

Abstract
High blood levels of α2-antiplasmin have been associated with failed tissue plasminogen activator (TPA) therapy for ischemic stroke. Yet, other data suggests that α2-antiplasmin may be protective in stroke, because it defends against bleeding and excitotoxicity. To address this paradox, we examined the effects of high α2-antiplasmin levels and α2-antiplasmin inactivation in mice treated with TPA 0.5-2.5 hour after middle cerebral artery (MCA) thromboembolism. Brain infarction, swelling, hemorrhage, blood brain barrier breakdown and neuronal apoptosis were measured by a blinded observer. Thrombus dissolution was determined by gamma counting. During TPA treatment, high α2-antiplasmin blood levels increased brain infarction (2.2-fold) and swelling (3.7-fold), but decreased MCA thrombus dissolution. Conversely, α2-antiplasmin inactivation during TPA treatment reduced brain infarction, hemorrhage and swelling, but increased MCA thrombus dissolution. Inactivation of α2-antiplasmin during TPA treatment reduced neuronal apoptosis and blood brain barrier breakdown. Inactivation of α2-antiplasmin also reduced short-term mortality. Taken together these data show that α2-antiplasmin opposes the effects of TPA therapy and contributes to enhanced brain injury after experimental thromboembolic stroke. Conversely, α2-antiplasmin inactivation during TPA treatment improves thrombus dissolution and reduces brain infarction, swelling and hemorrhage. Consistent with clinical observations, these data suggest that α2-antiplasmin exerts deleterious effects that reduce the efficacy and safety of TPA therapy for ischemic stroke.
Keywords: apoptosis, stroke, hemorrhage, edema, fibrinolysis, thrombosis, mortality
Introduction
Significant increases in ischemic stroke are projected in the next few decades as the population ages, but tissue plasminogen activator (TPA) remains the only proven therapy (Chimowitz, 2013). Most ischemic strokes are caused by obstruction of a brain artery by a fibrin-containing clot or thromboembolus that reduces blood flow below levels necessary for the viability of neurons and other key cells (Donnan, et al., 2011). TPA's therapeutic effects on reducing disability after ischemic stroke are linked to reperfusion of the cerebral artery. Still, the factors that limit TPA-induced reperfusion, and contribute to brain hemorrhage and adverse outcomes are poorly understood (Lees, et al., 2010)
TPA works by cleaving plasminogen to the enzyme plasmin in order to dissolve the culprit thrombus and reperfuse the ischemic brain. As such, factors that regulate plasmin activity would be predicted to affect the success of TPA therapy (Chimowitz, 2013). Plasmin activity is primarily regulated by a fast-acting inhibitor, α2-antiplasmin (a2AP) as well as by a slower inhibitor, α2-macroglobulin. Still, how a2AP modifies the safety and efficacy of TPA therapy for ischemic stroke is unclear. By generating high levels of plasmin, TPA therapy often consumes a2AP and lowers levels of coagulation factors such as fibrinogen; this has been implicated in TPA-induced bleeding (Weitz, et al., 1993). Moreover, administration of a2AP has been shown to decrease experimental bleeding after TPA therapy (Weitz, et al., 1993). Other studies indicate that a2AP may be neuroprotective during TPA therapy because in the brain tissue TPA enhances excitotoxicity and a2AP injection reduces excitotoxicity (Tsirka, et al., 1997). However, these findings are at odds with clinical studies linking high a2AP levels to increased stroke risk and the failure of TPA to reperfuse thrombotically-occluded vessels during stroke therapy (Marti-Fabregas, et al., 2005, Suri, et al., 2010) Therefore, in these studies we sought to determine whether a2AP modulates the therapeutic effects of TPA given after different intervals of ischemia in a thromboembolic model of stroke.
MATERIALS AND METHODS
Ischemic stroke
Adult male C57Black/6J mice (29 -35 g, Jackson Labs, Bar Harbor, ME) were anesthetized with isoflurane. Autologous radiolabeled thromboemboli were made with pooled fresh frozen mouse plasma and 125I-fibrinogen (~5,000 cpm). Mice were ventilated with 1.5- 2% isoflurane and O2. Temperature was maintained at 37 deg. C. The left common carotid artery was isolated, and the external carotid, thyroid, and occipital arteries were ligated. Microvascular clips were placed temporarily on the common and internal carotid arteries. A PE8 catheter containing the clot was inserted through a small arteriotomy in the external carotid artery for retrograde passage through the internal carotid artery to the origin of the middle cerebral artery (MCA); the thrombus was embolized at a speed of 0.45 mL/min in a volume of 200 μl saline. A laser Doppler flow probe confirmed appropriate embolization (see below). TPA (2 or 10 mg/kg, Genentech, South San Francisco, CA), saline (control infusion) or an a2AP inactivating monoclonal antibody (9.3 or 21.3 mg/kg, 4H9, Research Innovations, Novi, MI) were administered via the contralateral jugular vein at 0.5, 1 or 2.5 h after onset of ischemia. In some experimental groups, mice were treated with a bolus infusion of purified human a2AP (4.2 mg/kg, EMD Serono, Rockland, MA) after thromboembolism as noted below. TPA was given by bolus (20% of dose) followed by infusion over 30 min in saline in 200 μl; monoclonal antibody infusions were given as a bolus. Experimental groups were euthanized 6 h or 24 h after thromboembolism. Euthanasia was performed earlier if mice appeared moribund, in discomfort, etc. according to study criteria. After euthanization, blood was collected by cardiac puncture and saline tissue perfusion was performed as described (King, et al., 2009). These studies were approved by our Institutional Animal Care and Use Committee.
Cerebral Blood Flow
Cerebral blood flow in the MCA territory was monitored by a blood flow meter (ML-191, ADInstruments, Oxford Optonix, UK) using a laser Doppler probe (MSF 100XP, ADInstruments) through a fiberoptic filament attached by use of a tissue adhesive to the intact skull 2 mm caudal to bregma and 6 mm lateral to midline of the affected hemisphere.(Saleem, et al., 2007) Blood flow was recorded using a Power Lab 2/26 data acquisition system (ADInstruments) and successful MCA occlusion was confirmed by an ~80% drop in blood flow relative to baseline pre-embolism.
Analyses of Brain Infarction, Hemorrhage and Swelling
Immediately after the euthanasia and perfusion, the brains were isolated and sliced coronally into 2 mm sections in a rostral-caudal orientation. Both faces of the brain slices were immediately digitally photographed through a microscope (400X). Brain slices were promptly incubated in triphenyl tetrazolium chloride (TTC, 2%) to assess cellular viability followed by digital photography as above. Digital microscopic images were analyzed by a blinded observer using Image Pro Plus 6.2 software to measure areas of brain hemorrhage, TTC staining and hemisphere swelling. To determine the percent hemisphere infarction, the TTC-stained areas of the ischemic and non-ischemic hemispheres were measured on both faces of each brain slice. The percent infarction was calculated for each brain by the formula: infarct percentage = 100 × (VC-VL / VC), where VC = TTC-stained area in the control hemisphere × slice thickness, VL = TTC-stained area in the infarct hemisphere × slice thickness (Swanson, et al., 1990). The percent brain hemorrhage in the infarct hemisphere was determined by measuring the area of hemorrhage in digital microscopic images on both sides of each brain slice for the ischemic and contralateral, unaffected control hemisphere (in which there was no hemorrhage). The percent hemorrhage = 100 × (volume of hemorrhage in the infarcted hemisphere/ volume of the control hemisphere). The amount of swelling in the ischemic hemisphere was determined by comparing the volume of the ischemic hemisphere and the contralateral hemisphere for both faces of each brain slice. The percent swelling was determined for each brain by the formula: swelling percentage = 100 × (volume of the infarcted hemisphere – volume of the control hemisphere)/ volume of the control hemisphere).
Immunohistochemistry
Brain sections were fixed in 4% paraformaldehyde and paraffin embedded. The brain block that spanned the region of the rostral to caudal diencephalon was cut into 5 micron sections for immunostaining. Sections were deparaffinized (Safeclear II, Fisher Diagnostics, MI) and hydrated in serial dilutions of ethyl alcohol and water solutions. Antigen retrieval was performed with sodium citrate buffer (pH 6.0) using heat induction at 98°C for 20 minutes followed by another 20 minutes at room temperature. Sections were washed in PBS and blocked with 10% normal donkey serum PBS for 45 minutes at room temperature. Breakdown of the blood brain barrier was assessed by the amount of albumin leakage (goat anti-mouse albumin, Abcam) into the brain parenchyma around collagen IV (rabbit anti-mouse type IV collagen, Karlan Research Products Corporation) containing microvessels. Sections were incubated with primary antibody diluted in 2% normal donkey serum in PBS overnight at 4°C. After washing with PBS, secondary antibodies, DyLight549–conjugated donkey anti-goat, DyLight488-conjugated donkey anti-Rabbit IgG (Jackson ImmunoResearch Laboratories, PA) or FITC-conjugated donkey anti-mouse (Sigma-Aldrich) were applied for 45 minutes at room temperature. Apoptotic cells were identified by TUNEL staining (In Situ Cell Death Detection Kit, Fluorescein. Roche Applied Science) following manufacturer's protocol. Immunostaining for cleaved caspase 3 was performed with a specific antibody (cleaved caspase-3 Asp175, Cell Signaling, Beverly MA). Total nuclei were counterstained with DAPI using Vectshield mounting media (Vector Laboratories, Burlingame, CA). Slides were examined by fluorescence microscopy (Zeiss) with a computer-controlled Jenoptik ProgRes CF Microscope Research Camera using ProgRes CapturePro software under a constant setting for all slides, or by a Zeiss LSM 710 confocal microscope or by scanning fluorescence microscopy (Aperio ScanScope, Vista, CA). Digital image analyses were performed using ImagePro Plus 6.2 software (Media Cybernetics, Bethesda, MD). To detect albumin leakage, the merged images from the red channel (albumin) and green channel (collagen IV, basement membrane of blood vessel) were analyzed. In the normal mouse brain, albumin is restricted to the intravascular space and merged images show yellow coloration signifying colocalization of albumin and collagen IV. With breakdown of the blood brain barrier, merged images will show albumin (red) leakage outside the blood vessel border (collagen IV, green). For quantitative analysis of albumin immunostaining, red channel (albumin) images were used. Ten fields (40×) from each stroke hemisphere (primarily cortex and striatum) were analyzed, with brain sections from shams as controls. The albumin staining density in TPA and TPA+a2AP-I groups were expressed as a ratio to the staining density in sham (non-stroke group). Total TUNEL positive cells were counted in entire mouse brain at 5× magnification. Cleaved caspase 3 cells were counted in 12-15 fields (40×) from the stroke affected hemisphere using ImagePro Plus 6.2 software. All analyses were done in a blinded fashion.
Statistical Analyses
Normally distributed data were analyzed by an unpaired Student's t-test or a one way ANOVA using the Neuman-Keuls correction. Otherwise data were analyzed by a Mann-Whitney test or a one way Kruskal Wallis analysis using Dunn's correction for multiple statistical inference. Spearman's correlation was used to examine the relationships between two variables. Survival data were subjected to Kaplan Meier analyses. Data are expressed as the mean ± standard error. A two-tailed p <0.05 was considered statistically significant. Using the common standard deviation and other information derived from extensive pilot experimental data using this model, we specified a sample size of 5 mice in each experimental group to achieve an 80% power (two-tailed alpha =0.05), to detect a difference of 7% in stroke infarct size, 10% in swelling and 0.45% in hemorrhage. Survival studies were sized to detect a median survival ratio ≥ 3.9 with 80% power, two-tailed alpha = 0.05.
Analyses of Thrombus Dissolution
The percent dissolution of middle cerebral artery thrombi in vivo was determined as we have described by measuring the residual amount of 125I-fibrin thrombus activity in the brain (by gamma scintillation counting (Cobra II, Packard), by comparison to the amount of 125I-labeled fibrin in the clot immediately prior to placement in the MCA (Reed and Houng, 1999). The thrombus was formed with pooled fresh frozen mouse plasma as described with some modifications (Overgaard, et al., 1992). Pooled fresh frozen mouse plasma (5 ul) and 125I-fibrinogen (~12,500 cpm) were mixed with 1ul of premixed thrombin/CaCl2 (1U/ml thrombin 0.4mol/l CaCl2) on ice. Immediately 2 ul (5,000 cpm) of plasma mix was drawn into 60 cm PE-10 tubing and clotted overnight at 4°C. The PE-10 tubing containing the clot was cut into a 40 cm length and attached to two syringes filled with sterile PBS with 1% BSA. The clot was washed by back and forth movement in the tubing by alternate syringe aspiration for 5 minutes. The clot was expelled and stained in 0.5% Evans blue in PBS for 10 minutes for visualization. After washing twice with 1% BSA in PBS, the clot was cut into 6 pieces to facilitate loading back into the tubing and then gently compressed and washed with sterile saline. The clot was counted in gamma counter (COBRA II, Packard) and pulled into a 30cm segment of modified PE-08 tubing filled with saline for insertion to MCA. At the conclusion of the study, the brain was gamma counted and the amount of thrombus dissolution was determined by the formula, thrombus dissolution % = 100 × (cpm of the original clot – residual brain thrombus cpm)/ cpm of the original clot.
Inactivation of a2AP
We examined the ability of monoclonal antibodies to inactivate mouse a2AP as we have described. (Sazonova, et al., 2007) Briefly, an a2AP inactivating monoclonal antibody (4H9; 25 nM) or a control monoclonal antibody (anti-digoxin), or no antibody were mixed with human or mouse a2AP (25 nM) and then added to human plasmin (25 nM). After 10 min, a2AP-plasmin complex formation was examined by SDS-PAGE under non-reduced conditions followed by immunoblotting with rabbit anti-plasminogen antibody (1:4000, Boehringer Mannheim). They were detected with a secondary goat anti-rabbit IRDye-680 (Li-COR Biosciences, Lincoln, NE) by an infrared imager (LI-COR ODYSSEY).
Assessment of plasma plasminogen, fibrinogen and a2AP Levels
Plasma plasminogen and fibrinogen levels were measured as described (Reed, et al., 1990b, Sazonova, et al., 2007). We measured the relative concentrations of a2AP in plasma from mice supplemented with a2AP by comparison to a2AP in human plasma and normal mouse plasma in a competitive ELISA using a monoclonal antibody (451) that binds to both human and mouse a2AP. Purified monoclonal antibody 451 was immobilized in microtiter plates (50 ul, 10 ug/ml) for 1 hr at 37°C. Various dilutions of plasma (2-39,366) were mixed together (1:1, 35 ul each) with peroxidase labeled a2AP (1:500 dilution in 1% BSA in TBS). Peroxidase labeling was performed following manufacturer's instructions (EZ-Link TM Plus Activated Peroxidase Kit, Thermo Scientific, Rock ford Illinois). The plasma-a2AP peroxidase were added to wells and incubated at room temperature for 60 mins. Wells were washed 4 times with 0.05% Tween20/PBS, tapped dry and then developed with tetramethylbenzidine (1 step-Ultra TMB-ELISA, Thermoscientific) at 307 nm in a microtiter plate reader (Synergy-HT, Bio-TEK instruments Inc, Winooski VT). The data were analyzed with Graph Pad Prism to determine the dilution of plasma from each supplemented mouse that gave 50% inhibition of binding by comparison to pooled control plasmas.
RESULTS
Increased a2AP levels worsen stroke outcomes after TPA therapy
To examine whether elevated a2AP blood levels affected ischemic brain injury and other outcomes, we increased blood a2AP levels an average of 2.3 (±0.2)-fold in mice by intravenous administration of a2AP. When treated 1 h following thromboembolism with TPA, mice with increased a2AP levels had 2.2-fold larger strokes (Fig 1A, p<0.001) than mice with normal a2AP levels. Elevated levels of a2AP worsened brain swelling after TPA therapy (Fig 1B, p<0.001) by comparison to normal levels. Increased a2AP levels also decreased dissolution of the MCA thrombus (p<0.001) after TPA therapy (Fig. 1C) but did not significantly affect hemorrhage after TPA treatment (Fig. 1D).
Figure 1.
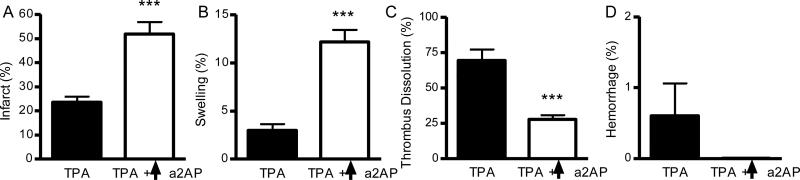
Increased a2AP levels (↑a2AP) worsen infarction and brain swelling after TPA therapy. Effects of increased a2AP levels and TPA treatment on A) brain infarction, B) brain swelling, C) dissolution of the culprit thromboembolus and D) brain hemorrhage. Mice with thromboembolic stroke were treated with TPA (10 mg/kg) after 1 h of ischemia; a2AP levels were increased by a2AP infusion immediately after thromboembolism. Mice were analyzed 6 h after thromboembolic stroke. The percent infarction was determined at the completion of the experiment by TTC-staining of serial brain slices followed by digital imaging analyses by a blinded observer. The percent swelling was determined at the completion of the experiment by digital imaging analyses of serial brain slices by a blinded observer. The percent dissolution was determined by gamma scintillation counting of the residual 125I-fibrin in the thrombus. The percent hemorrhage was determined at the completion of the experiment by digital imaging analyses of serial brain slices by a blinded observer. Data represent mean ± standard errors. N= 5 per group; ***p<0.001 vs. TPA
a2AP inactivation in normal mice reduces infarction and hemorrhage after standard dose TPA therapy
The finding that increased a2AP levels worsened stroke outcomes after TPA therapy suggested that normal, endogenous a2AP levels also may affect outcomes after ischemic stroke. To examine this, we selected a monoclonal antibody that inactivates mouse a2AP (Sazonova, et al., 2007). When mice were treated 2.5 h after thromboembolism, the combination of a2AP inactivation and standard dose TPA (10 mg/kg) reduced brain infarction by comparison to standard dose TPA alone (Fig. 2A, p<0.001). By comparison to mice treated with standard dose TPA alone, the combination of standard TPA and a2AP inactivation also significantly increased dissolution of the culprit MCA thromboembolus (Fig 2B, p<0.001). The combination of standard dose TPA and a2AP inactivation significantly reduced brain hemorrhage by comparison to standard TPA alone (Fig. 2C, p<0.05).
Figure 2.
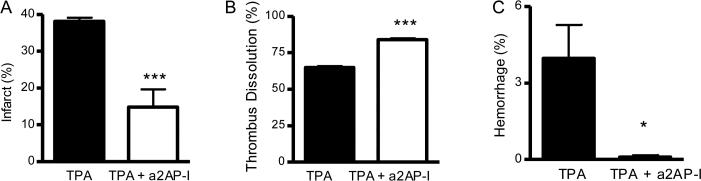
Standard dose TPA treatment with a2AP-inactivation affects infarction, dissolution of the culprit MCA thrombus and hemorrhage by comparison to standard dose TPA alone. Effects on A) brain infarction, B) dissolution of the culprit MCA thrombus and C) brain hemorrhage. Mice were treated with TPA (10 mg/kg) with or without a2AP inactivation 2.5 h after MCA thromboembolism and were analyzed 6 h after stroke. The percent hemorrhage was determined by imaging of brain slices at the completion of the experiment; similarly infarction was determined at the completion of the experiment by TTC staining of serial brain slices. Images were subjected to digital analysis by a blinded observer. Percent thrombus dissolution was determined as above. Data represent mean ± standard errors. N= 5 per group; *p<0.05 and ***p<0.001.
Effects of a2AP inactivation and low dose TPA treatment
To determine whether the protective effects of a2AP inactivation were simply related to improved dissolution of the culprit MCA thromboembolus, we treated mice with a 5-fold lower dose of TPA (2 mg/kg), with or without a2AP inactivation. By comparison to standard dose TPA, there was a 2.6-fold reduction in the dissolution of the culprit MCA thrombus by low dose TPA and a 1.6-fold reduction in dissolution by the combination of low dose TPA and a2AP inactivation. However, low dose TPA with a2AP inactivation significantly reduced brain infarction by comparison to low dose TPA alone (p<0.001, Fig. 3A) and standard dose TPA (p<0.01, Fig 2A). The combination of low dose TPA and a2AP inactivation also significantly enhanced thrombus dissolution vs. low dose TPA alone (Fig. 3B, p<0.05). There was no significant difference in brain hemorrhage in the low dose TPA treatment groups (Fig. 3C).
Figure 3.
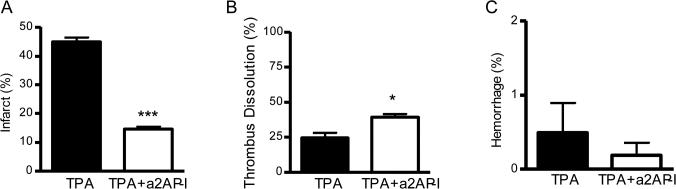
Low dose TPA with a2AP inactivation improves stroke outcomes vs. low dose TPA alone. Effects on A) brain infarction, B) dissolution of the culprit MCA thrombus and C) brain hemorrhage. Mice were treated with TPA (2 mg/kg) with or without a2AP inactivation 2.5 h after MCA thromboembolism and were analyzed 6 h after stroke. The percent hemorrhage was determined by imaging of brain slices at the completion of the experiment; similarly infarction was determined at the completion of the experiment by TTC staining of serial brain slices. Images were subjected to digital analysis by a blinded observer. Percent thrombus dissolution was determined as above. Data represent mean ± standard errors. N= 5 per group; *p<0.05 and ***p<0.001 vs. low dose TPA alone.
Since low dose TPA was associated with significantly lower dissolution of the thrombus than higher dose TPA, we examined systemic plasminogen and fibrinogen levels for evidence of plasminogen activation. Treatment of mice with low dose TPA caused a significant reduction in circulating plasminogen levels by comparison to control, untreated mice (68 ± 8 % vs. 100 ± 5 %, p<0.01), consistent with TPA-mediated plasminogen activation. The combination of TPA with a2AP inactivation also decreased plasminogen levels by comparison to controls (67 ± 4 % vs. 100 ± 5 %, p<0.01) to the same extent as TPA alone (67 ± 4 % vs. 68 ± 8%). Levels of the coagulation protein fibrinogen levels were also reduced by TPA vs. controls (1.3 ± 0.1 vs. 2.0 ± 0.1 mg/ml, p<0.01) to a greater extent than TPA with a2AP inactivation (1.7 ± 0.1 mg/ml, p<0.05). These data show that despite lower dissolution of the culprit thrombus, lower dose TPA treatment was associated with significant plasminogen activation and fibrinogen consumption.
Correlations between thrombus dissolution and outcomes
To examine the relationship between dissolution of the culprit thrombus, brain infarction and hemorrhage, a correlational analysis was performed. Greater dissolution of the culprit thrombus was associated with reduced brain infarction in mice treated with TPA alone (Spearman r =-.82, p=0.01) but not in mice treated with both TPA and a2AP inactivation (r= -0.43, p=0.14). There was no significant correlation between the percent dissolution of the MCA thrombus and hemorrhage (r= 0.04, p=0.85). Moreover, the size of brain infarction was not correlated with the magnitude of hemorrhage (r= 0.13, p=0.56) or with the severity of swelling (r=-0.06, p=0.79) in either group.
a2AP inactivation with TPA therapy reduces mortality, infarction brain swelling and hemorrhage
The previous experiments indicated that a2AP affected acute stroke outcomes when TPA was administered within up to 2.5 h after the onset of ischemia from MCA thromboembolism. However TPA therapy alone given shortly after the onset of ischemia was more effective than it was after prolonged ischemia, which may suggest that the adverse effects of a2AP on TPA are restricted to prolonged ischemia. To examine this, we evaluated survival and outcomes in mice treated within 30 min. of ischemia. Previous data (Fig. 3A) showed that the combination of lower dose TPA with a2AP inactivation was as effective at reducing infarct size as the combination of standard dose TPA with a2AP inactivation (Fig. 2A). Therefore we compared the effects of standard dose TPA alone vs. lower dose TPA with a2AP inactivation. Mice treated with lower dose TPA and a2AP inactivation had reduced mortality when compared to mice treated with standard dose TPA (Fig. 4A, p<0.01). When the brains of surviving mice were examined, brain infarction was significantly smaller in mice treated with TPA and a2AP inactivation vs. TPA alone (Fig. 4B, p<0.01) as was hemorrhage (Fig. 4C, p<0.01). There was also a significant reduction in brain swelling in mice treated with the combination of TPA and a2AP inactivation compared to those treated with TPA alone (Fig. 4D, p<0.05).
Figure 4.

Treatment with low dose TPA with a2AP inactivation reduces mortality, brain infarction, hemorrhage and swelling vs. standard dose TPA alone. Mice were treated 30 min after MCA thromboembolism with TPA (10 mg/kg) or TPA (2 mg/kg) with a2AP inactivation. Survival was monitored and brains of mice surviving at least 12 h were analyzed. A) Effect on 24 h survival. B) Effect on brain infarction in surviving mice. Percent infarction was determined by TTC staining as described above. C) Effect on brain swelling as described above. D) Effect on hemorrhage as described above. Data represent mean ± standard errors. Survival analyses N =17, otherwise N= 5 per group. *p<0.05, **p<0.01, vs. TPA alone.
Effects of a2AP inactivation with TPA therapy on apoptosis and the blood brain barrier
Combination treatment with TPA and a2AP inactivation markedly reduced TUNEL staining in non-vascular cells (collagen IV negative cells) by comparison to TPA treatment alone (Fig. 5A, p<0.01) consistent with reduced cell death in the stroke region. The combination of TPA and a2AP inactivation also markedly reduced cleaved caspase 3 staining vs. TPA alone (p<0.0005, not shown). In mice treated with TPA alone, there was leakage of albumin outside vascular spaces identified by collagen IV immunostaining (Fig. 5B), consistent with breakdown of the blood brain barrier. In contrast, albumin leakage was markedly reduced in mice treated with TPA and a2AP inactivation (Fig. 5B, p<0.01), signifying reduced barrier breakdown.
Figure 5.

TPA treatment with a2AP inactivation decreases breakdown of the blood brain barrier and apoptosis indicated by TUNEL-staining. A) TUNEL staining (green) around the area of collagen IV -stained blood vessels (red). DAPI-stained nuclei are in blue. Graph, image quantification of TUNEL positive non-blood vessel cells as described in Methods. B) Detection of breakdown of the blood brain barrier by leakage of albumin (red) outside of collagen IV-stained blood vessels (green) with DAPI-stained nuclei stained in blue. In mice treated with TPA + a2AP-I, albumin (red) co-localized with collagen IV-stained blood vessels as indicated by yellow coloration in the merged image. In mice treated with TPA there was significant leakage of albumin outside collagen IV-stained blood vessels as shown by the diffuse red staining and the minimal yellow coloration in the merged image. Graph, image quantification of blood brain barrier leakage of albumin described in Methods. Data represent mean ± standard errors. N= 4 per group; **p<0.01 vs. TPA alone.
DISCUSSION
While clinical studies have correlated high a2AP levels with the failure of TPA to reperfuse occluded cerebral vessels, these experiments provide the first experimental evidence that a2AP activity worsens outcomes after TPA treatment of thromboembolic ischemic stroke (Marti-Fabregas, et al., 2005). Increased a2AP levels magnified brain infarction and swelling after TPA treatment and reduced the ability of TPA to dissolve the culprit MCA thromboembolus. Conversely, inactivating a2AP, at various times (0.5, 1 and 2.5 h) after the onset of ischemia, improved the efficacy of TPA and extended its therapeutic time window for reducing brain infarction and brain swelling. Inactivation of a2AP improved acute dissolution of the culprit thrombus, reduced brain hemorrhage and improved short-term survival after TPA treatment.
Thrombus dissolution is controlled by plasminogen activators such as TPA as well as through several other molecular mechanisms (Cesarman-Maus and Hajjar, 2005). Plasminogen activator inhibitor-1 inhibits both endogenous and pharmacologic TPA. The susceptibility of fibrin to plasmin degradation is reduced by activated factor XIII and by thrombin-activated fibrinolysis inhibitor (Reed, et al., 1990a). Both a2AP and a2-macroglobulin inhibit plasmin in the circulation and, more slowly, plasmin bound to fibrin. In addition, the crosslinking of a2AP to fibrin by activated factor XIII also inhibits the initiation of fibrinolysis in vivo (Mutch, et al., 2007, Reed, et al., 1990a) Studies comparing the effects of PAI-1, thrombin-activated fibrinolysis inhibitor and a2AP indicated that all three molecules exert regulatory effects, but a2AP had the greatest influence on fibrinolysis (Mutch, et al., 2007). While the effects of a2AP inactivation on cerebrovascular thrombi have not been previously examined, a2AP inactivation synergistically amplifies fibrinolysis by endogenous and pharmacologic TPA in vitro and increases dissolution of venous and pulmonary emboli thrombi in vivo without causing bleeding (Butte, et al., 1997, Lijnen, et al., 1999, Matsuno, et al., 2003, Reed, et al., 1990a, Reed, et al., 1990b).
The therapeutic rationale for TPA treatment is to dissolve culprit thrombi to restore perfusion and reduce ischemic brain injury. As expected with TPA treatment alone, more extensive dissolution of the culprit thrombus was strongly correlated with greater reductions in stroke size. However, in mice treated with the combination of TPA and a2AP inactivation, thrombus dissolution and stroke size were not correlated. It is notable that, when compared to standard dose TPA alone, lower dose TPA with a2AP inactivation was less effective in dissolving the MCA thromboembolus (65.0 ± 1.1% vs. 39.5 ± 1.9%, p<0.01). Yet despite less thrombus dissolution, lower dose TPA with a2AP inactivation was significantly more effective at reducing mortality, brain infarction, hemorrhage and swelling in survivors, than standard dose TPA. This argues that a2AP inactivation exerts protective effects beyond simply increasing fibrinolysis by preventing plasmin inhibition. In addition, the fact that a2AP inactivation with TPA treatment consistently reduced brain hemorrhage, is not explained by previous models which indicate that a2AP acts solely to inhibit plasmin to protect against TPA-induced hemorrhage (Weitz, et al., 1993). Finally, the protective effects of a2AP inactivation were not simply attributable to reduced brain infarction because no correlation was found between the severity of brain infarction and brain hemorrhage or swelling. Recent studies have identified a new role for a2AP in the brain by showing that, independent of plasmin, a2AP plays a role in the growth of dendritic neurons through p38 mitogen activated protein kinase activation (Kawashita, et al., 2013). Additional studies are required to determine the potential non-fibrinolytic mechanisms responsible for the protective effects of a2AP-inactivation during ischemic stroke.
There are advantages to models of ischemic stroke induced by mechanical occlusion and thrombin injection with craniotomy. However, since important differences have been noted between mechanical and thromboembolic stroke, we used a thromboembolic model which some experts indicate has the greatest translational relevance to human stroke and is best suited for examining the overall physiologic effects of thrombus-dissolving agents like TPA (Aoki, et al., 2002, Mergenthaler and Meisel, 2012, Nagai, et al., 1999). A unique advantage of this model is that provides an integrated simultaneous assessment of the effects of TPA and a2AP on both dissolution of the culprit MCA thrombus and ischemic brain injury (Mergenthaler and Meisel, 2012, Nagai, et al., 1999). Proximal MCA stroke is associated with large brain infarctions, significant mortality and lower success rates for TPA; as such, this experimental model provides insights into factors that have potent, acute effects on stroke outcomes after TPA treatment, though longer term studies may be limited by the reduced survival of TPA-treated mice (del Zoppo and Koziol, 2007, Furuya, et al., 2004, Hara, et al., 2000, Kilic, et al., 2000, Orset, et al., 2007). Indeed, the diminished survival of mice treated with TPA alone limited our ability to assess effects on post-stroke disability. To determine the integrated effects of thrombus dissolution on ischemic brain outcomes we examined mice 6 h after thromboembolism when thrombus dissolution was still a dynamic process. Since ischemic injury may continue to evolve beyond the 6 h time point, we also examined the effects of TPA and a2AP inactivation on outcomes in 24 h survival experiments. To assess the time-related effects of TPA and a2AP inactivation on the ischemic brain, we treated mice at three different time points: 30 min, 1 h and 2.5 h after thromboembolism. The results were consistent: by comparison to standard TPA alone, treatment with TPA and a2AP inactivation reduced brain infarction and, as ischemia prolonged, diminished hemorrhage (Fig. 2- 4).
Taken together these studies suggest that a2AP contributes to TPA failure, brain swelling, apoptosis and hemorrhage in thromboembolic stroke. The finding that a2AP contributes to poor outcome in ischemic stroke is consistent with clinical studies that have associated high a2AP levels with TPA failure and with a greater risk of ischemic stroke (Marti-Fabregas, et al., 2005, Suri, et al., 2010). Further studies are warranted to determine the mechanisms of a2AP action and to explore whether a2AP inactivation may prove to be a useful approach to improving stroke treatment by reducing long term disability in addition to prolonging survival.
Highlights.
Tissue plasminogen activator (TPA) may fail due to high a2-antiplasmin (a2AP) levels.
In TPA therapy for experimental stroke, a2AP increased brain infarction and swelling.
TPA with a2AP inactivation (a2AP-I) decreased brain injury, hemorrhage and swelling.
TPA and a2AP-I also reduced apoptosis, blood brain barrier breakdown and mortality.
a2AP reduces the safety and efficacy of TPA for ischemic stroke.
Acknowledgements
This research was supported in part from research grants from the American Heart Association (10GRNT3870005) and National Institutes of Health (HL58496, HL9750 and NS073147) to GR.
Abbreviations
- a2AP
α2-antiplasmin
- TPA
tissue plasminogen activator
- TTC
triphenyl tetrazolium chloride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki T, Sumii T, Mori T, Wang X, Lo EH. Blood-brain barrier disruption and matrix metalloproteinase-9 expression during reperfusion injury: mechanical versus embolic focal ischemia in spontaneously hypertensive rats. Stroke. 2002;33:2711–2717. doi: 10.1161/01.str.0000033932.34467.97. [DOI] [PubMed] [Google Scholar]
- Butte AN, Houng AK, Jang IK, Reed GL. Alpha 2-antiplasmin causes thrombi to resist fibrinolysis induced by tissue plasminogen activator in experimental pulmonary embolism. Circulation. 1997;95:1886–1891. doi: 10.1161/01.cir.95.7.1886. [DOI] [PubMed] [Google Scholar]
- Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- Chimowitz MI. Endovascular treatment for acute ischemic stroke--still unproven. N Engl J Med. 2013;368:952–955. doi: 10.1056/NEJMe1215730. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Koziol JA. Recanalization and stroke outcome. Circulation. 2007;115:2602–2605. doi: 10.1161/CIRCULATIONAHA.107.698225. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Davis SM, Parsons MW, Ma H, Dewey HM, Howells DW. How to make better use of thrombolytic therapy in acute ischemic stroke. Nat Rev Neurol. 2011;7:400–409. doi: 10.1038/nrneurol.2011.89. [DOI] [PubMed] [Google Scholar]
- Furuya K, Kawahara N, Kawai K, Toyoda T, Maeda K, Kirino T. Proximal occlusion of the middle cerebral artery in C57Black6 mice: relationship of patency of the posterior communicating artery, infarct evolution, and animal survival. J Neurosurg. 2004;100:97–105. doi: 10.3171/jns.2004.100.1.0097. [DOI] [PubMed] [Google Scholar]
- Hara T, Mies G, Hossmann KA. Effect of thrombolysis on the dynamics of infarct evolution after clot embolism of middle cerebral artery in mice. J Cereb Blood Flow Metab. 2000;20:1483–1491. doi: 10.1097/00004647-200010000-00010. [DOI] [PubMed] [Google Scholar]
- Kawashita E, Kanno Y, Asayama H, Okada K, Ueshima S, Matsuo O, Matsuno H. Involvement of alpha2-antiplasmin in dendritic growth of hippocampal neurons. J Neurochem. 2013;126:58–69. doi: 10.1111/jnc.12281. [DOI] [PubMed] [Google Scholar]
- Kilic E, Hermann DM, Hossmann KA. Recombinant tissue-plasminogen activator-induced thrombolysis after cerebral thromboembolism in mice. Acta Neuropathol. 2000;99:219–222. doi: 10.1007/pl00007430. [DOI] [PubMed] [Google Scholar]
- King SM, McNamee RA, Houng AK, Patel R, Brands M, Reed GL. Platelet dense-granule secretion plays a critical role in thrombosis and subsequent vascular remodeling in atherosclerotic mice. Circulation. 2009;120:785–791. doi: 10.1161/CIRCULATIONAHA.108.845461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees KR, Bluhmki E, von Kummer R, Brott TG, Toni D, Grotta JC, Albers GW, Kaste M, Marler JR, Hamilton SA, Tilley BC, Davis SM, Donnan GA, Hacke W, Allen K, Mau J, Meier D, del Zoppo G, De Silva DA, Butcher KS, Parsons MW, Barber PA, Levi C, Bladin C, Byrnes G. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695–1703. doi: 10.1016/S0140-6736(10)60491-6. [DOI] [PubMed] [Google Scholar]
- Lijnen HR, Okada K, Matsuo O, Collen D, Dewerchin M. Alpha2-antiplasmin gene deficiency in mice is associated with enhanced fibrinolytic potential without overt bleeding. Blood. 1999;93:2274–2281. [PubMed] [Google Scholar]
- Marti-Fabregas J, Borrell M, Cocho D, Belvis R, Castellanos M, Montaner J, Pagonabarraga J, Aleu A, Molina-Porcel L, Diaz-Manera J, Bravo Y, Alvarez-Sabin J, Davalos A, Fontcuberta J, Marti-Vilalta JL. Hemostatic markers of recanalization in patients with ischemic stroke treated with rt-PA. Neurology. 2005;65:366–370. doi: 10.1212/01.wnl.0000171704.50395.ba. [DOI] [PubMed] [Google Scholar]
- Matsuno H, Okada K, Ueshima S, Matsuo O, Kozawa O. Alpha2-antiplasmin plays a significant role in acute pulmonary embolism. J Thromb Haemost. 2003;1:1734–1739. doi: 10.1046/j.1538-7836.2003.00252.x. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Meisel A. Do stroke models model stroke? Dis Model Mech. 2012;5:718–725. doi: 10.1242/dmm.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch NJ, Thomas L, Moore NR, Lisiak KM, Booth NA. TAFIa, PAI-1 and alpha-antiplasmin: complementary roles in regulating lysis of thrombi and plasma clots. J Thromb Haemost. 2007;5:812–817. doi: 10.1111/j.1538-7836.2007.02430.x. [DOI] [PubMed] [Google Scholar]
- Nagai N, De Mol M, Lijnen HR, Carmeliet P, Collen D. Role of plasminogen system components in focal cerebral ischemic infarction: a gene targeting and gene transfer study in mice. Circulation. 1999;99:2440–2444. doi: 10.1161/01.cir.99.18.2440. [DOI] [PubMed] [Google Scholar]
- Orset C, Macrez R, Young AR, Panthou D, Angles-Cano E, Maubert E, Agin V, Vivien D. Mouse model of in situ thromboembolic stroke and reperfusion. Stroke. 2007;38:2771–2778. doi: 10.1161/STROKEAHA.107.487520. [DOI] [PubMed] [Google Scholar]
- Overgaard K, Sereghy T, Boysen G, Pedersen H, Diemer NH. Reduction of infarct volume and mortality by thrombolysis in a rat embolic stroke model. Stroke. 1992;23:1167–1173. doi: 10.1161/01.str.23.8.1167. discussion 1174. [DOI] [PubMed] [Google Scholar]
- Reed GL, 3rd, Matsueda GR, Haber E. Inhibition of clot-bound alpha 2-antiplasmin enhances in vivo thrombolysis. Circulation. 1990a;82:164–168. doi: 10.1161/01.cir.82.1.164. [DOI] [PubMed] [Google Scholar]
- Reed GL, 3rd, Matsueda GR, Haber E. Synergistic fibrinolysis: combined effects of plasminogen activators and an antibody that inhibits alpha 2-antiplasmin. Proc Natl Acad Sci U S A. 1990b;87:1114–1118. doi: 10.1073/pnas.87.3.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed GL, Houng AK. The contribution of activated factor XIII to fibrinolytic resistance in experimental pulmonary embolism. Circulation. 1999;99:299–304. doi: 10.1161/01.cir.99.2.299. [DOI] [PubMed] [Google Scholar]
- Saleem S, Li RC, Wei G, Dore S. Effects of EP1 receptor on cerebral blood flow in the middle cerebral artery occlusion model of stroke in mice. J Neurosci Res. 2007;85:2433–2440. doi: 10.1002/jnr.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazonova IY, Thomas BM, Gladysheva IP, Houng AK, Reed GL. Fibrinolysis is amplified by converting alpha-antiplasmin from a plasmin inhibitor to a substrate. J Thromb Haemost. 2007;5:2087–2094. doi: 10.1111/j.1538-7836.2007.02652.x. [DOI] [PubMed] [Google Scholar]
- Suri MF, Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Novel hemostatic factor levels and risk of ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Cerebrovasc Dis. 2010;29:497–502. doi: 10.1159/000297966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz JI, Leslie B, Hirsh J, Klement P. Alpha 2-antiplasmin supplementation inhibits tissue plasminogen activator-induced fibrinogenolysis and bleeding with little effect on thrombolysis. J Clin Invest. 1993;91:1343–1350. doi: 10.1172/JCI116335. [DOI] [PMC free article] [PubMed] [Google Scholar]