

Abstract
Traumatic brain injury (TBI) can be associated with memory impairment, cognitive deficits, or seizures, all of which can reflect altered hippocampal function. Whereas previous studies have focused on the involvement of neuronal loss in post-traumatic hippocampus, there has been relatively little understanding of changes in ionic homeostasis, failure of which can result in neuronal hyperexcitability and abnormal synchronization. Because glia play a crucial role in the homeostasis of the brain microenvironment, we investigated the effects of TBI on rat hippocampal glia. Using a fluid percussion injury (FPI) model and patch-clamp recordings from hippocampal slices, we have found impaired glial physiology 2 d after FPI. Electrophysiologically, we observed reduction in transient outward and inward K+ currents. To assess the functional consequences of these glial changes, field potentials and extracellular K+ activity were recorded in area CA3 during antidromic stimulation. An abnormal extracellular K+accumulation was observed in the post-traumatic hippocampal slices, accompanied by the appearance of CA3 afterdischarges. After pharmacological blockade of excitatory synapses and of K+ inward currents, uninjured slices showed the same altered K+ accumulation in the absence of abnormal neuronal activity. We suggest that TBI causes loss of K+ conductance in hippocampal glia that results in the failure of glial K+ homeostasis, which in turn promotes abnormal neuronal function. These findings provide a new potential mechanistic link between traumatic brain injury and subsequent development of disorders such as memory loss, cognitive decline, seizures, and epilepsy.
Keywords: glial neuronal interactions, ion homeostasis, patch clamp, potassium selective microelectrodes, epilepsy, traumatic brain injury
Altered excitability of hippocampal neurons can be responsible for memory impairment, cognitive deficits, and seizures that are common outcomes of traumatic brain injury (TBI). These neurological outcomes have been well documented, but their pathogenic processes still remain largely unknown (Annegers et al., 1980; Temkin et al., 1990; Lowenstein et al., 1992; Wheal et al., 1998). Indeed, they can appear in patients who suffered either severe or only mild forms of TBI (Jennett, 1973; Rimel et al., 1981; Kraus, 1987; Temkin et al., 1996). Thus, the interplay of different pathogenic mechanisms may be involved in post-traumatic altered neuronal excitability, and consequently in memory and cognitive impairment, seizures, and their progression toward epilepsy.
The majority of the experimental research effort has been thus far focused on neuronal injury in animal models of TBI. Using lateral fluid percussion injury (FPI), a clinically relevant model of TBI, Lowenstein et al. (1992) first described the bilateral loss of hilar neurons and its association with dentate gyrus hyperexcitability. This lesion was shown by Smith et al. (1991) to be associated with spatial memory impairment. Lyeth et al. (1990) showed that midline FPI also causes spatial learning deficits in rats. Survival of specific neuronal populations and changes in synaptic circuitry or efficacy may all affect the neuronal excitability and synchronization (Schwartzkroin, 1993). Because, ultimately, neuronal firing is controlled by ionic gradients, the regulation of the extracellular environment and ionic gradients is fundamental for the maintenance of normal neuronal excitability and function, regardless of noxious effects on neurons. Previous work has demonstrated the sensitivity of in situneurons to elevated extracellular K+ and has lead to the acceptance of the “high-K+ ” model of epileptiform activity (Meltzer, 1899; Feldberg and Sherwood, 1957; Zuckermann and Glaser,1968; Traynelis and Dingledine, 1988; Jensen et al., 1994). Because potassium homeostasis is regulated by glia, neuronal excitability may, under certain circumstances, depend on non-neuronal mechanisms.
Glial membrane ion channels participate in the control of ionic homeostasis (Orkand et al., 1966; Newman, 1984; Ballanyi et al., 1987) and, under the condition of pharmacologically impaired K+ influx into glia, abnormal accumulation of K+ in the extracellular space and increase in neuronal excitability occur (Ballanyi et al., 1987; Janigro et al., 1997; D’Ambrosio et al., 1998b). Given this important role of glia, we were interested in exploring possible post-traumatic changes in glial membrane properties and their impact on neuronal excitability. In the present study, we investigated (1) whether TBI causes electrophysiological changes of hippocampal glia, with a special interest in alterations of potassium conductance that has been observed in other models of reactive glia (MacFarlane and Sontheimer, 1997;Bordey and Sontheimer, 1998), and (2) whether changes of glial membrane properties after TBI have functional consequences on extracellular K+ homeostasis and neuronal excitability. We compared the electrophysiological properties of uninjured and post-traumatic hippocampal glia. After FPI, glia demonstrated profound changes in their membrane K+ currents, particularly a loss of inward K+ current and IA. These changes were accompanied by impairment of extracellular K+homeostasis and an associated neuronal hyperexcitability.
MATERIALS AND METHODS
Fluid percussion injury. Male Sprague Dawley rats (postnatal days 26–31) were anesthetized with 4% halothane, intubated, and mechanically ventilated on 1.5% halothane and 30% O2. Core temperature was maintained at 37°C with a heating pad. The scalp was reflected to expose the skull, and a 4 mm midline burr hole was drilled 2 mm posterior to bregma, with care taken to not penetrate the dura. A female Luer-Lok cannula was then secured to the skull using a small anchoring screw (inserted into the skull adjacent to the cannula) and methyl-methacrylate cement. The animal was connected to the FPI device via the Luer-Lok, and a single 3–4 atm pressure pulse was delivered to the closed cranial cavity. The pressure pulse was measured by a transducer (Entran) located near the male–female connection. Animals receiving a sham injury were anesthetized and monitored in an identical manner, but the single pressure pulse was generated with the three-way stopcock closed so that no fluid entered the injury cannula. Animals were weaned from the ventilator and extubated. During the period of recovery from anesthesia and injury, neurological function was assessed by determining the presence or absence of reflexes, including paw-pinch withdrawal, corneal, pinna, respiratory drive, and righting reflex. Appropriate preinjury and postinjury management was maintained to insure that all guidelines established by the University of Washington Animal Care Committee were met.
GFAP immunoreactivity. At 2 d post-surgery, animals (naïve, sham, and FPI) were anesthetized (pentobarbital overdose) and perfused transcardially with 100 ml of 0.9% saline, followed by 200 ml of 4% paraformaldehyde (in 0.1 mphosphate buffer, pH 7.35) over a 30 min period. Brains were removed and post-fixed in the paraformaldehyde solution overnight, then placed in a 30% sucrose in 0.1 m phosphate buffer solution and allowed to sink (24–36 hr for cryoprotection). Tissue sectioning (35 μm) was performed on a freezing microtome. For GFAP immunocytochemistry, free-floating sections were incubated in a solution of 3% normal goat serum (NGS), 0.1% Triton X-100 (TX), and 1.0% bovine serum albumin (BSA) in 0.1 m Tris-buffered saline (TBS; pH 7.4) for 1 hr to block nonspecific staining. Sections were then transferred to the primary antisera containing 1:800 dilution of antibody against GFAP (Dako, Carpinteria, CA) in 0.1 mTBS with 3% NGS, 0.1% TX, and 1.0% BSA. After incubation for 24 hr, the tissue was rinsed in 0.1 m TBS, pH 7.4, five times for 10 min. Secondary antibody treatment included a 1.5 hr incubation in a 1:300 solution of biotinylated goat anti-rabbit IgG in 0.1m TBS with 3% NGS, 0.1% TX, and 1% BSA. A rinse cycle followed, then a 1 hr incubation in a 1:500 Elite avidin–biotin horseradish peroxidase complex (ABC; Vector Laboratories, Burlingame, CA) in 3% NGS, 0.1% TX, and 1% BSA in 0.1 m TBS. After three rinses in 0.1 m TBS, and three rinses in 0.1m TB, pH 7.6, the sections were immersed in 7.6 mm nickel ammonium sulfate and 0.05% 3,3-diaminobenzenidine (DAB) in 0.1 m TB solution, pH 7.6, for 5 min, followed by 15 min in 0.05% DAB containing 2%d-glucose, 0.04% ammonium-chloride, and 0.0003% glucose oxidase in TB. Four coronal sections of the hippocampus were visually examined per animal, and only the CA3 strata radiatum and pyramidale were considered.
We previously observed no difference either in GFAP reactivity or in neuronal excitability between CA1 subregion of naïve and sham-operated rats (D’Ambrosio et al., 1998a). No difference in the morphology or GFAP content of astrocytes was also observed in CA3 of sham-operated and naïve, uninjured animals. Thus, changes in glial electrophysiological properties and in extracellular K+ homeostasis are not likely to occur in sham rats. The electrophysiological experiments presented below were obtained by comparison of naïve versus post-FPI hippocampi.
Hippocampal slice preparation. Naïve or 24–48 hr post-FPI rats were anesthetized with halothane and decapitated. Brains were rapidly dissected out in ice-cold, oxygenated modified artificial CSF (aCSF) composed of (in mm): 120 NaCl, 3.1 KCl, 3 MgCl2, 1 CaCl2, 1.25 KH2PO4, 26 NaHCO3, and 10 dextrose. This low-calcium and high-magnesium solution was used to reduce cellular damage promoted by Ca2+ influx. The two hemispheres were separated by a medial sagittal cut. Each hemisphere was glued to the metal stage of a vibratome and bathed in the modified aCSF. Slices 400-μm-thick were obtained cutting perpendicularly to the longitudinal axes of the hippocampi. Slices were then gently transferred with a pipette to a holding chamber containing aCSF composed of (in mm): 120 NaCl, 3.1 KCl, 1 MgCl2, 2 CaCl2, 1.25 KH2PO4, 26 NaHCO3, and 10 dextrose. Slices were allowed to recover at room temperature (24–26°C) for at least 1 hr before they were transferred to the recording chamber. Saline solutions were equilibrated with 95% O2 and 5% CO2 to a final pH of 7.4.
Patch-clamp recordings. Slices were gently transferred to a submersion recording chamber in which they were constantly perfused with oxygenated aCSF at a rate of 1–2 ml/min. Complete solution exchange was achieved within 3 min. All the electrophysiological recordings were performed at room temperature (24–26°C); temperature fluctuated <1°C during each experiment. Glial cells were selected for recordings under visual control with a Nikon microscope equipped with Hoffman optics at 400× magnification. Patch-clamp recordings were obtained using an Axopatch 1-C (Axon Instruments, Foster City, CA) in current- or voltage-clamp mode. Whole-cell pipettes for ruptured patch were filled with (in mm): 140 K-gluconate, 1 MgCl2, 2 Na2-ATP, 0.3 NaGTP, 10 HEPES, and 0.5 EGTA, and adjusted to a final pH of 7.2 (with NaOH). The K+ reversal potential calculated for our pipette and aCSF solution was −88.7 mV. Pipettes had resistance of 5–6 MΩ. Series resistance (RS) was compensated at ∼80% (lag time, 10 μsec) and monitored during the experiment. Recordings were digitized at 48 kHz, filtered at 2–5 kHz, displayed on an oscilloscope, recorded on tape, and acquired on a 486 computer with Clampex 6 (Axon Instruments). Cell membrane capacitance was measured by applying voltage steps of ±5 mV from the holding potential of −70 mV and integrating the capacitive current for the time of the transient. Cell membrane potential was corrected for the tip potential recorded on withdrawal of the micropipette from the cell. Currents reported were not leak-subtracted. The cell input resistance (RIN) was measured in voltage clamp. From the holding potential, set to be equal to the resting membrane potential (RMP) (Iholding = 0), voltage steps of ±5 mV (duration, 100 msec) were applied. The input resistance was determined from the steady-state current response. Electrophysiological experiments were analyzed with Clampfit (Axon Instruments), and data were graphed and plotted with Origin 5.0 (MicroCal, Northampton, MA). Specific current densities (picoampere per picofarad) were calculated by dividing the ionic current by the cell capacitance. Unless otherwise specified, data are expressed as mean ± SEM.
Field recordings. Field potentials were recorded with extracellular pipettes filled with normal aCSF equilibrated with 95% O2 and 5% CO2. An Axopatch 1C (Axon Instruments) was used to amplify the signals. Slice stimulation was carried out using a constant current stimulator (WPI A365; World Precision Instruments). The stimuli were delivered through a bipolar concentric tungsten electrode. The antidromic stimulation of CA3 pyramidal cells was achieved by placing the electrode in CA2 stratum radiatum to activate Schaffer collaterals. Stimulation rate was set at 0.05, 1, or 3 Hz (400 μA; pulse duration, 100 μsec). In a subset of experiments, we blocked neuronal burst discharge by bath-applying the glutamatergic ionotrophic receptor antagonist kynurenic acid (1 mm; Sigma, St. Louis, MO). To assess the effect of Cs+ (1 mm) on inward K+ currents independent from the hyperpolarization-activated mixed cation conductance (Ih), we applied the selective blocker ZD 7288 (Zeneca). The experiments with Cs+were performed by adding CsCl (1 mm) to the bathing solution.
Extracellular potassium measurements by ion-selective microelectrodes. Double-barreled borosilicate capillaries were treated with sulfuric acid dissolved in 30% H2O, washed, and treated with increasing concentrations of acetone to displace water and improve drying. Pipettes were dried at 100°C, and were then pulled by a PB-7 vertical puller (Narishige, Tokyo, Japan). Microelectrodes with tip diameter of ∼2 μm were obtained. The ion-sensitive barrel was treated with trimethylchlorosylane, and its tip was back-filled with the potassium-selective solution (cocktail “B”; Fluka, Buchs, Switzerland). The rest of the potassium-selective barrel was filled with 140 mm KCl. The reference barrel was filled with aCSF. A WPI high-impedance dual-differential electrometer (WPI FD223) was used for potassium activity recordings. Signals were amplified with a voltage amplifier (AI2040; Axon Instruments), digitized, and stored on computer. The field potential was subtracted analogically from the potential recorded from the ion-selective barrel to dissect the contribution attributable to changes in K+activity. A set of microelectrodes was prepared the day before the experiments. Electrodes were calibrated before and after the experiments to verify their stability over time. We routinely performed tests for the selectivity of the electrode when the potassium channel blocker cesium was used during K+-activity recordings. A complete description of the methods used to compensate for the interfering ion can be found in a variety of references (Nicolsky, 1937; Eisenman, 1967; Ammann, 1986; Janigro et al., 1997). The relationship between the electromotive force read by the electrometer and the corresponding [K+] was obtained by fitting the Nicolsky–Eisenman equation to the experimental calibration points. We chose Fluka cocktail “B”, a valinomicin-based fluid exchanger, for its selectivity to potassium in the presence of the interfering ion cesium. We found that 1 mm Cs+ did not significantly affect the linearity of the response of our electrodes in the range of extracellular potassium levels that we were investigating (from 3 to 7 mm). Each electrode was calibrated using aCSF in which increasing K+ was compensated by removal of isomolar Na+. Potassium concentrations of 2, 4.35, 7, 12, and 43.5 mm, with or without CsCl (1 mm), were used for calibration. Only potassium-selective microelectrodes (KSM) showing slopes of 40–60 mV for a 10-fold change in [K+] were used. If, after recording, there was a decrease in responsiveness of the KSM (to a slope of <40 mV/decade), the results were discarded. Maximal care was taken during the experiments to measure the basal [K+]out. To record reliably changes of basal [K+]out, we eliminated DC shifts of potential during KSM insertion into the slice caused by the interaction of the ion-selective exchanger with the lipophylic matter of the tissue (Ammann, 1986); to this end, the electrode tip was twice lowered into an extraneous portion of the hippocampal slice (subiculum or entorhinal cortex). Using this protocol, we observed no further DC shifts during subsequent electrode insertions into the slice, and thus interpreted the DC shifts as attributable to changes in K+ activity (Ammann, 1986; Haglund and Schwartzkroin, 1990). KSM were consistently at a depth of ∼150 μm.
RESULTS
Hippocampal CA3 astrocytes are reactive after midline fluid percussion injury
Immunocytochemical studies on the GFAP+ cell population of the rat hippocampus were performed to characterize the histological nature of astrocytic changes after midline FPI (Fig.1). We were particularly interested in post-traumatic changes of the glial population in CA3 because we previously observed that synchronous neuronal activity arises in this region under the condition of impaired glial uptake of K+ through membrane ion channels (D’Ambrosio et al., 1998b). Sham sections showed no change in the overall GFAP immunoreactivity in CA3 strata radiatum and pyramidale (Fig. 1C,D). However, profound changes were observed in post-FPI sections, where an increase in GFAP immunostaining was evident in both CA3 stratum radiatum and pyramidale (Fig.1E,F). At higher magnification, post-FPI glia appeared thickened at visual examination because of the increased GFAP+ immunoreactivity (Fig.1F).
Fig. 1.
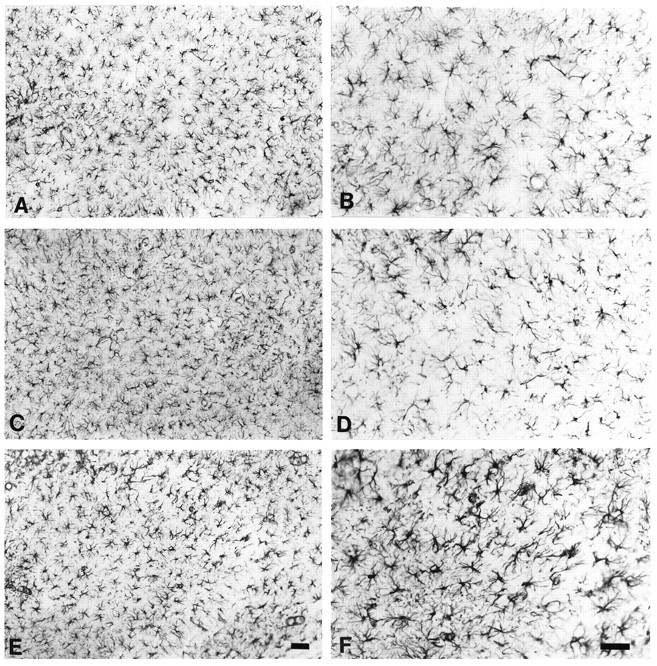
Reactive response of CA3 astrocytes after midline FPI. GFAP immunoreactivity in slices from control (A,B), sham (C, D), and injured animals (E, F). After midline FPI there is a marked increase in GFAP immunoreactivity of the single cells (100×, A, C,E; 200×, B, D,F). Scale bars, 50 μm.
Passive electrophysiological properties of post-FPI CA3 glia
The electrophysiological changes of post-traumatic glia were studied in acutely isolated hippocampal slices. Glial cells were visually identified in CA3 strata radiatum and pyramidale as cells with oval somata of <10 μm diameter. These cells never fired action potentials during seal formation (D’Ambrosio et al., 1998b). Identification of these cells as glia was confirmed by biocytin filling and subsequent morphological analysis of fixed sections (n = 16). Whole-cell patch-clamp recordings were obtained from a total of 49 glial cells from naïve or post-traumatic animals. The passive properties of control and post-FPI reactive glia are summarized in Table 1. With K+-gluconate-based intracellular solutions, RMPs spanned a wide range in both normal and post-traumatic glia (from −77 to −43 mV for control slices, and from −75 to −44 mV for post-traumatic slices). No differences were found between RMP distribution in control and post-traumatic CA3 glia: recordings obtained from control CA3 glia yielded an average RMP of −65.5 ± 2.6 mV (n = 28), whereas those obtained from post-FPI slices were −68 ± 2 mV (n = 21;p = 0.2). Mean input resistance (RIN) computed from all of the recorded control glial cells was 196 ± 21 MΩ (n= 28), whereas that for post-FPI glia was 163 ± 25 MΩ (n = 21; p = 0.3).
Table 1.
Passive electrophysiological properties of control and post-FPI CA3 glia
| Complex cells | Inwardly rectifying cells | |||||
|---|---|---|---|---|---|---|
| Control | FPI | Stat | Control | FPI | Stat | |
| RMP (mV) | −70 ± 4 (10) | −68 ± 3 (10) | p = 0.25 | −64 ± 10 (18) | −68 ± 4 (11) | p = 0.14 |
| RIN (MΩ) | 210 ± 74 (10) | 214 ± 138 (10) | p = 0.94 | 187 ± 128 (18) | 115 ± 71 (11) | p = 0.1 |
| Capacitance (pF) | 34 ± 14 (10) | 15 ± 7 (10) | p = 0.004* | 10.5 ± 5 (14) | 9.5 ± 4 (11) | p = 0.6 |
Values given for control and post-FPI glia are means ± SD, with number of recordings in parentheses. Asterisk indicates statistical significance.
Three electrophysiologically distinct types of glia are normally found in the hippocampus. These cells are recognizable by the expression of distinct whole-cell currents and by their sensitivity to extracellular Cs+ (D’Ambrosio et al., 1998b). These three types of glia (complex, inwardly rectifying, and linear cells) could also be found in CA3 of post-FPI slices. However, since the linear type was virtually absent even in control slices (n = 2), we did not consider these cells for this report. The RMP values for complex and inwardly rectifying cells did not differ from those of controls. Similarly control and post-FPI complex and inward rectifier cells yielded similar input resistance values (Table 1), and no statistically significant differences were found by comparing across control and injured groups.
No changes in membrane capacitance were observed in inwardly rectifying glia 24–48 hr after midline FPI. Conversely, a significant decrease in cell membrane capacitance was found in complex cells (Table 1). Control complex cells had a capacitance of 34 ± 5 pF (n = 10) whereas complex cells in post-FPI slices had 15 ± 7 pF (n = 10; p = 0.004). Biocytin filling and postelectrophysiological analysis of the fixed section revealed that all the recovered complex cells had oligodendrocyte-like morphology (n = 8 cells; Fig.2), whereas all the recovered inwardly rectifying cells showed astrocytic morphology (n = 4 cells). Cell-to-cell coupling among complex cells was never observed. Thus, the decrease in membrane capacitance could not be accounted for by changes in cell-to-cell coupling.
Fig. 2.

Post-FPI CA3 complex cells have oligodendrocyte-like morphology. Photomicrograph (A) and camera lucida drawings (B–D; C, same cell as inA) of biocytin-filled complex cells located in CA3 stratum radiatum. All the complex cells recovered for morphological analysis showed oligodendrocyte-like morphology (n= 8). These cells are characterized by fine long processes that show periodic swellings and that radiate from small somata. Scale bar, 50 μm.
Complex and inward rectifier glia lose inward potassium current after fluid percussion injury
Complex and inward rectifier glial cells are endowed with inward K+ currents that may be involved in buffering of extracellular K+ (Newman, 1984, 1995; Janigro et al., 1997; McKhann et al., 1997; D’Ambrosio et al., 1998b). Inward K+ current expressed in glia are sensitive to blockade by extracellular Cs+ (Ransom and Sontheimer, 1995; Janigro et al., 1997; D’Ambrosio et al., 1998b). We used bath application of this blocker during whole-cell glial recordings to measure the inward K+ current in control and FPI tissue. For both complex and inward rectifier glial cells, we found a significant loss of inwardly rectifying K+ currents after FPI. To quantify the expression of inward rectifier potassium currents in control and post-FPI slices, we used the same voltage command protocol previously employed to characterize these cell types (D’Ambrosio et al., 1998b). Voltage commands consisted of 750 msec duration ramps, from −170 to +100 mV (from a holding potential of −70 mV). Under these voltage command conditions, complex and inwardly rectifying glial cells normally showed Cs+-sensitive inwardly rectifying K+ currents. Bath application of Cs+ (1 mm) yielded a different degree of blockade depending on the cell type and the experimental condition (control vs FPI). For each cell, we computed the percentage of Cs+-sensitive current (ICs) present at any given membrane potential with respect to the total whole-cell current at that potential (Imax). This ratio is an indicator not only of the cell type endowed with inward potassium current (i.e., complex vs inward rectifier cells) but also of the effect of FPI. Figure 3 shows voltage-clamp recording and the pharmacological isolation of the inward current in complex cells from control and post-FPI hippocampal slices. Complex cells in normal hippocampal slices exhibited large inward Cs+-sensitive current components, particularly at hyperpolarized levels (ICs/Imax= 81 ± 11% at −140 mV; n = 7). In contrast, complex cells in post-FPI hippocampal slices displayed little Cs+ sensitivity i.e., exhibited only a small Cs+-sensitive component of the whole-cell inward currents (ICs/Imax= 28 ± 8% at −140 mV; n = 7). The inward current components that were Cs+-insensitive were small in control complex cells [(Imax−ICs)/Imax= 19 ± 11% at −140 mV; n = 7). Conversely, post-FPI complex cells displayed large Cs+-insensitive current component [(Imax −ICs)/Imax= 72 ± 8% at −140 mV; n = 7). We performed the same type of analysis from a total of 23 inward rectifier glial cells (Fig. 4). Inward rectifier glia in normal hippocampal slices exhibited proportionally more Cs+-sensitive inward current than those found in post-FPI hippocampal slices. Bath application of Cs+ (1 mm) revealed that inward rectifier cells in normal hippocampus were characterized by anICs/Imaxof 16 ± 1% (at −140 mV; n = 13). In contrast, inward rectifier cells from post-FPI hippocampus exhibited anICs/Imaxof 8 ± 2% (at −140 mV; n = 10). The inward current component that was Cs+-insensitive in control inward rectifier cells was 84 ± 1% (at −140 mV;n = 13) and 92 ± 2% in post-FPI inward rectifier cells (at −140 mV; n = 10).
Fig. 3.
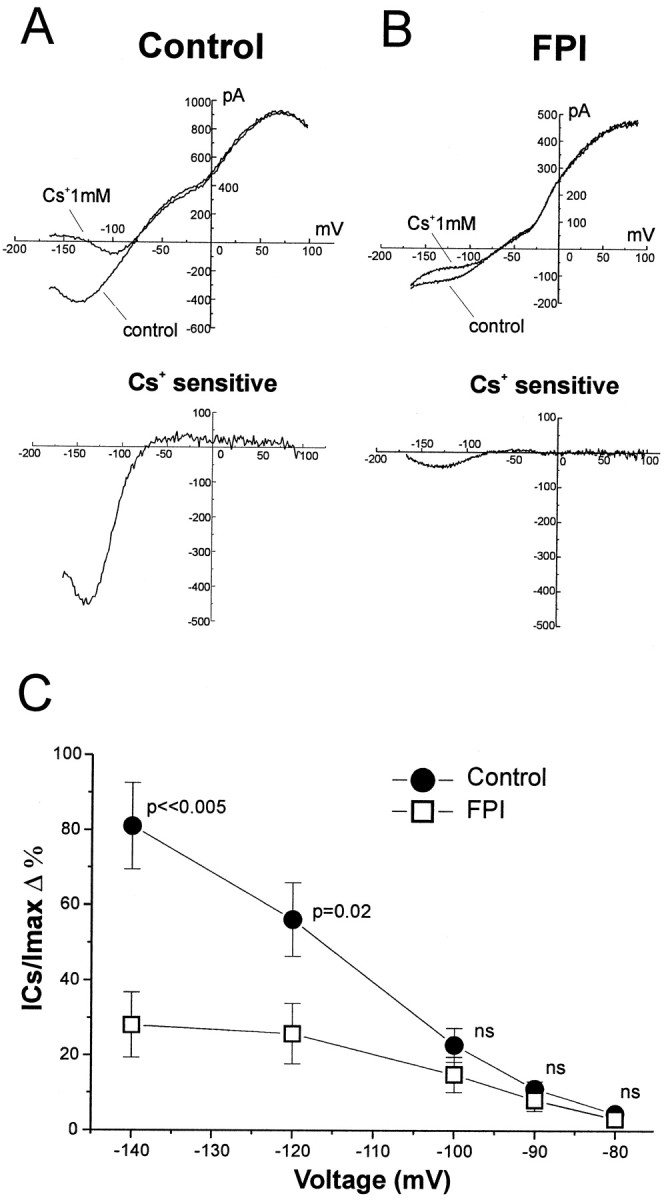
Complex glial cells lose inward K+ currents after midline FPI. Complex cells in control hippocampal slices exhibited large Cs+-sensitive currents (A,top and bottom panels) and were characterized by a large Cs+-sensitive component (81 ± 11% at −140 mV; n = 7). In contrast, complex cells in post-FPI hippocampal slices displayed little Cs+-sensitivity (B,top and bottom panels) and showed a decreased Cs+-sensitive component of the whole-cell inward currents (28 ± 8% at −140 mV; n = 7). C, The percentage of Cs+-sensitive currents (ICs) for complex cells in normal and post-FPI hippocampus is shown for membrane potentials from −140 to −80 mV. Voltage commands consisted of ramps from −170 to +100 mV over 750 msec from holding potential of −70 mV.
Fig. 4.
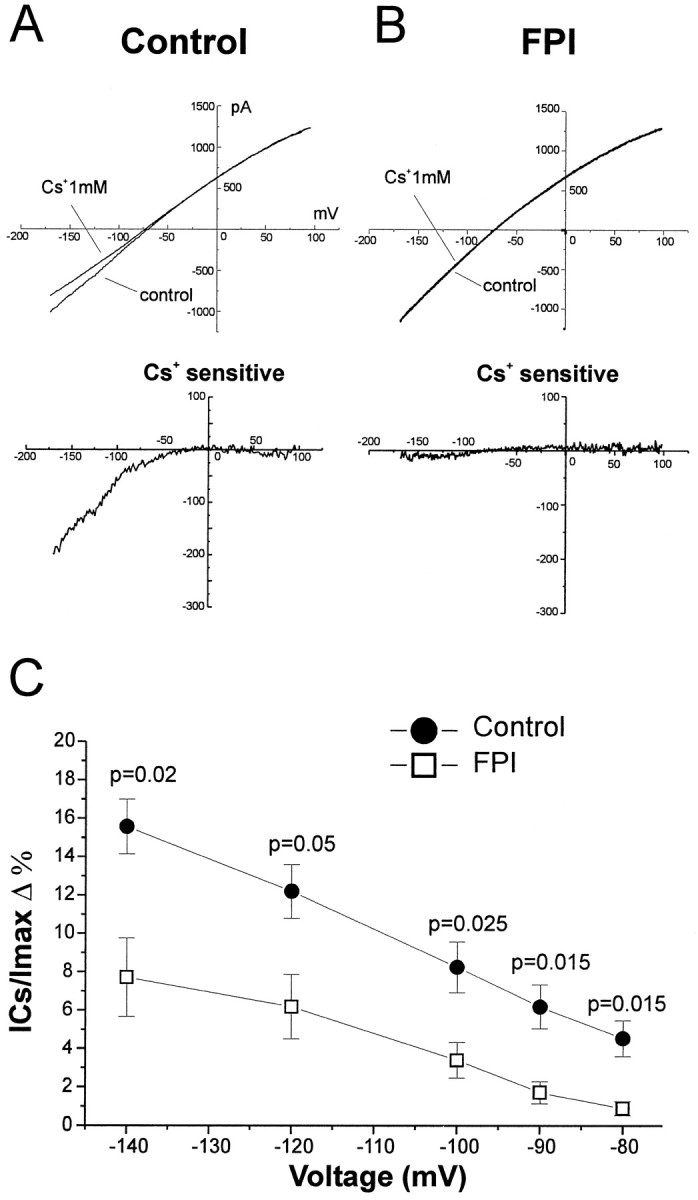
Inward rectifier glial cells lose inward K+ currents after midline FPI. Inward rectifier glia in normal hippocampal slices exhibited Cs+-sensitive currents (A, top and bottom panels) that were higher than those found in inward rectifier cells in post-FPI hippocampal slices (B,top and bottom panels). C,The percentage of Cs+-sensitive currents for inward rectifier cells in normal and post-FPI hippocampus is shown for membrane potentials from −140 to −80 mV. Bath application of Cs+ 1 mm revealed that inward rectifier cells in normal hippocampus were characterized by 16 ± 1% Cs+-sensitive current (at −140 mV;n = 13). In contrast, inward rectifier cells in post-FPI hippocampus showed decreased Cs+-sensitivity of the inward currents to 8 ± 2% (at −140 mV; n = 10). Voltage commands consisted of ramps from −170 to +100 mV over 750 msec from holding potential of −70 mV.
The discrepancy between loss of Cs+-sensitive K+ currents and no change inRIN may be accounted for by inward rectifier channels being located in fine distal processes of the glial membrane and thus undetectable by small voltage commands used to testRIN around cell RMP. We thus measured the absolute inward current evoked by the ramp command at −140 mV, a voltage command sufficient to cause potential changes even in distal processes. For this measurement we used only cells that appeared to be isolated, as determined by their membrane capacitance and by biocytin filling (D’Ambrosio et al., 1998b). Complex cells in normal hippocampal slices exhibited a mean inward current of −350 ± 33 pA (at −140 mV; n = 10). In contrast, complex cells in post-FPI slices exhibited significantly less inward current (−210 ± 43 pA at −140 mV; n = 10; p = 0.02). We performed the same type of analysis from isolated inward rectifier glial cells. In normal hippocampal slices, isolated inward rectifier cells had an inward current of −1130 ± 160 pA (at −140 mV; n = 12). In contrast, isolated inward rectifier cells in post-FPI slices had an inward current of only −620 ± 110 pA (at −140 mV; n = 10;p = 0.02). Thus, an increase in membrane resistance induced by FPI was detectable at hyperpolarized potentials.
Complex cells lose transient outward K+ current after fluid percussion injury
CA3 complex cells are endowed with transient outward potassium currents evoked by membrane depolarization (D’Ambrosio et al., 1998b). Owing to its activation properties, this current transiently activates at membrane potentials positive to −40 mV (Bordey and Sontheimer, 1997). Thus, this current may have a role in coupling glial cell membrane potential to pronounced changes in extracellular K+. We compared the density of this current in complex cells in control and post-FPI CA3.
Whole-cell recordings performed from complex cells revealed that the transient outward current is dramatically reduced after FPI (Fig.5). Complex cells were voltage-clamped at a holding potential of −70 mV. A conditioning voltage step to −80 mV (80 msec) was applied to deinactivate the transient current. When depolarizing voltage steps were applied (in 10 mV increments), a transient outward current that resembled the “A-type” current (IA) was activated in glia from normal slices (Fig. 5A). When the conditioning step was set to −40 mV, the transient outward current was inactivated completely, and only a sustained current was observed (Fig. 5B). Separation of the inactivating current was obtained as difference of whole-cell currents evoked by the deinactivating and inactivating protocols (Fig.5C). Complex cells in CA3 of control rats showed high density of transient outward current (30 ± 10 pA/pF at 100 mV; Fig. 5G, filled circles). In contrast, post-FPI complex cells showed a dramatic loss of the transient outward current (2 ± 1 pA/pF at 100 mV; p < 0.02; Fig.5G, open circles). No transient outward current was detectable with the activation protocol (Fig. 5D) or as the difference of the whole-cell currents evoked in presence and in absence of the deactivating conditioning step (Fig. 5C).
Fig. 5.
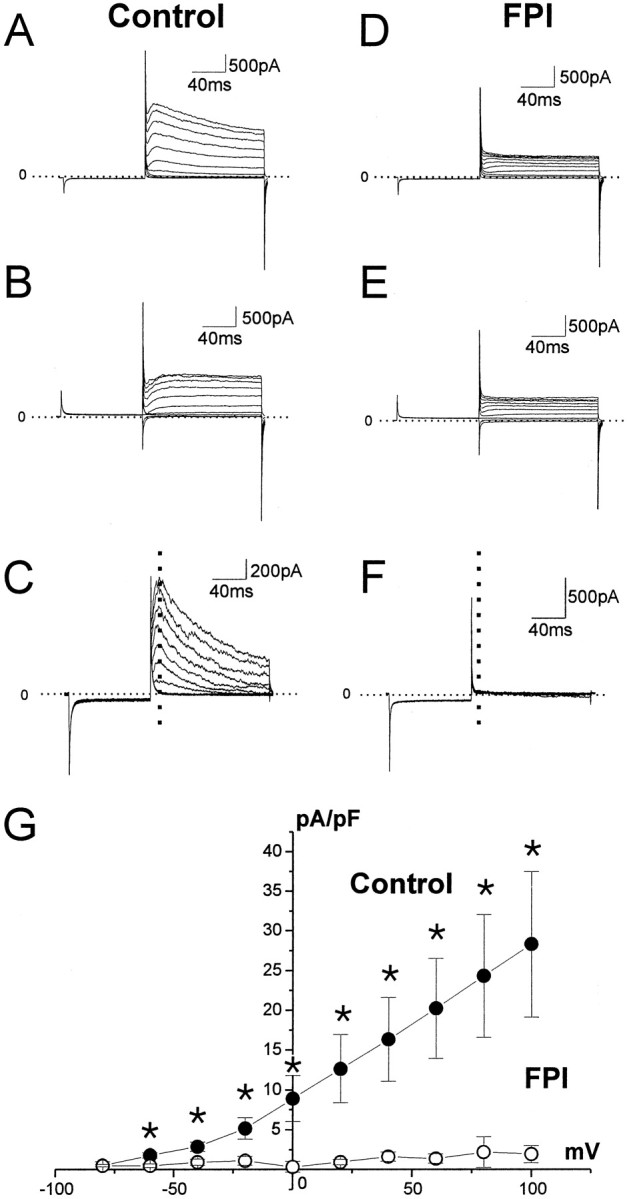
Complex glial cells lose K+transient outward currents after midline FPI. Complex cells are endowed with depolarization-induced transient outward currents that can be induced by voltage commands consisting of a conditioning step to −80 mV (holding potential, −70 mV) followed by depolarization in 10 mV increments (A). When the conditioning step was to −40 mV, the transient outward current was inactivated, and only a delayed rectifier current was observed (B).C, Current computed as difference between protocolA and B. The dashed linerepresents the time where the current is measured and plotted inG. Post-FPI complex cells showed a dramatic loss of the transient outward current (D, E), and no current is detectable as difference (F). G,Relationship between the peak evoked current density (dashed line) in normal and post-FPI complex cells at membrane potentials from −80 to +100 mV. The extrapolated reversal potential was −89.7 mV, which is near the predicted reversal potential for K+ currents (−88.7 mV). Asterisksrepresent statistical significance at p < 0.02.
Basal extracellular K+ is elevated in post-FPI slices
The data thus far show that expression of inward and transient outward K+ currents are decreased at early time points after FPI. To assess potential functional effects of decreasing glial K+ currents after trauma, we focused on activity in the CA3 subfield. This region of the hippocampus was found to be predominantly endowed with complex and inward rectifier cells (D’Ambrosio et al., 1998b). We first measured the K+-activity in CA3 strata radiatum and pyramidale (Fig. 6). The KSM was positioned just above the tissue, and the electrometer was set to a reference potential reflecting the level of K+ in the bath. The electrode was then gently inserted into the tissue at a depth of ∼150 μm, and after 1 min, it was repositioned into the bathing solution above the slice. For every slice, the subregions CA3a, CA3b, and CA3c were tested. In control slices, little or no changes in the reading of the electrometer were observed (Fig. 6A). When the same procedure was performed in post-FPI slices, positive DC shifts of the potential were detected, suggesting increased basal K+activity (Fig. 6B). The DC shifts in the potential recorded were −0.08 ± 0.06 mV in control (n = 12), and 2.06 ± 0.2 mV in post-FPI slices (n = 9;p < 0.01; Fig. 6C). The corresponding basal [K+]out was 4.34 ± 0.01 mm in control slices, and 4.78 ± 0.05 mm in post-FPI slices (Fig.6D). No differences in basal [K+]out were found within CA3 subregions in the same slice.
Fig. 6.

CA3 basal [K+]outis elevated after midline FPI. K+ activity recording in CA3 strata radiatum and pyramidale. The KSM was positioned just above the tissue, and the potential read was zeroed. The electrode was then gently inserted at a depth of ∼150 μm. A, In control slices, we observed either little or no DC shifts of the potential corresponding to little or no changes of potassium activity in the extracellular space of the slices. Top andbottom panels correspond to two experiments from two different slices. B, In post-FPI slices, the insertion of the electrode always elicited positive DC shifts, corresponding to increase of K+ activity. Top andbottom panels correspond to two experiments from two different post-FPI slices. C, The DC shifts in potential recorded were −0.08 ± 0.06 mV in control (n= 12) and 2.06 ± 0.2 mV in post-FPI slices (n= 9; p < 0.01). D, The corresponding basal [K+]out was 4.34 ± 0.01 mm in control slices and 4.78 ± 0.05 mm in post-FPI slices. Black andwhite bars represent duration of electrode insertion and extraction, respectively.
Accumulation of extracellular K+ during neuronal activity is abnormal in post-FPI slices
To examine the dynamics of extracellular K+ accumulation in control and post-FPI slices, field potentials and extracellular K+ activity were recorded during neuronal stimulation. Schaffer collaterals (SC) were stimulated for 4 min at 1 Hz to antidromically activate CA3 pyramidal cells. A low impedance field electrode and a KSM were placed in CA3 stratum radiatum to record activity in response of stimulation (Fig.7).
Fig. 7.

Neuronal stimulation induces abnormal accumulation of extracellular K+ and burst discharge in post-FPI slices. Field electrode and KSM were placed in CA3 stratum radiatum. Stimulating electrode was placed in CA2 stratum radiatum. K+ activity recordings were performed during 0.05 and 1 Hz antidromic stimulation. A, Control slices (filled circles) had a basal [K+]out similar to that of bathing aCSF. Antidromic stimulation at 1 Hz for 4 min induced a transient elevation of [K+]out to ∼5 mm and its recovery toward baseline values within the fourth minute. During the following 0.05 Hz, [K+]out transiently decreased to ∼4 mm and then recovered. Post-FPI slices (empty circles) had elevated basal [K+]out during stimulation at 0.05 Hz. When the high-frequency stimulation was performed [K+]out transiently increased to 5.4 mm and then decreased to 5 ± 0.05 mmwithout reaching the baseline value (asterisk;p < 0.001). During the following 0.05 Hz, [K+]out transiently decreased to ∼4.7 mm. B, Post-FPI CA3 develops frequency-dependent afterdischarges for antidromic stimulation. In control, only a small fraction of slices developed afterdischarges during antidromic 1 Hz stimulation (28%; 2 of 7 slices). Post-FPI slices showed a higher excitability. They did not show afterdischarges during 0.05 Hz stimulation, but afterdischarges appeared during 1 Hz stimulation (80%; 8 of 10 slices). Two traces obtained at the end of a minute of 1 Hz-stimulation are showed overlapped.
Control slices (Fig. 7, filled circles) had a basal [K+]out of 4.34 ± 0.01 mm, similar to that of bathing aCSF. Antidromic stimulation (1 Hz for 4 min) induced a transient elevation of [K+]out to ∼5 mm; recovered to baseline values within the fourth minute of stimulation (n = 4). During the following stimulation at 0.05 Hz, [K+]outtransiently decreased to ∼4 mm, and then recovered to baseline. Post-FPI slices (open circles) showed an elevated basal [K+]out during the initial stimulation at 0.05 Hz (4.78 ± 0.08 mm;n = 4). When the 1 Hz stimulation protocol was applied, [K+]outtransiently increased to ∼5.4 mm and then declined to 5 mm (5 ± 0.05 mm) without reaching its original pre-1 Hz baseline value (p < 0.001). During the subsequent stimulation at 0.05 Hz, [K+]outtransiently decreased to ∼4.7 mm and then recovered to the original baseline.
These results suggest that the mechanisms responsible for extracellular K+ homeostasis are impaired after traumatic injury. Consistent with that view, we observed that a higher percentage of post-FPI slices developed burst discharges during the 1 Hz stimulation protocol (Fig. 7B). Only a small fraction of control slices exhibited hyperexcitable discharges during antidromic 1 Hz stimulation (28%; two of seven slices), but 80% of post-FPI slices (8 of 10 slices) showed such activity.
The abnormal extracellular K+homeostasis is caused by post-traumatic glial impairment
The enhanced accumulation of extracellular K+ during 1 Hz stimulation in post-FPI slices could have been caused either by exaggerated release of K+ from abnormally hyperactive post-traumatic neurons, to a decreased uptake of K+ by glia, or to a cooperation of both mechanisms. To resolve this issue, we carried out the same stimulation protocol but in the presence of the ionotropic glutamatergic receptor antagonist kynurenic acid to block excitatory synaptic drive in CA3 pyramidal cells (Fig. 8). Blockade of glutamatergic synapses with kynurenic acid (1 mm) blocked synaptic excitation in CA3 via axon collaterals (Fig.8A) and abolished the activation of feedback interneurons (the activity of which could also be altered after FPI). Under these conditions, none of the slices bathed in kynurenic acid (control and FPI) developed burst discharges when SC were stimulated at frequencies ranging from 0.05 to 3 Hz, for 4–15 min. We then applied Cs+ (1 mm) to control slices to block the influx of K+into glia (Janigro et al., 1997; McKhann et al., 1997; D’Ambrosio et al., 1998). Because bath application of Cs+ also blocks neuronal h-type currents (Halliwell and Adams, 1982) and modifies neuronal excitability (Maccaferri et al., 1993; Maccaferri and McBain, 1996; Janigro et al., 1997), these experiments were performed in the presence of the selective blocker of h-type currents ZD 7288 (10 μm; BoSmith et al., 1993; Gasparini et al., 1996; Gasparini and DiFrancesco, 1997). The application of this selective blocker prevented the effect of Cs+ onIh, which would confound the effect of Cs+ on glial inward currents, without substantially modifying the profile of K+accumulation in the extracellular space.
Fig. 8.

Abnormal accumulation of extracellular potassium is caused by impaired glial homeostasis. Field electrode and KSM were placed in CA3 stratum pyramidale. Stimulating electrode was placed in CA2 stratum radiatum (A,bottom drawing). Baseline values were obtained at 0.05 Hz stimulation. Bath application of kynurenic acid (1 mm) abolished recurrent spikes (A, top traces; asterisk, artifact of the stimulus). Experiments were performed with bath application of kynurenic acid (1 mm) and ZD 7288 (10 μm), and only slices that had stable population spike and [K+]out were used. B,In control slices, 3 Hz antidromic stimulation induced the rise of [K+]out that peaked at 4.9 ± 0.1 mm. At the end of the 5 min period [K+]out was 4.5 ± 0.05 mm. In the following 5 min of stimulation at 0.05 Hz, [K+]out reached the value of at 3.9 ± 0.1 mm (n = 3).C, Cs+ (1 mm) added to the control bath solution increased baseline [K+]out to 4.9 mm. Three hertz antidromic stimulation induced the rise of [K+]out to 5.2 ± 0.1 mm. At the end of the 5 min period [K+]out was still 5.2 ± 0.1 mm. In the following 5 min of stimulation at 0.05 Hz, [K+]out reached the minimum value of 4.7 ± 0.05 mm (n = 3).D, In post-FPI slices, 3 Hz antidromic stimulation induced the rise of [K+]out that peaked at 5.2 ± 0.1 mm. At the end of the 5 min period [K+]out was 5.1 ± 0.1 mm. In the following 5 min of stimulation at 0.05 Hz, [K+]out reached the value of at 4.7 ± 0.05 mm (n = 3).
For this set of experiments, field recordings were paired to concomitant extracellular K+ activity recordings in CA3 stratum pyramidale. Baseline values of [K+]out and of the population spike amplitude were recorded for 5 min during SC stimulation at 0.05 Hz. In slices that had stable population spike and extracellular K+ activity for at least 5 min, we then antidromically activated CA3 with SC stimulation at 3 Hz for 5 min. In control slices, this protocol induced a transient increase of [K+]out that peaked at 4.9 ± 0.2 mm and recovered to 4.5 ± 0.1 mm (n = 3) at the end of the 5 min period. In the ensuing 5 min of stimulation at 0.05 Hz, there was an undershoot of [K+]out of 0.45 ± 0.05 mm, reaching the value of 3.9 ± 0.1 mm (Fig. 8B). After a 30 min recovery period, 1 mmCs+ was added to the bathing solution containing ZD 7288 and kynurenic acid. Ten minutes of Cs+ perfusion established a new baseline of [K+]out at 4.9 ± 0.1 mm. Five minutes of 3 Hz antidromic activation under these conditions induced a persistent increase of [K+]out (5.2 ± 0.1 mm). At the end of the 5 min period, [K+]out was consistently elevated (5.2 ± 0.1 mm;n = 3). In the following 5 min of stimulation at 0.05 Hz, an undershoot of [K+]out of 0.2 ± 0.05 mm (significantly smaller than that observed in absence of Cs+;p < 0.01) occurred to a value of 4.7 ± 0.05 mm (Fig. 8C).
These same experiments were performed in slices obtained from post-FPI animals. As in control slices, bath application of ZD 7288 and kynurenic acid prevented the appearance of afterdischarges. Five minutes of 3 Hz CA3 antidromic stimulation induced an increase of [K+]out that peaked at 5.2 ± 0.1 mm and did not recover toward baseline values (similar to control slices bathed in 1 mmCs+; Fig. 7). At the end of the 5 min period, [K+]outwas 5.1 ± 0.1 mm (n = 3). The following 5 min of stimulation at 0.05 Hz we observed an undershoot of [K+]out of 0.15 ± 0.05 mm that was significantly smaller than that observed in control (p < 0.01) and comparable to that in Cs+-treated slices (p = 0.6). The undershoot reached a [K+]out of 4.7 ± 0.05 mm (Fig. 8D). Neuronal activity was monitored throughout the experiment, and no abnormal synchronous firing was observed. These results suggest that abnormal accumulation of extracellular potassium during sustained neuronal firing occurs even in the absence of enhanced released of K+ from post-traumatic hyperactive neurons.
DISCUSSION
To our knowledge this is the first report demonstrating impairment of glial potassium homeostasis in post-traumatic hippocampus. The present study takes advantage of a clinically relevant in vivo model of TBI (Dixon et al., 1987), combined with the study in acutely isolated hippocampal slices. This approach allows us to study at the single-cell level the events accompanying the pathological progression of TBI in vivo. We have asked whether post-traumatic electrophysiological changes in hippocampal glia can promote abnormal neuronal function and have focused on the CA3 subregion of the hippocampus for our analysis. FPI induces reactive gliosis in this hippocampal region, as well as significant electrophysiological changes in CA3 glia. Trauma was associated with loss of inwardly rectifying and transient outward potassium currents. Furthermore, post-FPI slices exhibited elevated baseline [K+]out and altered neuronal activity-induced extracellular K+ accumulation; the latter did not appear to be caused by enhanced K+ release from neurons. The results suggest that post-FPI glia in CA3 have impaired uptake of potassium. These findings parallel numerous reports describing post-traumatic neuronal remodeling leading to hyperecitability and present a new non-neuronal process that can contribute to post-traumatic changes in neuronal function.
Properties of post-FPI hippocampal glia
Despite the loss of inwardly rectifying K+ current in post-traumatic glia, their RMPs did not differ significantly from control values. This result is consistent with our previous finding in normal tissue, that there were no differences in RMPs among the three types of glia differently endowed with Cs+-sensitive inwardly rectifying K+ currents (D’Ambrosio et al., 1998b). At least two possibilities exist: (1) post-FPI glia express Cs+-insensitive conductance regulating RMP; or (2) the Cs+-sensitive inwardly rectifying channels do not contribute significantly to the RMP recorded at the cell soma because they are located in the fine distal processes. Although reactive glia have been associated with depolarized RMPs (MacFarlane and Sontheimer, 1997; Bordey and Sontheimer, 1998),Burnard et al. (1990) found unaltered RMPs in reactive glia in hippocampal slices from kainic acid-treated rats. Further experiments are thus required to clarify this issue.
No changes in RIN measured around RMP were observed in post-FPI reactive glia. This may depend on the distal sites of the inwardly rectifying channels that cannot be probed by the protocol used to test RIN. Indeed, because of poor space-clamp associated with recordings from cells with highly intricate morphology, voltage steps of ±5 mV applied to the glial cell soma are not likely to influence the membrane potential of fine distal processes where the channels are likely to be predominantly located (Mi et al., 1996). That changes in membrane conductance occur after lesion was confirmed when the cell soma was voltage-clamped at significantly more negative potentials.
There was a dramatic decrease in membrane capacitance of complex cells after FPI. Interestingly, all complex cells showed oligodendrocyte-like morphology at the light microscope level. Since these cells showed no cell-to-cell coupling even in normal tissue (D’Ambrosio et al., 1998b), we interpret the loss of capacitance not as a result of uncoupling, but rather as a disruption of cell processes. This disruption may have consequences on the K+homeostasis in the region of the axons ensheathed by those oligodendroglia, modifying axonal excitability and conduction. Indeed, it has been suggested that oligodendroglia and Schwann cells are capable of passive K+-uptake through ion channels in corpus callosum and spinal cord (Chvátal et al., 1997, 1998), and at peripheral nodes of Ranvier (Mi et al., 1996).
After FPI, complex and inward rectifier cells experienced a loss of Cs+-sensitive inwardly rectifying K+ currents (Figs. 3, 4). This finding is consistent with previous report by others showing loss of inward K+ currents in reactive glia from human epileptic foci (Bordey and Sontheimer, 1998) or in an in vitro model of injury-induced reactive gliosis (MacFarlane and Sontheimer, 1997). The inward rectifier K+ current provides an efficient passive mechanism for buffering extracellular K+(Newman, 1984, 1995) and contributes to K+influx into hippocampal glia in situ (D’Ambrosio et al., 1998b). In addition to loss of inward K+currents, complex cells had a dramatic loss of transient outward currents; because this current is activated by depolarizations, it could contribute to RMP maintenance after changes in extracellular K+ and thus be involved in K+ homeostasis (Chvátal et al., 1997, 1998).
The observation of post-traumatic changes of glial K+ conductances that are active at RMP have led us to conclude that post-traumatic glia have impaired passive uptake of K+ through ion channels. This impairment would occur whether K+buffering is accomplished by the spatial buffer mechanism or by passive influx of KCl through ion channels (Newman, 1995).
Abnormal extracellular K+ accumulation occurs in post-FPI hippocampus
Because glial K+ channels are involved in ion homeostasis, one would predict that post-FPI-mediated loss of K+ conductances would have functional effects similar to drug-mediated blockade of these conductances in normal slices. Because Cs+treatment blocks those channels and leads to an alteration in [K+]out regulation and neuronal excitability, we investigated these features in post-FPI slices.
Whereas in CA3, control slices exhibited baseline [K+]out equivalent to the [K+] in medium bathing the tissue, post-FPI CA3 had a baseline [K+]out that was ∼0.5 mm higher. Albeit small, this difference is significant and can affect hippocampal function. In fact, it has been shown that CA3 pyramidal cell firing is exquisitely sensitive to small changes of [K+]out(Jensen et al., 1994). Furthermore, our measure is likely to be an underestimate of the actual increase in baseline [K+]out occurringin vivo after FPI because removal of K+ is facilitated by bath flow in the hippocampal slice in vitro.
We chose to study the efficacy of [K+]out regulation during neuronal activity by antidromically activating CA3 pyramidal cells. This approach bypassed the likely involvement of the hilar neuronal network that is affected by FPI (Lowenstein et al., 1992;Grady et al., 1996). In control slices, 1 Hz stimulation induced a transient increase of [K+]out that returned to baseline toward the end of the 4-min-long stimulation protocol. However, in post-FPI slices, [K+]out did not return to baseline during this time period, exhibiting a much longer time course recovery. These post-FPI slices were also more prone to burst discharge generation than control slices during this stimulation protocol, suggesting that CA3 pyramidal cell excitability is abnormal at a time when regulation of baseline [K+]out, and of neuronal-activity-induced K+ accumulation, is impaired. In a previous study, we found that post-FPI CA1 pyramidal cells are hypoexcitable when orthodromically stimulated. We assessed this change in excitability as higher stimulating currents required to evoke the postsynaptic field EPSP and population spike response (D’Ambrosio et al., 1998a). We didn’t perform similar experiments in the present study because we were interested in the generation of burst discharges after purely antidromic population spikes, and antidromic stimulation was performed to avoid confounding effects of post-traumatic changes in synaptic transduction.
Abnormal accumulation of extracellular K+ is caused by impaired glial K+ homeostasis
However, the above mentioned experiment does not directly address the question whether the glial impairment is contributing to the abnormal neuronal stimulation-induced extracellular K+ accumulation. Indeed, both enhanced neuronal firing and abnormal glial function could result in greater amounts of K+ accumulated in the extracellular compartment. A dissociation between the neuronal and the glial contribution is difficult to establish, and has therefore delayed our understanding of the role of glia in [K+]outregulation. Almost 30 years ago, Pollen and Trachtenberg (1970)hypothesized a critical role for reactive glia in abnormal neuronal function through impairment of K+homeostasis. However, without control of neuronal excitability, the central role of glia malfunction could not be determined (Glötzner, 1973; Pedley et al., 1976; Lewis et al., 1977).
We investigated this issue by taking advantage of pharmacological manipulations allowed by the hippocampal slice preparation. CA3 recurrent synaptic excitation was abolished by bath application of kynurenic acid (1 mm), thus preventing burst discharges in all slices (whether obtained from control rats or from post-FPI rats). Using a combination of theIh-selective blocker ZD 7288 (BoSmith et al., 1993; Gasparini et al., 1996; Gasparini and DiFrancesco 1997) and Cs+, we selectively targeted the influx of K+ through glial ion channels. Furthermore, potassium currents through ion channels cannot be inward in neurons because the average neuronal RMP (approximately −60 mV) is much more depolarized than EK(approximately −90 mV) (Hille, 1993). Under these experimental conditions in normal slices (i.e., in the presence of Cs+) baseline [K+]out increased to 4.9 mm, similar to baseline values after FPI. During 3 Hz stimulation, potassium levels peaked at 5.2 mm and did not return to baseline values (and there was almost no undershoot when the 0.05 Hz stimulation was restored), again, similar to the K+response in post-FPI slices. Thus, extracellular Cs+ in normal slices reproduced the impairment of K+ regulation observed in post-FPI slices, under a condition in which network excitability in neurons was not a contributing factor.
Conclusions and implications
It has been long suspected that reactive glia may have improper K+-buffering capabilities (Pollen and Trachtenberg, 1970), and it has been observed that glial reactivity is accompanied by loss of inward K+ current (Francke et al., 1997; MacFarlane and Sontheimer, 1997; Bordey and Sontheimer, 1998). However, the question of whether reactive glia in general, and post-traumatic glia in particular, are capable of adequate ion homeostasis has not been directly addressed. We have now shown that 2 d after FPI neuronal function is altered, and that the resultant hyperexcitability is caused by an impairment of K+ homeostasis that follows the loss of glial K+ conductances. This finding may be relevant to an understanding of learning and memory impairments, or seizure susceptibility, that can follow TBI. In fact, each of these neurological deficits can be caused by altered CA3 pyramidal cell excitability (Traynelis and Dingledine, 1988; Thompson, 1991;Hetherington and Shapiro, 1997). Our results provide evidence of the functional consequence of the loss of K+inwardly rectifying currents in reactive glia and add an additional level of complexity to the interplay of mechanisms that are involved in the development of neurological disorders after TBI.
Footnotes
This work was supported by a grant from the Epilepsy Foundation (R.D.) and by National Institutes of Health Grants NIEHS ES 07033, NS 18895, NS 51614 (D.J.), and NS 33107 (M.S.G.). We would like to thank W. Shawn Carbonell for proofreading, and Guy M. McKhann II and Philip A. Schwartzkroin for helpful comments and review of this manuscript. We are also grateful to Lesnick E. Westrum and H. Jurgen Wenzel for help with the camera lucida.
Correspondence should be addressed to Dr. Damir Janigro, Cleveland Clinic Foundation NB20, Neurosurgery, 9500 Euclid Avenue/Desk S80, Cleveland, OH 44195.
REFERENCES
- 1.Ammann D. Ion selective microelectrodes. Springer; Berlin: 1986. [Google Scholar]
- 2.Annegers JF, Grabow JD, Groover RV, Laws ER, Elvebeck LR, Kurland LT. Seizure after head traumas: a population study. Neurology. 1980;30:683–689. doi: 10.1212/wnl.30.7.683. [DOI] [PubMed] [Google Scholar]
- 3.Ballanyi K, Grafe P, Bruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol (Lond) 1987;382:159–174. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bordey A, Sontheimer H. Postnatal development of ionic currents in rat hippocampal astrocytes in situ. J Neurophysiol. 1997;78:461–477. doi: 10.1152/jn.1997.78.1.461. [DOI] [PubMed] [Google Scholar]
- 5.Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- 6.BoSmith RE, Briggs I, Sturgess NC. Inhibitory actions of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br J Pharmacol. 1993;110:343–349. doi: 10.1111/j.1476-5381.1993.tb13815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnard DM, Chrichton SA, MacVicar BA. Electrophysiological properties of reactive glial cells in the kainate-lesioned hippocampal slice. Brain Res. 1990;510:43–52. doi: 10.1016/0006-8993(90)90725-q. [DOI] [PubMed] [Google Scholar]
- 8.Chvátal A, Berger T, Vorisek I, Orkand RK, Kettenmann H, Syková E. Changes in glial K+ currents with decreased extracellular volume in developing rat white matter. J Neurosci Res. 1997;49:98–106. doi: 10.1002/(sici)1097-4547(19970701)49:1<98::aid-jnr11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Chvátal A, Anderová M, Ziak D, Syková E. Rat spinal cord glial tail currents are affected by the size of the extracellular space. Soc Neurosci Abstr. 1998;24:126.15. [Google Scholar]
- 10.D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 1998a;786:64–79. doi: 10.1016/s0006-8993(97)01412-1. [DOI] [PubMed] [Google Scholar]
- 11.D’Ambrosio R, Wenzel J, Schwartzkroin, McKhann G, Janigro D., II Functional specialization and topographic segregation of hippocampal astrocytes. J Neurosci. 1998b;18:4425–4438. doi: 10.1523/JNEUROSCI.18-12-04425.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 13.Eisenman G. Glass electrodes for hydrogen and other cations: principles and practice. Dekker; New York: 1967. [Google Scholar]
- 14.Feldberg W, Sherwood SL. Effects of calcium and potassium injected into the cerebral ventricles of the cat. J Physiol (Lond) 1957;139:408–416. doi: 10.1113/jphysiol.1957.sp005901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francke M, Pannicke T, Biedermann B, Faude F, Wiedemann P, Reichenbach A, Reichelt W. Loss of inwardly rectifying potassium currents by human retinal glial cells in diseases of the eye. Glia. 1997;20:210–218. doi: 10.1002/(sici)1098-1136(199707)20:3<210::aid-glia5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 16.Gasparini S, Maccaferri G, D’Ambrosio R, DiFrancesco D. The hyperpolarization-activated current (Ih/Iq) in rat hippocampal neurons. In: Torre V, Conti F, editors. Neurobiology: ionic channels, neurons and the brain. Plenum; New York: 1996. pp. 63–74. [Google Scholar]
- 17.Gasparini S, DiFrancesco D. Action of the hyperpolarization-activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflügers Arch. 1997;435:99–106. doi: 10.1007/s004240050488. [DOI] [PubMed] [Google Scholar]
- 18.Glötzner FL. Membrane properties of neuroglia in epileptogenic gliosis. Brain Res. 1973;55:159–171. doi: 10.1016/0006-8993(73)90495-2. [DOI] [PubMed] [Google Scholar]
- 19.Grady MS, Charleston JS, Maris DO. Quantitation of rat hippocampal cells using the optical volume fractionator: a comparison of midline to lateral fluid percussion injury. J Neurosurg. 1996;13:599. [Google Scholar]
- 20.Haglund MM, Schwartzkroin PA. Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J Neurophysiol. 1990;63:225–239. doi: 10.1152/jn.1990.63.2.225. [DOI] [PubMed] [Google Scholar]
- 21.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 22.Hetherington PA, Shapiro ML. Hippocampal place fields are altered by the removal of single visual cues in a distance-dependent manner. Behav Neurosci. 1997;111:20–34. doi: 10.1037//0735-7044.111.1.20. [DOI] [PubMed] [Google Scholar]
- 23.Hille B. Ionic channels of excitable membranes, Ed 2. Sinauer; Sunderland, MA: 1993. [Google Scholar]
- 24.Janigro D, Gasparini S, D’Ambrosio R, McKhann G, DiFrancesco D., II Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci. 1997;17:2813–2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jennett B. Epilepsy after non-missile head injuries. Scott Med J. 1973;18:8–13. doi: 10.1177/003693307301800103. [DOI] [PubMed] [Google Scholar]
- 26.Jensen M, Azouz R, Yaari Y. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol. 1994;71:831–839. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- 27.Kraus JF. Epidemiology of head injury. In: Cooper PR, editor. Head injury, Ed 2. Williams & Wilkins; Baltimore: 1987. pp. 1–19. [Google Scholar]
- 28.Lewis DV, Mutsuga N, Schuette WH, Van Buren J. Potassium clearance and reactive gliosis in alumina gel lesion. Epilepsia. 1977;18:499–506. doi: 10.1111/j.1528-1157.1977.tb04997.x. [DOI] [PubMed] [Google Scholar]
- 29.Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyeth B, Jenkins L, Dixon R, Dixon C, Phillips L, Clifton G, Young H, Hayes R. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. [DOI] [PubMed] [Google Scholar]
- 31.Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurones. J Physiol (Lond) 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maccaferri G, Mangoni M, Lazzari A, DiFrancesco D. Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J Neurophysiol. 1993;69:2129–2136. doi: 10.1152/jn.1993.69.6.2129. [DOI] [PubMed] [Google Scholar]
- 33.MacFarlane SN, Sontheimer H. Electrophysiological changes that accompany reactive gliosis in vitro. J Neurosci. 1997;17:7316–7329. doi: 10.1523/JNEUROSCI.17-19-07316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKhann GM, D’Ambrosio R, Janigro D. Heterogeneity of astrocyte resting membrane potentials revealed by whole-cell and gramicidin-perforated patch recordings from cultured neocortical and hippocampal slice astrocytes. J Neurosci. 1997;17:6850–6863. doi: 10.1523/JNEUROSCI.17-18-06850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meltzer SJ. On the toxicology of potassium chlorate, with a demonstration of the effects of intracerebral injections. Am J Physiol. 1899;3:9. [Google Scholar]
- 36.Mi H, Deerinck TJ, Jones M, Ellisman MH, Schwarz T. Inwardly rectifying K+ channels that may participate in K+ buffering are localized in microvilli of Schwann cells. J Neurosci. 1996;16:2421–2429. doi: 10.1523/JNEUROSCI.16-08-02421.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman EA. Glial cell regulation of extracellular potassium. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 717–731. [Google Scholar]
- 38.Newman EA. Regional specialization of retinal glial cell membrane. Nature. 1984;309:155–157. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicolsky BP. Theory of glass electrodes. Zh Fis Khim. 1937;10:495. [Google Scholar]
- 40.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the CNS of amphybia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 41.Pedley TA, Fisher RS, Prince D. Focal gliosis and potassium movement in mammalian cortex. Exp Neurol. 1976;50:346–361. doi: 10.1016/0014-4886(76)90010-8. [DOI] [PubMed] [Google Scholar]
- 42.Pollen DA, Trachtenberg MC. Neuroglia: gliosis and focal epilepsy. Science. 1970;167:1252–1253. doi: 10.1126/science.167.3922.1252. [DOI] [PubMed] [Google Scholar]
- 43.Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying potassium currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–346. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- 44.Rimel R, Giordani B, Barth J, Boll T, Jane J. Disability caused by minor head injury. Neurosurgery. 1981;9:221–228. [PubMed] [Google Scholar]
- 45.Schwartzkroin PA. In: The treatment of the epilepsy: principles and practice (Wyllie E, ed), pp 83–98. Lea and Febiger; Philadelphia: 1993. [Google Scholar]
- 46.Smith D, Okiyama K, Thomas M, Claussen B, McIntosh T. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- 47.Temkin NR, Dikmen S, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497–502. doi: 10.1056/NEJM199008233230801. [DOI] [PubMed] [Google Scholar]
- 48.Temkin NR, Haglund MM, Winn HR. Post-traumatic seizures. In: Youmans JR, editor. Neurological surgery, Ed 4. Saunders; Philadelphia: 1996. pp. 1834–1839. [Google Scholar]
- 49.Thompson RF. Are memory traces localized or distributed? Neuropsychologia. 1991;29:571–582. doi: 10.1016/0028-3932(91)90011-v. [DOI] [PubMed] [Google Scholar]
- 50.Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- 51.Wheal HV, Bernard C, Chad JE, Cannon RC. Pro-epileptic changes in synaptic function can be accompanied by pro-epileptic changes in neuronal excitability. Trends Neurosci. 1998;21:167–174. doi: 10.1016/s0166-2236(97)01182-x. [DOI] [PubMed] [Google Scholar]
- 52.Zuckermann EC, Glaser GH. Hippocampal epileptic activity induced by localized ventricular perfusion with high-potassium cerebrospinal fluid. Exp Neurol. 1968;20:87–110. doi: 10.1016/0014-4886(68)90126-x. [DOI] [PubMed] [Google Scholar]