

Abstract
Dysregulation of lipid homeostasis is intimately associated with obesity, type 2 diabetes, and cardiovascular diseases. Sterol regulatory-element binding proteins (SREBPs) are the master regulators of lipid biosynthesis. Previous studies have shown that the conserved transcriptional cofactor Mediator complex is critically required for the SREBP transcriptional activity, and recruitment of the Mediator complex to the SREBP transactivation domains (TADs) is through the MED15-KIX domain. Recently, we have synthesized several boron-containing small molecules. Among these novel compounds, BF175 can specifically block the binding of MED15-KIX to SREBP1a-TAD in vitro, resulting in an inhibition of the SREBP transcriptional activity and a decrease of SREBP target gene expression in cultured hepatocytes. Furthermore, BF175 can improve lipid homeostasis in the mouse model of diet-induced obesity. Compared with the control, BF175 treatment decreased the expression of SREBP target genes in mouse livers and decreased hepatic and blood levels of lipids. These results suggest that blocking the interaction between SREBP-TADs and the Mediator complex by small molecules may represent a novel approach for treating diseases with aberrant lipid homeostasis.
Introduction
The current prevalence of obesity substantially increased the incidence of several comorbidities, including type 2 diabetes, cardiovascular diseases, and some types of cancer (1,2). Strikingly, ∼70% of diabetic patients are also diagnosed with nonalcoholic fatty liver disease (NAFLD) (3), which is often associated with hepatic insulin resistance (4). The most common feature of NAFLD is excessive fat accumulation in hepatocytes. Although fatty acids from diets and adipose tissue lipolysis support re-esterification in the liver to drive triglyceride synthesis, up to 30% of hepatic fatty acids are from de novo lipogenesis in NAFLD, but <5% in normal individuals (5,6). In addition, increased hepatic de novo lipogenesis may lead to dyslipidemia and atherosclerosis, the primary risk factors for heart disease.
Among the known lipogenic regulators, sterol regulatory-element binding protein (SREBP) transcription factors are master regulators of lipid homeostasis (7–9). Through activating the expression of rate-limiting lipogenic and cholesterogenic genes, such as fatty acid synthase (FAS) and HMG-CoA reductase (HMGCR), SREBPs promote the biosynthesis of fatty acids, triglycerides, and cholesterol (7–9). Therefore, suppressing the SREBP pathway may efficiently inhibit lipid biosynthesis. The three mammalian SREBP isoforms (SREBP-1a, -1c, and -2) are synthesized as inactive precursors that are tethered to the endoplasmic reticulum membrane when cellular levels of sterols or fatty acids, the end products of SREBP target genes, are high. Decrease of intracellular sterols or specific fatty acids can result in the transportation of SREBPs to the Golgi apparatus, where they undergo proteolytic maturation. The N-terminal mature forms of SREBP transcription factors then migrate into the nucleus and activate the transcription of their target genes (7–9). Moreover, insulin is known to stimulate de novo lipogenesis through SREBP-1c (10) by increasing SREBP-1c gene transcription (11,12), proteolytic maturation from SREBP-1c precursor (13,14), and nuclear SREBP-1c protein stability (15).
Recently, we synthesized a group of novel boron-containing compounds and found that some of them had inhibitory effects on lipogenic gene expression and lipid biosynthesis (16). Here, we further studied one of the compounds, BF175, in vitro and in vivo. We show that BF175 specifically inhibits SREBP-mediated transcription by blocking the binding to the Mediator complex. BF175 has inhibitory effects on the expression of SREBP target genes in vitro and in vivo. In addition, BF175 displayed several beneficial effects on lipid metabolism in diet-induced obesity (DIO). These results suggest for the first time that the SREBP transcriptional activity can be targeted by small molecules for inhibiting lipid biosynthesis.
Research Design and Methods
Antibodies and Synthesis of BF175
Anti-SREBP1 (2A4; Santa Cruz Biotechnology, Inc.), anti-FAS (Cell Signaling Technology, Inc.), anti–Flag M2 (Sigma-Aldrich), anti–β-actin (Sigma-Aldrich), and anti–β-tubulin (Life Technologies) antibodies were purchased in this study. The boron-containing compounds BF175 and BF62 were synthesized and purified according to the method we reported previously (16).
Plasmids
SREBP1c-TAD and SREBP2-TAD in pcDNA3-HA-Gal4DBD were generated by subcloning the transactivation domains (TADs) from pGEX-2TN (17). Wild-type and SRE mutant pSREBP1c-luc were gifts (18). Other plasmids were described previously (17).
Tissue Culture and Quantitative RT-PCR assay
HEK293, HepG2, and primary rat hepatocytes were cultured as described previously (19). Extraction of total RNA from cells or mouse livers and real-time RT-PCR have been reported previously (19).
Transfection and Luciferase Assay
For luciferase assays, 5 × 105 cells per well were plated into 24-well plates and transfected with 100 ng of firefly luciferase plasmids that contain the promoters of either FAS, SREBP-1c, or 2xGal4-binding elements in a basal promoter, using Lipofectamine 2000 (Life Technologies) as the transfection reagent. For the transfection control, cells were also cotransfected with 5 ng of renilla luciferase plasmids that only contain a basal promoter. In the Gal4 experiments, 10 ng of HA-Gal4-SREBP1a-TAD (VP16-TAD or Myb-TAD) plasmids were also included. Transfected cells were lysed after 24 h and analyzed using the Dual-Luciferase System (Promega) according to the manufacturer's instructions.
Glutathione S-transferase Pull-Down Assay
Glutathione S-transferase (GST) fusion proteins (GST only, MED15-KIX, SREBP-1a-TAD, or VP16-TAD) were expressed in Escherichia coli BL21 cells and purified by glutathione Sepharose (Amersham Pharmacia) according to the manufacturer’s protocol. The quality and quantity of GST fusion proteins were analyzed by Coomassie staining. Purified Flag-tagged SREBP-1a or nuclear extracts from cultured cells were prepared as previously described (17). Flag-tagged MED15 or SREBP-1a proteins were expressed in HEK293 cells by transient transfection and extracted into binding buffer containing 20 mmol/L Tris-HCl at pH 8.0, 150 mmol/L NaCl, 0.1 mmol/L EDTA, 10% glycerol, 0.05% NP-40, 1 mmol/L DTT, 1 mmol/L benzamidine, 0.25 mmol/L PMSF, and 2 µg/mL aprotinin. Nuclear extracts or cell lysates were applied to 25 μL of beads containing GST fusion proteins and incubated at 4°C for 3 h. Beads were washed five times with 1 mL each of the binding buffer containing 250 mmol/L NaCl and once with the binding buffer. Bound proteins were eluted with 0.3% sarkosyl and analyzed by immunoblotting.
Protein Extraction, Immunoblotting, and Oil Red O Staining of Drosophila Larvae
Protein extraction from cells or mouse livers and immunoblotting were described previously (19). After feeding with regular fly food containing either 0% (control) or 0.2% BF175 for 2 days, the larvae of w1118 wild-type Drosophila strain were analyzed by Oil Red O staining as reported previously (19).
Animals and Animal Care
Male C57BL/6J mice were purchased from The Jackson Laboratory at 8 weeks of age and kept in the Animal Facility of Albert Einstein College of Medicine for 1 week before they were supplied with a high-fat diet (HFD, 60% kcal from fat, D12492; Research Diets) for 4 weeks. Then, the treatment with BF175 was performed for either 1 week in solution by osmotic pumps or eight weeks in HFD. Some mice were placed individually in metabolic cages for analyses of energy expenditure and physical activity. Plasma levels of triglycerides and free fatty acids were measured with kits from Sigma-Aldrich. Total cholesterol levels in plasma were determined with a kit from Cayman Chemical. Blood glucose levels were measured using a glucose meter. All animal procedures were in accordance with Albert Einstein College of Medicine research guidelines for the care and use of laboratory animals.
Statistical Analyses
Data are presented as the mean ± SD. The significance of differences between two groups was evaluated using Student t test. P value <0.05 was considered significant.
Results
BF175 Specifically Inhibits SREBP Transcriptional Activity
Using a novel synthetic strategy, we recently synthesized several boron-containing compounds (16). When tested in cultured hepatocytes, some of the compounds displayed an inhibitory effect on the biosynthesis of palmitate and cholesterol through inhibiting lipogenic gene expression, although some compounds were either inactive or toxic at higher doses to cultured cells (16). Based on those results, we chose one compound, named BF175 (Fig. 1A), for further analysis, as it was less toxic to hepatocytes in tissue culture (data not shown).
Figure 1.

A: Chemical structures of BF175 and BF62. B: Effects of the indicated doses of BF175 or BF62 on the binding of MED15-KIX to purified Flag-tagged SREBP-1a by GST pull-down assays. C: Effects of BF175 (50 µmol/L) on the binding of SREBP1a-TAD to overexpressed Flag-tagged MED15 in HEK293 cell lysates by GST pull-down assays. D: Effects of 50 µmol/L BF175 or BF62 on the activity of SREBP1a-TAD in HepG2 cells by dual-luciferase assays. Data represent the mean ± SD (n = 3). *P < 0.05; #P < 0.01 vs. control (DMSO).
Previous studies have demonstrated that the SREBP transcription factors are master regulators of lipid biosynthesis (7–9). Our previous work has shown that the multisubunit cofactor complex, Mediator (20,21), is critically required for the activity of the SREBP TADs in vitro and in vivo, and the KIX domain in the ARC105/MED15 subunit of the Mediator complex is responsible for the interaction with the SREBP-TADs (17). We then examined whether BF175 could inhibit the interaction between MED15-KIX and SREBP1a-TAD. For that purpose, we performed GST pull-down experiments. As shown in Fig. 1B, in the presence of BF175, the ability of MED15-KIX to pull-down purified Flag-tagged SREBP-1a was decreased in a dose-dependent manner. Conversely, BF175 also significantly inhibited the interaction between GST-SREBP1a-TAD and Flag-tagged MED15 or endogenous MED15 in HEK293 cell extracts (Fig. 1C and data not shown). These data demonstrate that BF175 directly blocks the interaction between MED15-KIX and SREBP-1a-TAD. To determine the specificity of BF175, we also studied another boron-containing compound, BF62 (Fig. 1A), which is highly similar to BF175 in chemical structure but could not inhibit lipogenic gene expression (16). Consistent with its biological functions, BF62 also did not affect MED15-KIX binding to the purified SREBP-1a (Fig. 1B), suggesting that the inhibitory effect of BF175 on MED15-KIX binding to SREBP1a-TAD depends on the chemical structure.
Since the Mediator complex recruitment to SREBP1a-TAD through MED15-KIX is essential for SREBP-1a activation (17), BF175 may inhibit the transcriptional activity of SREBP1a-TAD. To test this possibility, the TAD of SREBPs, VP16 or Myb, was fused to the Gal4 DNA-binding domain to generate an artificial transcription factor, and the transcriptional activity of this fusion protein was assayed using the luciferase system whose expression is under the control of two Gal4-binding elements (22). Using this luciferase system, we found that BF175 significantly decreased the activity of SREBP1a-TAD in HepG2 cells (Fig. 1D), consistent with the fact that BF175 blocks the interaction between MED15-KIX and SREBP1a-TAD. As a control, BF62 did not display a similar effect, but through unknown mechanism(s) slightly increased the activity of SREBP1a-TAD (Fig. 1D). In addition, BF175 also inhibited the TAD activities of SREBP-1c and -2 but with much lower efficiency (Fig. 2A), consistent with our previous results showing that all three SREBPs bind to MED15-KIX but with a lower affinity for SREBP-1c and -2 (17).
Figure 2.
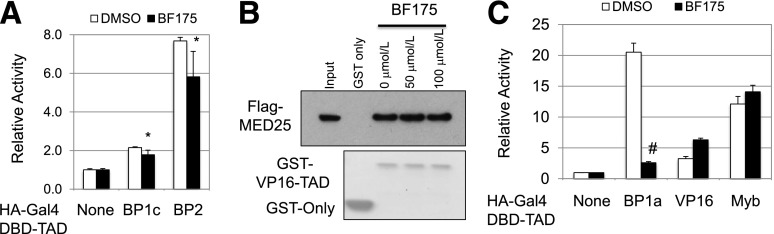
A: Effects of BF175 (50 µmol/L) on the activity of the SREBP-1c and SREBP-2 TADs in HepG2 cells by dual-luciferase assays. B: Effects of the indicated doses of BF175 on the binding of VP16-TAD to overexpressed Flag-tagged MED25 in HEK293 cell lysates by GST pull-down assays. C: Effects of BF175 (50 µmol/L) on the activity of the indicated TADs in HEK293 cells by dual-luciferase assays. Data represent the mean ± SD (n = 3). *P < 0.05; #P < 0.01 vs. control (DMSO).
Our previous study (22) and another report (23) have shown that VP16-TAD functionally recruits the Mediator complex through the VBD domain of the MED25 subunit, whereas MED15 is not involved in regulating VP16-TAD. Moreover, Myb-TAD does not recruit the Mediator complex (17). Furthermore, although MED15-KIX and CBP-KIX are similar in structure, unlike MED15-KIX, which is only known to bind to SREBPs so far, CBP-KIX interacts with the TADs of SREBPs, VP16, Myb, and many other transcription factors (17). To further determine the specificity of BF175 inhibiting the interaction between MED15-KIX and SREBPs, we examined the effects of BF175 on two other transcription factors, VP16 and Myb. As shown in Fig. 2B, BF175 had no effects on MED25-VBD binding to VP16-TAD. In agreement with the protein-protein interaction data, whereas BF175 strongly inhibited the activity of SREBP1a-TAD, it had no effects on the TAD activities of VP16 or Myb in HEK293 cells (Fig. 2C). These data also clearly eliminated any effects of BF175 on other common transcriptional cofactors, including CBP/p300, and other subunits of the Mediator complex. Together, multiple lines of evidence have demonstrated that BF175 inhibition of MED15-KIX and SREBP-TAD interaction is relatively specific.
BF175 Inhibits Lipogenic Gene Expression
The FAS gene is a major target of SREBP-1a and -1c and is essential for the biosynthesis of fatty acids from malonyl-CoA and NADPH. To examine the effect of BF175 on the FAS promoter activity, we performed luciferase reporter assays. As shown in Fig. 3A, BF175 significantly inhibited the human FAS promoter activity in HEK293 cells, consistent with the effect of BF175 on SREBP-1a transcription activity. We then examined the effect of BF175 on the mRNA levels of endogenous FAS gene. In HepG2 cells, BF175 treatment resulted in a dose-dependent decrease of FAS mRNA levels with an IC50 of ∼50 µmol/L as measured by quantitative RT-PCR (qRT-PCR) (Fig. 3B). Thus, BF175 inhibits FAS gene expression at the transcriptional level.
Figure 3.

A: Effects of BF175 (50 µmol/L) on the FAS promoter activity in HEK293 cells by dual-luciferase assays. B: Relative mRNA levels of the FAS gene, as detected by qRT-PCR, in HepG2 cells treated with indicated doses of BF175 for 18 h, and cyclophilin B was used as the invariant control. C: Relative mRNA levels of the indicated SREBP genes, as detected by qRT-PCR, in HepG2 cells treated with BF175 (50 µmol/L) for 18 h. D: Effects of BF175 (50 µmol/L) on the SREBP-1c promoter activity in HEK293 cells by dual-luciferase assays. MU, SRE sites mutated; WT, wild-type. All data represent the mean ± SD (n = 3). *P < 0.05; #P < 0.01 vs. control (DMSO).
Next, we examined whether BF175 treatment affects the mRNA levels of SREBP genes. As shown in Fig. 3C, BF175 treatment only caused a significant decrease of SREBP-1c mRNA levels in HepG2 cells by qRT-PCR but had no effects on the mRNA levels of either SREBP-1a or SREBP-2. Thus, BF175 specifically inhibits SREBP-1c gene expression. Along with FAS gene, SREBP-1c is often examined as a lipogenic marker, and it is known that SREBP-1a or -1c can activate the transcription of SREBP-1c, forming a feed-forward regulatory loop (18). Consistent with the qRT-PCR data, BF175 inhibited the luciferase activity driven by the wild-type SREBP-1c promoter but had no effects when the SREBP-1–binding sites in the promoter were mutated (Fig. 3D), suggesting a SREBP-dependent effect of BF175 on gene expression. This result is also consistent with the inhibitory effect of BF175 on SREBP-1a transcriptional activity. Together, our data suggest that BF175 specifically inhibits SREBP-dependent gene expression in cultured cells.
We next examined whether BF175 also inhibits other SREBP target genes and whether BF175 has similar functions in primary rat hepatocytes. As shown in Fig. 4A, treatment with 50 µmol/L BF175 also decreased the expression of stearoyl-CoA desaturase-1 (SCD1) and the LDL receptor (LDLR) (both are SREBP target genes in HepG2 cells). Interestingly, in isolated primary rat hepatocytes, 30 µmol/L BF175 displayed a robust inhibition on the mRNA levels of FAS (Fig. 4B). Consistent with the gene expression data, BF175 treatment in isolated primary rat hepatocytes also resulted in a significant reduction of FAS proteins as detected by immunoblotting (Fig. 4C). Furthermore, BF175 also inhibited the expression of other SREBP target genes, such as acetyl-CoA carboxylase (ACC1), ATP citrate lyase (ACLY), and SCD1, in primary rat hepatocytes (Fig. 4D). The more robust effects of BF175 in primary rat hepatocytes may be due to lower multidrug resistance in primary cells as compared with tumor cells, HepG2. Together, our data demonstrate that BF175 is a potent inhibitor of lipogenic gene expression in cultured hepatocytes.
Figure 4.

A: Relative mRNA levels of the indicated genes, as detected by qRT-PCR, in HepG2 cells treated with 50 µmol/L BF175 for 18 h. Effects of BF175 (30 µmol/L) on the mRNA levels of the FAS gene (B), FAS protein levels (C), and mRNA levels of the indicated genes (D) in primary rat hepatocytes that were treated for 18 h. Data represent the mean ± SD (n = 3). *P < 0.05; #P < 0.01 vs. control (DMSO).
Effects of Short-term Treatment with BF175 in Animal Models
The inhibitory effects of BF175 on SREBP-dependent lipogenic gene expression in cultured hepatocytes prompted us to determine whether BF175 inhibits feeding-induced lipid accumulation in the fat body of Drosophila, because SREBP plays a pivotal and conserved role in the process (19). As shown in Fig. 5A, short-term feeding Drosophila larvae with BF175 significantly decreased lipid levels in the fat body, as quantitatively measured after Oil Red O staining.
Figure 5.

A: Effects of BF175 on lipid accumulation in Drosophila larvae as detected by Oil Red O staining followed by isopropanol extraction. B–E: C57BL/6J mice were first fed with HFD for 4 weeks and then treated with BF175 (0.3 mg/g body weight per week) or buffer (containing 0.5% DMSO) for a total of 7 days by subcutaneously implanted osmotic pumps. All mice were continuously provided with HFD during the treatment. At the end of the treatment, mice were killed and the livers were collected. B: Effects of BF175 on the triglyceride levels in the mouse liver. C and E: Relative mRNA levels of the indicated genes as detected by qRT-PCR in the mouse liver. D: Immunoblots of hepatic SREBP-1 proteins. Three representative mice from each group are shown. Data are the mean ± SD (n = 6). *P < 0.05; #P < 0.01 vs. control (DMSO).
To determine whether BF175 also has beneficial effects in disease models of dysregulated lipid metabolism in mammals, we decided to use the well-established mouse models of DIO. Eight-week-old C56BL/6J mice were first fed with HFD for 4 weeks, and then we administrated BF175 (0.3 mg/g body weight/week) or control buffer using subcutaneously implanted osmotic pumps. Mice were treated for 1 week. All mice were continuously fed with HFD during the experiments. Similar to the data from Drosophila, BF175 treatment significantly reduced lipid accumulation in the liver, as measured by the amount of triglycerides (Fig. 5B), indicating that BF175 has inhibitory effects on hepatic lipid accumulation in mice in vivo. As shown in Fig. 5C, 1-week treatment with BF175 significantly decreased the hepatic mRNA levels of lipogenic genes, such as ACC, acetyl-CoA synthetase (ACS), ACLY, and FAS, similar to the effect of BF175 in cultured hepatocytes. Interestingly, BF175 treatment had no significant effects on the mRNA levels of L-pyruvate kinase (L-PK) (Fig. 5C), which is not regulated by SREBPs (24) but by another lipogenic transcription factor, carbohydrate regulatory element binding protein (ChREBP) (25,26). This result suggests that the inhibitory effects of BF175 on lipogenic gene expression in vivo are at least partially SREBP-1 specific. In addition, BF175 treatment resulted in a significant reduction of SREBP-1 mRNA (Fig. 5C). Consistent with this, the protein levels of SREBP-1 precursors were decreased by BF175 in mouse livers as detected by immunoblotting, whereas the nuclear forms of SREBP-1 had a trend of decrease but with some variations (Fig. 5D). Interestingly, BF175 treatment also significantly decreased the expression of genes involved in cholesterol biosynthesis and uptake, including HMGCR, HMG-CoA synthase (HMGCS), and LDLR (Fig. 5E). In contrast to the effect of BF175 in HepG2 cells, it also decreased the mRNA levels of SREBP-2 in vivo (Fig. 5E). Thus, our data suggest that the SREBP pathways are targeted by BF175 in vivo.
Effects of Chronic Treatment With BF175 in the Mouse Model of DIO
Next, we examined the long-term effects of BF175 in the mouse model of DIO. For this purpose, we mixed BF175 into HFD. After feeding 8-week-old C57BL/6J mice with HFD for 4 weeks to establish the disease model, mice were fed either with the original HFD or HFD containing 0.2% (by weight) of BF175 for an additional 8 weeks. As shown in Fig. 6A, compared with the control HFD-fed mice, oral administration of BF175 significantly inhibited HFD-induced body weight gain, indicating an antiobesity function of BF175 in this animal model. Importantly, the decreased body weight gain was not due to a decrease of food intake. In fact, BF175-treated mice consumed modestly more food (Fig. 6B), suggesting that such dose of BF175 was not toxic to mice at least during the 8-week period. Calorimetry data showed that BF175 treatment significantly increased energy expenditure (Fig. 6C) and physical activity (Fig. 6D) in HFD-fed mice.
Figure 6.

C57BL/6J mice were first fed with HFD for 4 weeks and then continuously treated with HFD in the absence (control) or presence of BF175 (0.2% in HFD) for an additional 8 weeks. A: Body weight gains during the treatment period of 8 weeks. B: Food intake in the last week of treatment. Effects of BF175 by subcutaneous administration for a week under HFD on energy expenditure (C) and physical activity (D) in mice as analyzed by calorimetry. Data represent the mean ± SD (n = 6, or n = 4 for calorimetry experiments). *P < 0.05; #P < 0.01 vs. control (HFD only).
To examine the effects of BF175 on lipogenic gene expression after 8 weeks of oral administration with HFD, we measured the hepatic mRNA levels of SREBP target genes. As shown in Fig. 7A, although some changes were not statistically significant, most of those genes, including ACC, ACS, FAS, HMGCR, HMGCS, and LDLR, were expressed at significantly lower levels in response to BF175 treatment. In addition, 8 weeks of treatment with BF175 resulted in a significant decrease in plasma levels of triglycerides (Fig. 7B), free fatty acids (Fig. 7C), and cholesterol (Fig. 7D) in addition to a decrease in hepatic levels of triglycerides (data not shown), indicating a beneficial role of BF175 on obesity-induced hyperlipidemia. Such effects of BF175 were at least in part due to its inhibitory functions on SREBP target gene expression. Moreover, 8 weeks of treatment with BF175 also caused a decrease in fasting blood glucose levels (Fig. 7E), suggesting that BF175 was also effective in controlling diet-induced hyperglycemia. Together, these results have demonstrated the beneficial effects of BF175 on fatty liver and dyslipidemia in the mouse model of obesity.
Figure 7.

C57BL/6J mice were first fed with HFD for 4 weeks and then continuously treated with HFD in the absence (control) or presence of BF175 (0.2% in HFD) for a total of 8 weeks. At the end of the treatment, mice were killed and plasma samples were collected. A: Relative mRNA levels of the indicated genes as detected by qRT-PCR in mouse livers. Plasma levels of triglycerides (TG) (B), free fatty acids (NEFA) (C), total cholesterol (D), and fasting blood glucose (E). All data represent the mean ± SD (n = 6). *P < 0.05; #P < 0.01 vs. control (HFD only).
Discussion
Dysregulation of lipid homeostasis is a risk factor for developing conditions linked to metabolic syndrome in humans, including obesity, hyperlipidemia, insulin resistance, fatty liver, and hypertension (27–29). Metabolic syndrome is associated with increased risks of several diseases, including type 2 diabetes and cardiovascular disease (27–29). As a key activator of fatty acid biosynthesis, SREBP-1c has been implicated in type 2 diabetes (28,29). Genetically, single nucleotide polymorphisms and other sequence variations of the SREBP-1 gene are linked to type 2 diabetes in humans (30–32). Moreover, SREBP-1c expression is correlated with insulin resistance in morbid obesity (33), and SREBP-1c levels are often elevated in the livers of animal models of insulin resistance, including Zucker obese fa/fa rats (34), ob/ob mice (35), insulin receptor substrate-2 knockout mice (36), and HFD-induced obese mice (37). In addition, increased expression and genetic polymorphisms of SREBP genes have been linked to cardiovascular disease, consistent with the fact that hypertriglyceridemia and hypercholesterolemia are important risk factors for cardiovascular disease (33,38,39). Thus, SREBP transcription factors are potential targets for treating metabolic diseases. Indeed, recent studies have shown that blocking the SREBP maturation from precursors by small molecules inhibited lipogenic gene expression and attenuated obesity and atherosclerosis in mouse models (40,41). In addition to the well-studied SREBP maturation process, nuclear SREBPs are also regulated by transcriptional cofactors and protein stability (16,42). Particularly, the KIX domains of CBP/p300 and the MED15 subunit of the Mediator complex provide the functionally critical interaction surfaces for the SREBP-TADs (17,43,44).
The introduction of boron atoms into small molecules is expected to enhance their binding to the target molecules, such as proteins, DNA, or RNA, through not only hydrogen bonds but also sometimes covalent bonds (45). For that reason, the boron-containing compounds are predicted to be more potent in modulating biological targets, and the use of boron atoms in pharmaceutical drug design represents a novel approach. Due to the high affinity with oxygen, boron can interact with the serine residues on the protein surface (45). We have recently developed a novel synthetic strategy to synthesize a group of novel boron-containing stilbene derivatives (16). Interestingly, some of them, including BF175, could inhibit SREBP target gene expression and lipid biosynthesis in cultured cells, whereas the analog compound BF62 displayed no effects (16). The presence of two chlorine groups in BF175 may explain the difference (Fig. 1A). Chlorine is frequently used in enzyme inhibitors and is known to interact with the benzene rings in amino acids tyrosine, tryptophan, and phenylalanine (46). Therefore, it is conceivable that BF175, but not BF62, can bind more stably to proteins that have a motif containing a serine and a nearby tyrosine, tryptophan, or phenylalanine on the surface. Interestingly, among the known functional domains of key lipogenic regulators, we identified one such motif (Y55L56S57) in the third helix of the MED15-KIX domain (17). Importantly, this region makes direct contacts with the SREBP-TADs (17), and the CBP/p300-KIX domain lacks any similar motifs. Supporting this hypothesis, we show that BF175, but not BF62, blocks the interaction between SREBP1a-TAD and MED15-KIX in a purified system (Fig. 1B and C), whereas BF175 has no effect on another interaction between VP16-TAD and MED25-VBD (Fig. 2B). Moreover, BF175 only inhibits the activity of SREBP1a-TAD, but not the TADs of VP16 and Myb (Fig. 2C), essentially eliminating the possibility that BF175 can also target the CBP/p300-KIX domain, as all these TADs can bind to the CBP/p300-KIX domain. Although we cannot exclude the possibility that BF175 may have other targets in the cell, the current data strongly suggest a specific effect of BF175 on SREBP-TAD binding to MED15-KIX. In addition, unlike the CBP/p300-KIX domain, which binds to many transcription factors, MED15-KIX does not bind to most of the transcription factors in mammalian cells, and so far it is only known to interact with SREBPs (17). Thus, it is reasonable to expect that BF175 can affect only a limited number of transcription factors, and thus our results have suggested a novel strategy to inhibit SREBP-mediated gene expression.
Consistent with the model of BF175 action and the fact that only SREBP-1c transcription can be activated by SREBP-1a/1c, this compound can repress the expression of only SREBP-1c, but not SREBP-1a or SREBP-2 in HepG2 cells, although BF175 decreases the transcripts of both SREBP-1 (presumably SREBP-1c) and SREBP-2 genes in the mouse liver in vivo. Importantly, this inhibition leads to significant beneficial effects in a DIO model by decreasing the expression of SREBP-1 target genes. Interestingly, BF175 also inhibits SREBP-2 target genes in the cholesterol pathway in vivo, likely due to the inhibition of both SREBP-2 transcriptional activity and its expression. The mechanism(s) underlying the inhibition of SREBP-2 gene expression by BF175 in vivo is currently unclear, but it is less likely from a direct effect of BF175 based on our results from HepG2 cells. In addition, although the significant inhibition of the LDL receptor gene expression supports the model of BF175 action, it may have revealed a side effect of BF175 in vivo, as lowering the LDL receptor may be deleterious to the cardiovascular system. However, previous studies by blocking the SREBP maturation process have shown that inhibiting SREBP target gene expression improves lipid profiles and has antiatherosclerosis effects in mouse models (40,41). Thus, it is likely that SREBP inhibition–caused decrease of cholesterol biosynthesis can override the effects of lowering the LDL receptor in vivo.
In addition to reducing lipid accumulation in liver and blood, long-term treatment with BF175 also displayed the antiobesity effect. This is likely because of the increase of energy expenditure. Interestingly, previous studies of SREBP inhibition by blocking SREBP maturation have shown a similar effect in other mouse models (40,41). Although the underlying mechanism(s) may not be solely through SREBP inhibition, we also cannot exclude the role of SREBPs. It would be ideal to examine the SREBP-dependent effects of BF175 in vivo using SREBP-knockout mice. Unfortunately, whole-body knockout of SREBPs is embryonic lethal (47). Nevertheless, consistent with the close association of lipid metabolism with glucose metabolism, we found that BF175 could also modestly improve glucose profiles in mice of DIO.
In summary, we have identified BF175 as a novel bioactive boron-containing compound. Multiple lines of evidence suggest that BF175 inhibits the SREBP pathways by blocking SREBP-TAD binding to MED15-KIX. BF175 has beneficial effects in treating HFD-induced obesity in mice. Our findings support the role of BF175 as a potential lead compound to develop therapeutic agents for treating human diseases caused by dysregulation of lipid homeostasis. Although it may target other biological processes, BF175 represents the first small molecule blocker of the physical interaction between SREBP-TAD and MED15-KIX. In future studies, it would be interesting to examine whether BF175 also has beneficial effects in other models of metabolic disorders, such as atherosclerosis.
Article Information
Acknowledgments. The authors thank other members of the Yang and Pessin laboratories.
Funding. B.C.D. was supported by grants from the National Institutes of Health (NIH) (AA-020630 and AI-093220). F.Y. was supported by the Diabetes Research and Training Program (P60-DK-020541), the American Diabetes Association (7-11-BS-173), and the NIH/National Institute of Diabetes and Digestive and Kidney Diseases (DK-093623). A patent partially based on this study is pending.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. X.Z., X., H.Z., and J.-Y.J. performed experiments and contributed to discussion. A.A., E.S.T.Y., and Q.W. performed experiments. J.E.P. contributed to discussion and edited the manuscript. B.C.D. synthesized the compounds used in this study and contributed to discussion. F.Y. designed the experiments, interpreted data, and wrote the manuscript. F.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am 2011;95:875–892 [DOI] [PubMed] [Google Scholar]
- 2.Kaidar-Person O, Bar-Sela G, Person B. The two major epidemics of the twenty-first century: obesity and cancer. Obes Surg 2011;21:1792–1797 [DOI] [PubMed] [Google Scholar]
- 3.Targher G, Bertolini L, Padovani R, et al. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007;30:1212–1218 [DOI] [PubMed] [Google Scholar]
- 4.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 Diabetes. Hepatology 2014;59:713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals With Nonalcoholic Fatty Liver Disease. Gastroenterology 2014;146:726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes Dev 2009;23:2578–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 2004;86:839–848 [DOI] [PubMed] [Google Scholar]
- 9.Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 2012;23:65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferré P, Foufelle F. SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Horm Res 2007;68:72–82 [DOI] [PubMed] [Google Scholar]
- 11.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 1999;96:13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasty AH, Shimano H, Yahagi N, et al. Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level. J Biol Chem 2000;275:31069–31077 [DOI] [PubMed] [Google Scholar]
- 13.Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci U S A 2003;100:3155–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A 2002;99:12753–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundqvist A, Bengoechea-Alonso MT, Ye X, et al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 2005;1:379–391 [DOI] [PubMed] [Google Scholar]
- 16.Das BC, Zhao X, Tang XY, Yang F. Design, synthesis and biological study of pinacolyl boronate-substituted stilbenes as novel lipogenic inhibitors. Bioorg Med Chem Lett 2011;21:5638–5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang F, Vought BW, Satterlee JS, et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 2006;442:700–704 [DOI] [PubMed] [Google Scholar]
- 18.Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J 2006;400:179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X, Feng D, Wang Q, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest 2012;122:2417–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 2010;11:761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiaoli, Yang F. Mediating lipid biosynthesis: implications for cardiovascular disease. Trends Cardiovasc Med 2013;23:269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F, DeBeaumont R, Zhou S, Näär AM. The activator-recruited cofactor/Mediator coactivator subunit ARC92 is a functionally important target of the VP16 transcriptional activator. Proc Natl Acad Sci U S A 2004;101:2339–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittler G, Stühler T, Santolin L, et al. A novel docking site on Mediator is critical for activation by VP16 in mammalian cells. EMBO J 2003;22:6494–6504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stoeckman AK, Towle HC. The role of SREBP-1c in nutritional regulation of lipogenic enzyme gene expression. J Biol Chem 2002;277:27029–27035 [DOI] [PubMed] [Google Scholar]
- 25.Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A 2004;101:15597–15602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A 2004;101:7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005;111:1448–1454 [DOI] [PubMed] [Google Scholar]
- 28.Vergès B. New insight into the pathophysiology of lipid abnormalities in type 2 diabetes. Diabetes Metab 2005;31:429–439 [DOI] [PubMed] [Google Scholar]
- 29.Girard J, Lafontan M. Impact of visceral adipose tissue on liver metabolism and insulin resistance. Part II: Visceral adipose tissue production and liver metabolism. Diabetes Metab 2008;34:439–445 [DOI] [PubMed] [Google Scholar]
- 30.Laudes M, Barroso I, Luan J, et al. Genetic variants in human sterol regulatory element binding protein-1c in syndromes of severe insulin resistance and type 2 diabetes. Diabetes 2004;53:842–846 [DOI] [PubMed] [Google Scholar]
- 31.Eberlé D, Clément K, Meyre D, et al. SREBF-1 gene polymorphisms are associated with obesity and type 2 diabetes in French obese and diabetic cohorts. Diabetes 2004;53:2153–2157 [DOI] [PubMed] [Google Scholar]
- 32.Felder TK, Oberkofler H, Weitgasser R, et al. The SREBF-1 locus is associated with type 2 diabetes and plasma adiponectin levels in a middle-aged Austrian population. Int J Obes (Lond) 2007;31:1099–1103 [DOI] [PubMed] [Google Scholar]
- 33.Mingrone G, Rosa G, Greco AV, et al. Intramyocitic lipid accumulation and SREBP-1c expression are related to insulin resistance and cardiovascular risk in morbid obesity. Atherosclerosis 2003;170:155–161 [DOI] [PubMed] [Google Scholar]
- 34.Kakuma T, Lee Y, Higa M, et al. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc Natl Acad Sci U S A 2000;97:8536–8541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem 1999;274:30028–30032 [DOI] [PubMed] [Google Scholar]
- 36.Tobe K, Suzuki R, Aoyama M, et al. Increased expression of the sterol regulatory element-binding protein-1 gene in insulin receptor substrate-2(-/-) mouse liver. J Biol Chem 2001;276:38337–38340 [DOI] [PubMed] [Google Scholar]
- 37.Deng QG, She H, Cheng JH, et al. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology 2005;42:905–914 [DOI] [PubMed] [Google Scholar]
- 38.Védie B, Jeunemaitre X, Mégnien JL, Atger V, Simon A, Moatti N. A new DNA polymorphism in the 5′ untranslated region of the human SREBP-1a is related to development of atherosclerosis in high cardiovascular risk population. Atherosclerosis 2001;154:589–597 [DOI] [PubMed] [Google Scholar]
- 39.Salek L, Lutucuta S, Ballantyne CM, Gotto AM, Jr, Marian AJ. Effects of SREBF-1a and SCAP polymorphisms on plasma levels of lipids, severity, progression and regression of coronary atherosclerosis and response to therapy with fluvastatin. J Mol Med (Berl) 2002;80:737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang JJ, Li JG, Qi W, et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab 2011;13:44–56 [DOI] [PubMed] [Google Scholar]
- 41.Kamisuki S, Mao Q, Abu-Elheiga L, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol 2009;16:882–892 [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Xiaoli, Zhao X, Yang F. The Mediator Complex and Lipid Metabolism. J Biochem Pharmacol Res 2013;1:51–55 [PMC free article] [PubMed] [Google Scholar]
- 43.Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 1997;91:741–752 [DOI] [PubMed] [Google Scholar]
- 44.Giandomenico V, Simonsson M, Grönroos E, Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol 2003;23:2587–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das BC, Thapa P, Karki R, et al. Boron chemicals in diagnosis and therapeutics. Future Med Chem 2013;5:653–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matter H, Nazaré M, Güssregen S, et al. Evidence for C-Cl/C-Br...pi interactions as an important contribution to protein-ligand binding affinity. Angew Chem Int Ed Engl 2009;48:2911–2916 [DOI] [PubMed] [Google Scholar]
- 47.Shimano H, Shimomura I, Hammer RE, et al. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Invest 1997;100:2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]