous OTOF mutations were studied (Figures 1 and 2). Details of molecular studies have been previously reported [19]. Each mutation co-segregated with hearing loss as a fully penetrant autosomal recessive trait.
Figure 1.
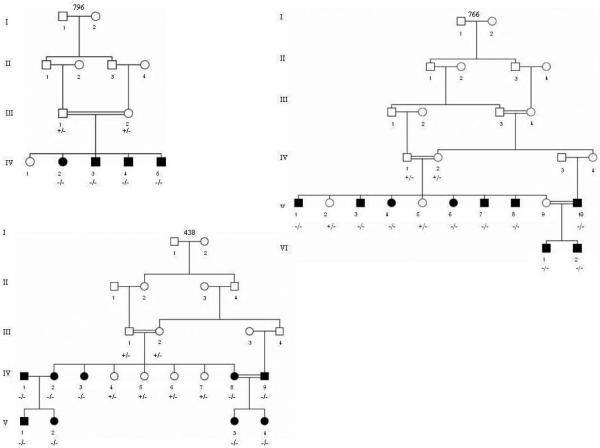
Pedigrees of families with OTOF mutations. Closed symbols: affected individuals. Circles are females squares are males. −/−, +/−, and −/− are homozygous mutant, heterozygous, and homozygous wild type, respectively.
Figure 2.
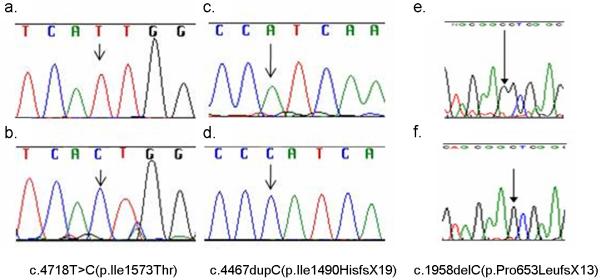
Electropherograms showing homozygous OTOF mutations.
a. wild type.
b. homozygous c.4718T>C(p.Ile1573Thr) in family 796.
c. wild type.
d. homozygous c.4467dupC(p.Ile1490HisfsX19) in family 766.
e. wild type.
f. homozygous c.1958delC(p.Pro653LeufsX13) in family 438.
The clinical details of the affected children are summarized in Table 1. We determined homozygous p.Pro653LeufsX13 (c.1958delC) mutation in affected members of family 438. The audiological characteristics of this truncating mutation was studied in one affected individual and were prelingual-onset, bilateral symmetric moderate sensorineural hearing loss with flat audiogram configuration (Table 1). Otoacoustic emission tests were evaluated in one person with a history of hearing device usage and were absent.
Table 1.
Summary of clinical and audiological data of studied patients
| Subject | Age (Year) |
Hearing level |
Audiogram shape |
OAE | Onset | Mutation | |
|---|---|---|---|---|---|---|---|
| Family 438 |
IV-9 | 22 | Bilateral- moderate |
Flat | Absent | C/P | c.1958delC (p.Pro653LeufsX13) |
|
| |||||||
| Family 766 |
V-1 | 35 | Bilateral- profound |
Flat | Absent | C/P | c.4467dupC (p.Ile1490HisfsX19 |
| V-3 | 30 | Bilateral- severe |
Flat | Absent | C/P | ||
| V-4 | 16 | Right profound, left severe |
Right scoop, Left flat | Absent | C/P | ||
| V-6 | 14 | Bilateral- profound |
Fragmentary shape |
Absent | C/P | ||
|
| |||||||
| Family 796 |
IV-2 | 17 | Bilateral- severe | Flat | Present | C/P | c.4718T>C (p.Ile1573Thr) |
| IV-3 | 13 | Bilateral- moderate |
Sloping | Present | C/P | ||
| IV-4 | 11 | Bilateral- moderate |
Sloping | Present | C/P | ||
| IV-5 | 9 | Bilateral- mild |
Flat | Present | C/P | ||
C/P: Congenital or prelingual
We found the p.Ile1490HisfsX19 (c.4467dupC), another truncating mutation, in nine individuals from family 766. Audiological findings were studied in four affected subjects. Hearing loss was prelingual-onset, bilateral symmetric profound and sensorineural with flat and fragmentary shape audiogram configuration. Transient evoked otoacoustic emissions were absent. Tympanometry results were normal. Acoustic middle ear muscle reflexes were absent (Table 1). Computed tomography of the temporal bone was normal in one affected person.
We found homozygous p.Ile1573Thr (c.4718T>C), a missense mutation in four individuals from family 796, who were audiologically characterized. Hearing loss was prelingual-onset, bilateral symmetric mild to severe sensorineural with flat-sloping audiogram configuration. ABR performed on one patient who is 9 years old. The hearing levels of all children were consecutively as follows: mild hearing loss in a 9-year-old child, moderate hearing loss in an 11-year and a 13-year-old, and severe hearing loss in a 17-year-old child. Transient evoked otoacoustic emissions were present at least three frequencies in both ear in all children. Also tympanometry results were normal and acoustic middle ear muscle reflexes were absent in all children (Table 1). No ABR waves were obtained in both ears by using click stimuli in the 9 years old child.
Computed tomography of the temporal bone was normal in one affected person from each family.
4. Discussion
In this study we provide details of the auditory phenotype for three OTOF mutations. A large number of affected individuals were available with all homozygous mutations, making comparison between different mutations easier. The OAEs were present in all four children of family 796 with the p.Ile1573Thr mutation showing that this missense mutation typically causes AN/AD. Another interesting finding in this family was that the level of hearing worsened as children got older which suggests that the hearing loss due to p.Ile1573Thr is progressive. Progressive hearing loss has been reported associated with another missense mutation, p.Glu1700Gln, in OTOF [13]. In contrast to what we observed in family 796, none of the five affected subjects in the other two families with homozygous p.I1490HfsX19 or p.Pro653LeufsX13 mutations had AN/AD.
OTOF mutations were identified in 56% of patients with AN/AD [18]. However, it is not clear whether the preservation of OHC function is a constant phenotype for hearing loss caused by OTOF mutations. It has been reported that OTOF-related deafness appears to be an AN/AD in the first years of life and with time OAEs disappear and electrophysiologic testing becomes more consistent with a cochlear defect [8,13, 20]. The reasons behind this finding may be environmental factors, such as usage of hearing aids, and/or genetic factors [8, 21]. All four affected members of family 766 in this study denied using hearing aids but their audiological tests showed OHC loss invariably. Thus, the presence of OAEs in all affected members who were homozygous for a potentially less disruptive mutation (p.Ile1573Thr in family 796) and the absence of OAEs in all affecteds with a severely disrupting mutation (p.Ile1490HisfsX19 in family 766) might suggest that the OHC dysfunction is a direct consequence of the identified OTOF mutation. Audiological phenotypes of previously reported homozygous OTOF mutations are shown in Table 2. It is interesting to note that the oldest patient with preserved OAEs was 18 years old with a missense mutation [9]. The oldest reported patient with preserved OAEs and a homozygous truncating mutation was 11 years old [8]. Hence, it is possible that truncating mutations cause loss of OAEs at an earlier age, while potentially milder mutations promote the preservation of OAEs at older ages.
Table 2.
Audiological phenotypes of previously reported homozygous OTOF mutations
| Mutations | Age tested/ Number of Cases |
Hearing level | OAE | Reference | |
|---|---|---|---|---|---|
| Missense | p.Leu1011Pro | 6y / 1 | Severe-profound | Bil (+) | Tekin et al. [9] |
| 17y/ 1 | Severe-profound | Bil (−) | |||
| 18y/ 1 | Severe-profound | Bil (+) | |||
| p.Phe1795Cys | 12-20m/ 1 | Profound | Bil (+) | Santeralli et al. [22] |
|
| 2y6m/ 1 | Profound | Bil (+) | Zadro et al. [23] | ||
| p.Glu1700Gln | 1y7m-2y7m/4 | Progressive moderate | (+) | Chiu et al. [13] | |
| 2y/ 1 | Progressive profound | (−) | |||
| Birth→2y/1 | Progressive profound | (+)→(−) | |||
| p.G541S | 26/1 | Temperature sensitive hearing loss |
(−) | Varga et al. [6] | |
| p.R1939Q | 1y7m-2y6m/5 | Profound | (−) | Matsunaga et al. [18] |
|
| 1y9m-2y10m/2 | Severe | (−) | |||
| 4m-10m/3 | Profound | (+) | Iwasa et al. [25] | ||
|
Other
Mutations |
c.1981dupG p.D661GfsX2 |
na/1 | Profound | na | Mahdieh et al. [26] |
| p.Gln829Ter | 1-11y/3 | Profound | Bil (+) | Rodriguez- Ballesteros et al. [8] |
|
| 2-9y/3 | Profound | Uni (+) | |||
| 2y/ 1 | Profound | Bil (+)→ Bil (−) |
|||
| 4-85y/7 | Profound | Bil (−) | |||
| c.1886_1887dupA (p.Pro630SerfsX9) |
16m-4y/ 2 | Profound | Bil (+) | Varga et al. [22] | |
| IV18+1G>T | 3-6y/2 | Moderate to severe | Bil (+) | ||
| IVS28-2A>C | 15m-3y/ 2 | Profound | Bil (+) | ||
| IVS9-2T>A | 3y/ 1 | Profound | Bil (+) | Zadro et al. [23] | |
| p.Glu1804Del | 7-10y/3 | Temperature sensitive hearing loss |
Bil (+) | Marlin et al. [24] | |
(+)→(−): Present both ears in the first test became absent later on
5. Conclusion
In this study we summarize the audiological phenotype associated with three OTOF mutations. The c.4718T>C (p.Ile1573Thr) mutation is associated with the AN/AD phenotype and with progressive SNHL. The 4467dupC (p.I1490HfsX19) mutation is associated with severe to profound SNHL with no evidence of AN/AD. The c.1958delC (p.Pro653LeufsX13) mutation is associated with moderate SNHL. Further studies are needed to conclude on the genotype-phenotype correlation regarding the mutations in the OTOF gene.
Acknowledgements
This work was supported by National Institutes of Health grant R01DC009645 to M.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu. Rev. Genet. 2001;35:589–646. doi: 10.1146/annurev.genet.35.102401.091224. [DOI] [PubMed] [Google Scholar]
- 2. [accessed on November 6, 2013];Hereditary Hearing Loss Homepage. http://hereditaryhearingloss.org;
- 3.Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, et al. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- 4.The Human Gene Mutation Database Professional Edition. [accessed on October 21, 2013]. [Google Scholar]
- 5.Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, et al. OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet. 2000;67(3):591–600. doi: 10.1086/303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Nonsyndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40(1):45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–689. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez-Ballesteros M, del Castillo FJ, Martín Y, Moreno-Pelayo MA, Morera C, Prieto F, et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum Mutat. 2003;22(6):451–456. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- 9.Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A. 2005;138(1):6–10. doi: 10.1002/ajmg.a.30907. [DOI] [PubMed] [Google Scholar]
- 10.Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, et al. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43(7):576–581. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodríguez-Ballesteros M, Reynoso R, Olarte M, Villamar M, Morera C, Santarelli R, et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat. 2008;29(6):823–831. doi: 10.1002/humu.20708. [DOI] [PubMed] [Google Scholar]
- 12.Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditory neuropathy. Brain. 1996;119:741–753. doi: 10.1093/brain/119.3.741. [DOI] [PubMed] [Google Scholar]
- 13.Chiu YH, Wu CC, Lu YC, Chen PJ, Lee WY, Liu AY, Hsu CJ. Mutations in the OTOF Gene in Taiwanese Patients with Auditory Neuropathy. Audiol Neurootol. 2010;11(15):364–374. doi: 10.1159/000293992. [DOI] [PubMed] [Google Scholar]
- 14.Choi BY, Ahmed ZM, Riazuddin S, Bhinder MA, Shahzad M, Husnain T, et al. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75:237–243. doi: 10.1111/j.1399-0004.2008.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirghomizadeh F, Pfister M, Apaydin F, Petit C, Kupka S, Pusch CM, et al. Substitutions in the conserved C2C domain of otoferlin cause DFNB9, a form of nonsyndromic autosomal recessive deafness. Neurobiol Dis. 2002;10:157–164. doi: 10.1006/nbdi.2002.0488. [DOI] [PubMed] [Google Scholar]
- 16.Romanos J, Kimura L, Favero ML, Izarra FA, de Mello Auricchio MT, Batissoco AC, et al. Novel OTOF mutations in Brazilian patients with auditory neuropathy. J Hum Genet. 2009;54:382–385. doi: 10.1038/jhg.2009.45. [DOI] [PubMed] [Google Scholar]
- 17.Rouillon I, Marcolla A, Roux I, Marlin S, Feldmann D, Couderc R, et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol. 2006;70:689–696. doi: 10.1016/j.ijporl.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Matsunaga T, Mutai H, Kunishima S, Namba K, Morimoto N, Shinjo Y, et al. A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clin Genet. 2012;82:425–432. doi: 10.1111/j.1399-0004.2012.01897.x. [DOI] [PubMed] [Google Scholar]
- 19.Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomarkers. 2011;15(1-2):1–5. doi: 10.1089/gtmb.2010.0120. [DOI] [PubMed] [Google Scholar]
- 20.Smith RJH, Gurrola JG, Kelley PM. OTOF-Related Deafness. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews™ [Internet] University of Washington, Seattle; Seattle (WA): Feb 29, 2008. pp. 1993–2013. updated 2011 Jun 14. [PubMed] [Google Scholar]
- 21.Rogers R, Kimberling WJ, Starr A, Kirschhofer K, Cohn ES, Kenyon JB, Keats BJB. The genetics of auditory neuropathy. In: Sininger Y, Starr A, editors. Auditory neuropathy: a new perspective on hearing disorders. Singular Publishing Co; San Diego: 2001. pp. 165–82. [Google Scholar]
- 22.Santarelli R, Del Castillo I, Rodríguez-Ballesteros M, Scimemi P, Cama E, Arslan E, et al. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J Assoc Res Otolaryngol. 2009;10:545–56. doi: 10.1007/s10162-009-0181-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zadro C, Ciorba A, Fabris A, Morgutti M, Trevisi P, Gasparini P, Martini A. Five new OTOF gene mutations and auditory neuropathy. Int J Pediatr Otorhinolaryngol. 2010;74:494–8. doi: 10.1016/j.ijporl.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Marlin S, Feldmann D, Nguyen Y, Rouillon I, Loundon N, Bonnet C, et al. Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: Deafening fever. Biochem Biophys Res Commun. 2010;394(3):737–42. doi: 10.1016/j.bbrc.2010.03.062. [DOI] [PubMed] [Google Scholar]
- 25.Iwasa Y, Nishio SY, Yoshimura H, Kanda Y, Kumakawa K, Abe S, et al. OTOF mutation screening in Japanese severe to profound recessive hearing loss patients. BMC Med Genet. 2013 Sep 22;14:95. doi: 10.1186/1471-2350-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahdieh N, Shirkavand A, Rabbani B, Tekin M, Akbari B, Akbari MT, Zeinali S. Screening of OTOF mutations in Iran: a novel mutation and review. Int J Pediatr Otorhinolaryngol. 2012 Nov;76(11):1610–5. doi: 10.1016/j.ijporl.2012.07.030. [DOI] [PubMed] [Google Scholar]