

Abstract
Organic dust samples from swine confinement facilities elicit pro-inflammatory cytokine/chemokine release from bronchial epithelial cells and monocytes, dependent, in part, upon dust-induced activation of the protein kinase C (PKC) isoform, PKCε. PKCε is also rapidly activated in murine tracheal epithelial cells following in vivo organic dust challenges, yet the functional role of PKCε in modulating dust-induced airway inflammatory outcomes is not defined. Utilizing an established intranasal inhalation animal model, experiments investigated the biologic and physiologic responses following organic dust extract (ODE) treatments in wild-type (WT) and PKCε knock-out (KO) mice. We found that neutrophil influx increased by greater than two-fold in PKCε KO mice following both a one-time challenge and three weeks of daily challenges with ODE as compared to WT mice. Likewise, lung pathology revealed that bronchiolar and alveolar inflammation and lymphoid aggregates were significantly increased in ODE-treated PKCε KO mice. Airway hyper-responsiveness to methacholine increased in PKCε KO + ODE to a greater magnitude than WT + ODE animals. There were no significant differences in cytokine/chemokine release elicited by ODE treatment between groups. However, ODE-induced nitric oxide (NO) production differed in that ODE exposure increased nitrate levels in WT animals but not in PKCε KO mice. Moreover, ODE failed to upregulate NO from ex vivo stimulated PKCε KO lung macrophages. Collectively, these studies demonstrate that PKCε-deficient mice were hypersensitive to organic dust exposure, and suggest that PKCε is important in the normative lung inflammatory response to ODE. Dampening of ODE-induced NO may contribute to these enhanced inflammatory findings.
Keywords: protein kinase C epsilon, airway inflammation, airway hyper-responsiveness, nitric oxide, neutrophil, lung
INTRODUCTION
Chronic inhalation of organic dusts or bioaerosols from agriculture environmental settings, particularly industrial animal farms, can result in the development of adverse respiratory diseases including rhinosinusitis, workplace exacerbated asthma, chronic bronchitis, and obstructive pulmonary disease [1]. Organic dust is inherently complex, containing a wide diversity of microbial components and particulate matter that activate innate immune responses that subsequently lead to significant airway diseases. Neutrophil influx and increased IL-6 and CXCL8/IL-8 levels in bronchoalveolar lavage fluid have been observed in previously non-exposed persons following a one-time swine confinement facility exposure [1, 2]. In addition, a first-time response to an organic dust environmental challenge is associated with an increase in airway hyper-responsiveness (AHR) to methacholine, cross-shift changes in pulmonary function, and increased exhaled nitric oxide (NO) [3-5]. Although this intense acute inflammatory response dampens with repeated exposures, persons repetitively exposed to organic dust environments are at an increased risk of developing chronic lung disease [5, 6]. Animal models have now been developed that resemble this acute and repetitive organic dust-induced airway inflammatory response and lung injury [7, 8]. However, the mechanistic regulation of the in vivo airway response following organic dust exposures is not well-defined.
Given the complexity of organic dust, understanding key intracellular signaling pathways activated by organic dust exposure could be important in disease modification strategies. Our previous work has implicated protein kinase C (PKC) isoforms, particularly PKCε, as important signal transducers in mediating swine confinement facility and cattle feedlot organic dust extract-induced pro-inflammatory mediator production in vitro [9-12]. Namely, organic dust extracts rapidly activate PKCε in bronchial epithelial cells, and moreover, reduction in PKCε activity resulted in significant inhibition of organic dust-induced IL-8/CXCL8 release [9, 11]. In addition, PKCε is rapidly activated following organic dust exposure in human phagocytes whilst inhibition of PKCε resulted in a modest but significant reduction in dust-induced monocyte TNF-α production [12]. Although we have also shown that PKCε is activated in tracheal epithelial cells following an in vivo organic dust challenge [7], it is not known what functional role PKCε plays in modulating organic dust-induced airway inflammatory responses. There is evidence to suggest that PKCε might be a critical signaling pathway in vivo because PKCε-deficient mice demonstrated an increase in sensitivity and mortality following Gram-negative and Gram-positive bacterial challenges [13]. Moreover, PKCε may play a role in organic dust induced NO because PKCε has been shown to regulate nitric oxide synthase in murine studies of chronic hypoxic pulmonary hypertension [14].
We initially hypothesized that in the absence of PKCε, organic dust-induced airway inflammatory consequences would be reduced because our prior in vitro studies demonstrated that inhibition of PKCε resulted in a reduction in organic dust-induced cytokine/chemokine release [9-12]. To test this hypothesis, we investigated organic dust-induced airway disease with mice deficient in PKCε utilizing an established animal model. Wild-type control (WT, Prkce+/+) and deficient (Prkce−/−, PKCε knock-out, KO) mice were intranasally treated with organic dust extracts (ODE) or saline once (single exposure model) or daily for three weeks (repetitive exposure model). Quantitative and qualitative measures in airway and lung inflammation and airway hyperresponsiveness were assessed. In contrast to our initial hypothesis that mice would be protected from organic dust-induced airway inflammation, we found that PKCε-deficient mice displayed a striking and significant increase in neutrophil influx, airway hyper-responsiveness, and lung pathology. These findings were not associated with a dysregulated TNF-α, IL-6, or neutrophil chemoattractant response, but a derangement in nitric oxide response. Collectively, these data implicate PKCε as a functional component in the normative response to organic dust-induced airway inflammation and lung injury.
METHODS
Animals
C57BL/6 wild-type (WT) and PKCε heterozygote (Prkce+/−) mice on C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME). Prkce+/− mice were pair-bred to generate PKCε knock-out (KO, Prkce−/−) animals for experiments. Mice were genotyped by PCR analysis of tail DNA (data not shown). PKC epsilon deficient mice were free of any noticeable abnormality. Mice were a minimum of 6 wk old and a maximum of 12 wk old for all studies. Although we found no significant differences in the endpoints for individual animals of younger age versus slightly older aged animals, ages and sex of mice were matched with purchased WT (Prkce+/+) mice in all studies. Food and water were provided ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center according to NIH guidelines for the use of rodents.
Organic dust extract (ODE)
Organic dust extract (ODE) was prepared as previously described [15]. Briefly, settled surface dust samples were collected 3 feet above floor level from local confinement swine feeding operation facilities, which house approximately 500-700 animals. Dust (10 g) was placed into HBSS (10 ml), centrifuged for 20 minutes at 2000g, and the final supernatant was filter sterilized (0.22 μm), a process that also removes coarse particles. The aqueous ODE was diluted to a final concentration of 12.5% (vol/vol) in sterile phosphate buffered saline (PBS) for in vivo experiments. The final diluted 12.5% ODE, which has been previously shown to elicit optimal lung inflammation in mice [7], contained 2.91-3.88 mg/ml of total protein as measured by nanodrop spectrophotometry (NanoDrop Technologies, Wilmington, DE). Endotoxin levels in ODE were assayed using the limulus amebocyte lysate assay commercially available (Sigma, St. Louis, MO) and shown to not be responsible for detectable levels of PKC activation or cytokine release [11].
Preparation of murine lung slices and protein kinase C activity
Precision-cut murine lung slices from WT (Prkce+/+) and KO (Prkce−/−) mice were prepared in cultures as previously described [16] using the method of Sanderson et al. [17]. WT and KO lung slices were stimulated with the PKC activator, phorbol 12-myristate 13-acetate (PMA, 200 ng/ml, positive control), ODE or media alone for 1-24 h. Determination of PKCε catalytic activity was accomplished in fractionates from tissues by direct specific substrate peptide phosphorylation assays as previously described [16] with data expressed as picomoles of phosphate incorporated per minutes per milligram.
Murine Model of ODE exposure and tissue collection
Utilizing an established intranasal inhalation murine model of ODE-induced lung inflammation [7], mice were treated with 50μl of 12.5% ODE or sterile phosphate buffered saline (PBS; pH 7.4, diluent) once (single exposure) or daily for 3 weeks (repetitive exposure).
Bronchoalveolar lavage fluid (BALF) was collected as previously described [7]. Briefly, the total cell number recovered from pooled lavages (3 × 1 ml lavages) was enumerated and differential cell counts determined using cytospin-prepared slides (Cytopro Cytocentrifuge, Wescor Inc, Logan, UT) stained with DiffQuick (Dade Behring, Newark, DE). Cell-free supernatant for cytokine/chemokine and nitric oxide (NO) from the first lavage was collected and frozen at −80°C for later analysis.
For histology studies, whole lungs were excised after lavage and inflated to 15 cm H2O pressure with 10% formalin for 24 hours (Sigma, St. Louis, MO) to preserve pulmonary architecture as previously described [7]. Briefly, lungs were processed, embedded in paraffin, and sections (4-5 μm) were cut and stained with hematoxylin and eosin (H&E) by the institution's Tissue Science Core facility whereby standard lung slice sections are initiated at tissue branching (approximately 500 μM deep). Two H&E stained slides of the lung from each animal were microscopically reviewed and semi-quantitatively assessed for the degree and distribution of the lung inflammation (i.e. intrapulmonary cellular aggregates and alveolar and bronchiolar compartment inflammation) by a pathologist (WWW) who was blinded to the treatment conditions utilizing a previously published scoring system [7]. Each slide was entirely reviewed at scanning magnifications (X2, X4, and X10 objectives; Nikon Eclipse model E600 microscope) and the semiquantitative evaluation included assessment of all lung tissue present on each slide. Each inflammatory parameter was independently assigned a value from 0 to 3 with the greater the score, the greater the inflammatory changes in the lung.
Murine primary lung macrophages were obtained as previously described [18]. Briefly, after perfusion of the right ventricle with sterile PBS to remove circulating blood cells from the pulmonary vasculature, lung tissues were dissociated in an automated fashion using the gentleMAC Dissociator instrument according to manufacturer's instructions (Miltenyi Biotec, Auburn, CA). Mononuclear cells were collected by lymphocyte separation technique (Fisher Scientific, Pittsburgh, PA), incubated for 2 h in culture medium, and enriched for lung macrophages by removal of nonadherent cells. Lung macrophage yield was > 92% as determined by Giemsa staining [19].
Immunohistochemistry
Formalin-fixed, paraffin-embedded lung section slides were analyzed for CD3 (pan-T cell marker), CD45R/B220 (pan-B cell murine marker), and Mac-3 (phagocytes) by immunohistochemistry methods as previously described [7]. Briefly, after deparaffinization, antigen unmasking was performed using the heat-induced epitope retrieval method (DIVA Decloaker solution; Biocare Medical, Concord, CA) [20]. Next, endogenous peroxidase activity was quenched with 3% hydrogen peroxide. Slides were blocked before application of primary antibodies: rabbit anti-CD3 (dilution 1:300; Dako, Carpinteria, CA), rat anti-CD45R/B220 (clone RA3-6B2, dilution 1:200; BD Pharmingen, San Jose, CA), and anti-Mac-3 (clone M3/84, dilution 1:200; BD Pharmingen). Slides were then incubated with appropriate biotinylated IgG, and primary antibody binding was detected using the avidin-biotin-immunoperoxidase method (Vectastain Elite ABC, ready-to-use kit, Vector). Chromogen substrate (IMMPACT DAB, Vector) developer was used, and slides were counterstained with 1% Mayer's hematoxylin Slides were scanned (Ventana Roche Coreo Scanner AU, Tucson, AZ) and images were taken by Ventana Roche Image viewer computer software. Positive staining areas for CD3, B220, and Mac3 cells were analyzed using Image Pro image analysis software version 7.0 (Media Cybernetics, Bethesda, MD) for Windows. An average of 20 representative photos to capture peri-bronchiolar infiltrates and cellular aggregates (avoiding areas with large blood vessels) were taken, and the total percentage of positive cell staining per image was calculated and averaged. Color segmentation was used to identify total positive stained cells per airway area
Cytokine/Chemokine Assays
Cell-free supernatants from BALF were harvested for quantitation of TNF-α, IL-1β, IL-6, keratinocyte chemoattractant (KC; CXCL1), and macrophage inflammatory protein-2 (MIP-2; CXCL2) using commercially available ELISA kits (R&D Systems, Minneapolis, MN) with a sensitivity of 10.9, 15.625, 7.8, 15.6, 7.8 pg/mL, respectively.
Invasive pulmonary function measurement
Airway hyper-responsiveness (AHR) was invasively assessed by direct airway resistance and compliance using a computerized small animal ventilator (Finepointe, Buxco Electronics, Wilmington, NC) as previously described [7]. Briefly, 3 h following intranasal treatment with ODE (12.5%) or saline (PBS), mice were anesthetized, tracheostomized, and mechanically ventilated at a rate of 160 breaths/min and tidal volume of 0.15 ml. Once animals stabilized, dose responsiveness to aerosolized methacholine (0-96 mg/ml) was obtained and reported as total lung resistance (RL).
Nitric Oxide Analysis
Nitric oxide (NO) production in BALF was assessed via the total nitric oxide and nitrate/nitrite parameter assay kit (R&D Systems) according to manufacturer's instructions.
Real-time quantitative RT-PCR (qRT-PCR)
Inducible nitric oxide synthase (iNOS, NOS2) and endogenous NOS (eNOS, NOS3) mRNA expression was determined from lung tissues by qRT-PCR at 24 h following treatment with saline or ODE. RNA was extracted from lung tissue using the Magmax 96 kit according to the manufacturer's instructions. Concentration and purity of the RNA were determined using the NanoDrop spectrophotometer (Thermo Fisher, Wilmington, DE). All RNA samples had A260/A280 ratio of 1.8-2.0. For qRT-PCR, cDNA was synthesized using 100 ng of template RNA and a Taqman reverse transcription kit as previously described [18]. Real-time PCR reactions were prepared in triplicate using 1x TaqMan Master Mix and primers and probes for NOS2 (Mm01309902_m1) and NOS3 (Mm00435217_m1) and ribosomal (18s) RNA was used as an endogenous control. All qRT-PCR regents were purchased from Applied Biosystems (Foster City, CA). PCR was performed using ABI PRISM 7700 Sequence Detection System (Applied Biosystems). Threshold values were normalized to the expression of ribosomal RNA. qRT-PCR results are expressed as fold increase in induction (normalized copy number of ODE-treated divided by normalized copy of number of PBS treated).
Statistical Methods
Data are presented as the mean ± standard error of mean (SEM). Statistics were performed using a two-tailed, non-paired t-test to determine significant differences between treatment groups. To analyze the methacholine dose response curves, we used a two-way ANOVA (because there are 2 independent variables: treatment group and dose of methacholine) followed by Bonferonni post hoc tests when group differences were significant, p<0.05). In all analyses, GraphPad (version 5.01) software was used, and p values less than 0.05 were considered statistically significant.
RESULTS
Absence of PKCε activity confirmed in PKCε deficient mice
Whereas all mice were genotyped by PCR analysis of tail DNA (data not shown), absence of functional PKCε activity in PKCε KO mice was confirmed in ex vivo stimulated precision-cut lung slices (Figure 1). Lung slices were chosen because they provide insight into tissue-structured multicellular lung responses. Consistent with our previous work in isolated epithelial cells [9] and monocytes [12], ODE rapidly activated PKCε in a time-dependent manner in lung slices from WT mice. However, activatable PKCε was not detected upon treatment with ODE or PMA (general PKC activator) in PKCε KO mouse lung slices, confirming the absence of PKCε activity in the PKCε-deficient mice.
Figure 1. Absence of PKCε activity confirmed in lung slices from PKCε deficient mice.
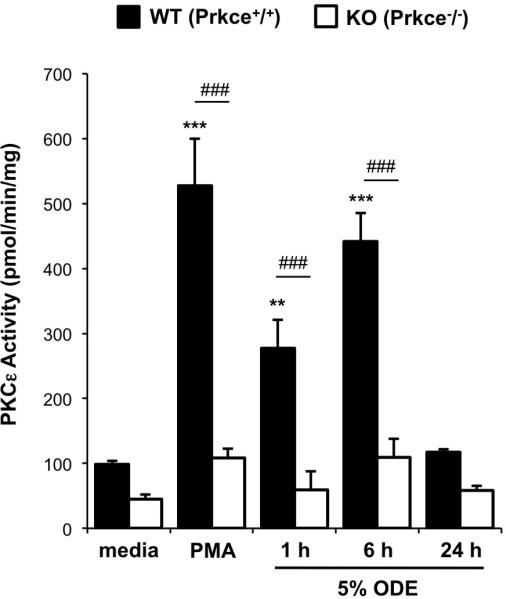
Precision-cut lung slices from wild-type (WT, Prkce+/+) and knock-out (KO, Prkce−/−) mice were ex vivo treated with phorbol 12-myristate 13-acetate (PMA, 200 ng/ml, positive control), organic dust extract (5% ODE), or media alone in submerged in vitro culture. PKCε activity was assayed from 1-24 hr. Results represent mean ± SEM (N=5-6 per condition) with statistical significance denoted by asterisks (**p<0.01, ***p<0.001) versus media alone, and hatch marks (###p<0.001) denotes difference between matched WT and KO conditions.
Acute dust-induced neutrophil influx, but not cytokine/chemokine production, is increased in PKCε-deficient mice
To determine whether there were any functional consequences of PKCε in mediating ODE-induced airway inflammatory responses in vivo, PKCε KO and WT mice were treated with saline or ODE once (single exposure model) using an established intranasal inhalation protocol [6]. Consistent with our previous work, ODE induced a rapid influx of airway neutrophils by 5 h that peaks at 24 h post-exposure (Figure 2A). Interestingly, airway neutrophils were significantly increased (two-fold), as opposed to decreased, in PKCε KO mice at both 5 and 24 h following a one-time exposure to ODE as compared to WT mice (Figure 2A, N=5-6 mice per treatment groups, p<0.05).
Figure 2. PKCε deficient mice demonstrate enhanced influx of neutrophils, but not cytokine/chemokine release, following a one-time challenge with ODE.

Wild-type (WT, Prkce+/+) and PKCε knock-out (KO, Prkce−/−) mice were intranasally treated with saline (PBS) or ODE (12.5%) once and BALF was collected at 5 and 24 h following exposure. Results represent the mean ± SEM (N=5-8 mice/group) of the total cells and cell differential (A) and cell-free levels of lung lavage fluid supernatant cytokines/chemokines (B). Statistical significance is denoted by asterisks (*p<0.05, **p<0.01, **p<0.001) versus respective saline-treated groups, and hatch mark denotes statistical significance (#p<0.05, ##p<0.01) between WT and KO treated mice.
Organic dust-induced human and murine airway disease has also been associated with increased TNF-α, IL-1β, IL-6, and neutrophil chemoattractants (human IL-8 and murine homologs KC/CXCL1 and MIP-2/CXCL2) [6]. ODE increased TNF-α, IL-1β, IL-6, CXCL1 and CXCL2 production at 5 and 24 h post-exposure in WT and PKCε KO mice as compared to saline alone (Figure 2B, N=5-6 mice per treatment groups). However, there was no significant difference in the magnitude of ODE-induced cytokine/chemokine production between WT and PKCε KO mice (Figure 2B). TNF-α, IL-6, CXCL1, CXCL2 levels, but not IL-1β, were overall less at 24 h as compared with 5 h following ODE treatment, which is consistent with the kinetics of these mediators. These studies demonstrate that PKCε KO mice have enhanced inflammatory cellular influx, but not specific cytokine/chemokine release, following a one-time challenge with organic dust.
Repetitive dust-induced inflammatory cellular influx is dependent on PKCε
Because singular and repetitive ODE treatment can affect cellular influx differently, we next investigated if airway inflammatory outcomes would be modulated following repetitive ODE treatment in PKCε KO mice. PKCε KO and WT mice were treated with ODE (12.5%) daily for 3 weeks whereupon BALF and lung tissues were collected 24 h following final treatment. As compared to baseline control, repetitive ODE treatment induced an increase in airway inflammatory cell influx in WT and PKCε KO mice (p<0.01 WT and p<0.001 KO animals). Repetitive ODE treatment resulted in a significant increased influx of neutrophils (approximately two-fold increase) and also macrophages in the PKCε KO mice as compared to WT (Figure 3A, N=6 mice/group). BALF inflammatory cell influx is lower after repeated ODE exposures as compared to single ODE exposure, consistent with the described adaptation-like response [7, 8]. Consistent with the adaptation response, cytokine/chemokine release in BALF following repetitive exposures returns to baseline [7, 8], and in this current study there was no increase in cytokine/chemokine release following repetitive ODE exposures as compared to control and there was no difference between KO and WT treatment groups (data not shown).
Figure 3. PKCε is important in mediating repetitive ODE-induced lung inflammation.

Wild-type (WT, Prkce+/+) and PKCε knock-out (KO, Prkce−/−) mice were intranasally treated with saline or ODE (12.5%) daily (repetitive exposure) for 3 weeks whereupon BALF and lung tissue were collected 24 h following final exposure. A, Results represent the mean ± SEM (N=6 mice/group) of the total cells and cell differential recovered from the BALF of mice. B, Semi-quantitative inflammatory score (mean ± SEM, N=6 mice/group) of the degree and distribution of cellular aggregates, alveolar compartment, and bronchiolar compartment lung inflammation is shown. C, A representative 4- to 5-μm-thick section, H&E stained, of one of six mice per treatment group is shown at 20× magnification and 40× magnification where noted. All lung specimens were inflated to 15 cm H2O pressure during fixation to avoid atelectasis artifact. Representative lung sections from saline control treated for comparison are also shown. Scale bar line represents 30 μm. Statistical significance is denoted by asterisks (*p<0.05, **p<0.001, ***p<0.001) between WT and KO-treated groups and hatch marks denote statistical significance between saline and ODE treated groups (##p<0.01, ###p<0.001).
It has been established that repetitive ODE treatment induces inflammatory cell influx within the bronchiolar and alveolar compartment and that perivascular and peribronchiolar mixed mononuclear cellular aggregates develop [7]. Consistent findings were demonstrated in this current study in that repetitive ODE treatment significantly induced inflammatory cell influx into the lung parenchyma and that cell aggregates developed, and these findings were not observed with saline treatment (Figure 3). Importantly, histological examination of lung tissues demonstrated striking differences in lung parenchymal inflammation between PKCε KO + ODE and WT + ODE treatment groups (Figure 3B-C). Specifically, PKCε KO mice treated repetitively with ODE demonstrated increased size and distribution of cellular aggregates throughout the lung sections by microscopic review. Semi-quantitative analysis of ODE-induced histopathologic changes was assessed using an established lung pathology scoring system. There were significant increases in the inflammatory scores in all parameters assessed (i.e. cellular aggregates, alveolar compartment and bronchiolar compartment) in PKCε KO + ODE as compared to WT + ODE mice (Figure 3B). Collectively, these studies suggest that PKCε is important in regulating inflammatory cellular influx and lung pathologic injury following repetitive ODE exposure.
Composition of dust-induced cellular aggregates is similar between WT and PKCε-deficient mice following repetitive ODE treatment
It has been demonstrated that repetitive ODE treatment results in the formation of mononuclear aggregates representing a mixture of T lymphocytes, B lymphocytes, and phagocytes with the majority of aggregates containing T lymphocytes and a smaller proportion containing B lymphocytes [7]. Although present in the aggregates, phagocytes are mainly observed in the alveolar compartment. Since there was no evidence of cellular aggregates and alveolar and bronchiolar compartment inflammation in saline treated WT or KO mice (Figure 3), lung sections from saline treated mice were not further investigated. Because there was a striking enhancement in the distribution, number and size of cellular aggregates in the lung parenchyma in the PKCε KO + ODE as compared to WT + ODE, we sought to determine whether the composition of the dust-induced mixed cellular aggregates differed between groups. However, no difference was observed in the composition of T lymphocytes, B lymphocytes, and phagocytes between the PKCε KO + ODE as compared to WT + ODE. Namely, the majority of cellular aggregates induced by ODE contained T lymphocytes with a smaller proportion containing B lymphocytes in both PKCε KO and WT animals following repetitive challenges (Figure 4). However, increased T lymphocyte influx was observed in the PKCε KO mice following ODE treatment as compared to WT + ODE (Figure 4B).
Figure 4. Repetitive organic dust exposure induces the influx of phagocytes, T and B cells in PKCε deficient mice.

Wild-type (WT, Prkce+/+) and PKCε knock-out (KO, Prkce−/−) mice were intranasally treated with ODE (12.5%) daily for 3 weeks. Lung sections (4- to 5-μm thick) were stained with anti-Mac-3 antibody for phagocytes, anti-CD3 antibody for T cells, and anti-murine CD45R/B220 antibody for B cells. Representative lung sections shown depict phagocyte influx into alveolar compartment and predominately T and B cell influx within cellular aggregates in both WT and KO animals repetitively treated with ODE at 40X magnification with scale bar line representing 30 μm. Isotype control antibody staining (negative) control is also shown. B, Quantification of positive staining of mouse lung tissue representing mean ± SEM of 20 fields of representative mouse per group. Statistical significance is denoted by asterisks (*p<0.05) between WT and KO-treated groups.
Dust-induced airway hyperresponsiveness (AHR) is increased in PKCε KO mice
It is established that an acute exposure to organic dust elicits an increase in AHR in mice [7, 8] and humans [5]. However, the mechanisms underlying this response are not known. In this current study, we confirmed that ODE treatment increases AHR as determined by invasive lung function measurements of lung resistance (RL)(Figure 5). Importantly, there was a significant increase in AHR in PKCε KO mice following ODE treatment as compared to WT mice + ODE at methacholine doses of 48 mg/ml (p<0.05) and 96 mg/ml (p<0.001) (Figure 5). Interestingly, PKCε KO treated with saline control demonstrated a significant increase in AHR to methacholine as compared to WT animals treated with saline (Figure 5). Together, these findings suggest that not only does PKCε play an important role in ODE-induced AHR to methacholine, but there may also be a role for PKCε in modulating AHR independent of the inflammatory stimulus.
Figure 5. PKCε deficient mice demonstrate increased airway hyper-responsiveness (AHR).

Wild-type (WT, Prkce+/+) and PKCε knock-out (KO, Prkce−/−) mice were intranasally treated (tx) with ODE or saline (PBS) and total lung resistance (RL) was directly measured using a mechanically ventilated mouse system 3 h following exposure. Data are expressed as means with standard error bars (N=3-4 mice/group). Letters denote statistical significance difference between groups. a: AHR is increased in KO + ODE (closed square, dotted line) as compared to WT + ODE (closed circle, solid line) at MCh 48 mg/ml (p<0.05) and 96 mg/ml (p<0.0001). b: AHR is increased in KO + ODE as compared to KO + PBS (open square, dotted line) at MCh 24 mg/ml (p<0.01) and 96 mg/ml (p<0.001) mg/ml. c: AHR is increased in WT + ODE as compared to WT + PBS (open circle, solid line) at MCh 48 (p<0.001) and 96 (p<0.001) mg/ml. d: AHR is increased in KO + PBS as compared to WT + PBS at methacholine (MCh) 48 (p<0.01) and 96 (p<0.01) mg/ml.
The NO pathway is dysregulated in PKCε-deficient mice following ODE challenge
Because exhaled NO (humans) and BALF nitrates (mice) are increased following swine confinement facility organic dust exposures [15, 21-23], we investigated if differences occurred between WT and PKCε KO mice following ODE exposure. There was a significant increase in BALF NO levels at 24 h following ODE exposure in WT animals as compared to saline exposure (Figure 6A). However, this normative response was not observed with the PKCε KO mice as there was no difference in BALF NO levels between saline and ODE-treated PKCε KO mice (Figure 6A). Next, we isolated primary murine lung macrophages and ex vivo stimulated the macrophages with 1% ODE to determine NO release. Consistent with the BALF findings, ODE elicited a significant increase in NO release from WT lung macrophages, which was not observed with PKCε KO lung macrophages (Figure 6B). Because others have linked derangements in nitric oxide synthase (NOS) isoenzymes in the lung (i.e. NOS2 and NOS3) with PKCε activity [13, 14], we investigated lung tissue NOS2 and NOS3 gene expression following ODE exposure (24 h post exposure). There was a significant increase in lung NOS2 (inducible [iNOS]) mRNA following ODE exposure in both WT and PKCε KO animals as compared to saline, but the magnitude of ODE-induced NOS2 expression increase was significantly lower in the PKCε KO mice as compared to WT (Figure 6B). Significant increases in lung NOS3 (endothelial [eNOS]) were also observed in both WT and KO animals, but no significant difference was observed between groups (Figure 6C). These studies suggest that NO dysregulation might be important in the enhanced airway inflammatory response following ODE treatment in the PKCε-deficient mice.
Figure 6. Nitric oxide pathway is dysregulated in PKCε deficient mice following ODE challenge.

Wild-type (WT, Prkce+/+) and PKCε knock-out (KO, Prkce−/−) mice were treated (tx) with ODE (12.5%) or saline (PBS) once, and bronchoalveolar lavage fluid (BALF) and lung tissue were collected 24 h following exposure. Primary lung macrophages from WT and KO mice were ex vivo stimulated with 1% ODE or saline for 24 hr. A, Results represent the mean ± SEM (N=5 mice/group) of cell-free levels of lung lavage fluid supernatant total nitrate. B, Results represent the mean ± SEM (N=7/group) of total nitrate level in lung macrophage cell-free supernatants. The mean (± SEM) fold change in inducible nitric oxide synthase, NOS2 (B) and exogenous nitric oxide synthase, NOS3 (C) following ODE treatment as compared to PBS treatment from lung tissue are shown (N=5 mice/group, independent, matched experiments). Statistical significance is denoted by asterisks (*p<0.05, **p<0.001, ***p<0.001) between respective saline vs. ODE-treated groups, and hatch mark denotes statistical significance (#p<0.05) between WT and KO ODE-treated groups.
DISCUSSION
These studies indicate that PKCε plays an important functional role in regulating organic dust-induced airway inflammation and lung injury. Although we hypothesized that inflammatory consequences would be less in PKCε-deficient animals following organic dust exposures based upon our prior in vitro work [9, 11, 12], we instead found that the absence of PKCε resulted in increased ODE-stimulated neutrophil influx, airway hyper-responsiveness, and lung injury. We interpret these findings to mean that PKCε is instrumental in regulating a normative response to organic dust exposure. Interestingly, we found no differences in the magnitude of ODE-induced cytokine/chemokine release between WT and KO mice, but there was evidence for dysregulation in the nitric oxide pathway. Our findings of heightened inflammatory consequences are consistent with other in vivo studies whereby PKCε-deficient mice demonstrated enhanced sensitivity and mortality to bacterial lung infection [13]. Moreover, a potential protective role for PKCε has also been proposed by others for differing disease states. Namely, loss of PKCε was associated with increased pulmonary vascular tone and subtle cardiovascular defects following hypoxic injury [14], and in kidney disease, increased renal hypertrophy and fibrosis was directly linked to deficiencies in PKCε activity [24].
Neutrophil influx is a hallmark of organic dust-induced airway disease [1]. The control and regulation of neutrophil recruitment following lung injury can be induced and regulated by a number of complex factors and networks including (but not limited to) chemokines, cytokines, integrins, selectins, proteases, elastase, and reactive oxygen species [25]. Although neutrophil influx in the context of organic dust exposure remains to be fully elucidated, there are several known important contributing factors. The most relevant chemokines for neutrophil recruitment in the rodent lung are CXCL1 and CXCL2, and it has been repeatedly demonstrated that these organic dusts induce CXCL1 and CXCL2 release in BALF and also in isolated epithelial and phagocytic cells [7, 9, 11]. In the current studies, neutrophils were strikingly increased in the BALF of ODE-challenged PKCε-deficient mice as compared to WT mice. However, there was no significant difference in the magnitude of CXCL1 and CXCL2 induced by ODE between WT and KO animals to explain why loss of PKCε would result in an enhancement of ODE-induced neutrophilia. These observations underscore that, although important, CXCL1 and CXCL2 do not completely explain ODE-induced neutrophil recruitment. Future lines of study should investigate mediator signals within the lung parenchyma as it is also possible that lung cytokine/chemokine release following an ODE may be compartmentalized.
Organic dusts are recognized as complex, containing a wide diversity of microbial cell wall components enriched in peptidoglycans and endotoxins from Gram-positive and Gram-negative bacteria, respectively [4, 26, 27]. Both endotoxin and peptidoglycan are directly chemotactic for neutrophils [25, 28, 29]. Moreover, extracts of organic dusts from a variety of agricultural environments exhibit direct chemotactic activity in vitro, and this response has been shown to be independent of endotoxin [30-32]. Interestingly, porcine IL-8 and a leukotriene B-like component (both neutrophil chemoattractants) have been directly measured in organic dusts, albeit in low concentrations [23, 30]. Because the enhancement and difference in neutrophil influx was observed rapidly (i.e. 5 h post exposure), it is possible that PKCε might be involved in a direct ODE-induced chemotactic process as opposed to an ODE-induced CXCL1 and CXCL2 production to explain the neutrophil recruitment. T and B cells were also recruited to the lung parenchyma following repetitive ODE exposures, consistent with previous reports [7]. In this study, there was increased T cell influx in PKCε KO mice treated with ODE as compared to WT, and therefore, further studies should focus on the phenotype and potential contribution of T cells in mediating the organic dust-induced lung inflammatory response.
Others have reported that PKCε-deficient mice demonstrate a dysregulated NOS pathway, which might explain findings of a compromised immune response [13, 14]. Consistent with these reports, we found that the normative increased NO response to ODE did not occur in PKCε-deficient mice. NO is recognized as important in airway responses and lung injury with both anti-inflammatory and pro-inflammatory properties described. However, its role in regulating organic dust airway disease is not well-defined. Previously, we have detected ODE-stimulated increases in NO from airway epithelial cells [23]. In addition, exhaled NO is slightly, but significantly, increased in healthy subjects and agricultural workers following exposure to swine confinement facility organic dusts [3, 22]. In rodents, significant, but small increases in BALF nitrates are found following acute ODE exposure [15]. In this study, ODE did not induce significant increases in nitrate release in BALF or from ex vivo-stimulated lung macrophages from PKCε-deficient mice. Thus, the lack of an appropriate NO response to ODE challenge suggests an impairment in host defense function. This observation may explain why loss of PKCε was associated with a hypersensitive airway inflammatory response. The regulation of NO generation is thought to be dependent on NOS2, and to a lesser extent NOS3, in the lung [33]. There is evidence by others that loss of PKCε is associated with derangements in the NOS pathway that were postulated to explain a heightened immune response to bacterial infection and hypoxic injury [13, 14]. It is also recognized that future lines of study should characterize NOS2 and NOS3 expression in the lung tissue. Nonetheless, our findings would also support a role for NOS2 dysregulation in lung-associated responses in the PKCε-deficient animal.
Acute organic dust exposures evoke an increase in AHR to methacholine and marked decline in post-exposure peak flows and lung function [1]. The mechanisms to explain ODE-induced increases in AHR are not clear. Interestingly, swine confinement facility organic dust-induced AHR is not dependent on the Toll-like receptor 4 (TLR4) [34] or TLR2 [15] signaling pathway. Namely, these previous studies have demonstrated no difference with AHR to methacholine in mice deficient in TLR4 or TLR2 following challenge with organic dust despite other indicators of a reduced airway inflammatory response. In other pro-inflammatory models, neutrophil recruitment and TNF-α have been implicated in the etiology of AHR, but conversely, AHR can occur independently of neutrophil recruitment or TNF [35-37]. In this study, PKCε was noted to play an important role in organic dust-induced AHR since AHR to methacholine was enhanced in PKCε-deficient mice treated with ODE, which corresponded to increases in airway neutrophil influx, but not significant changes in TNF-α, IL-1β, IL-6, CXCL1 or CXCL2 levels. Moreover, AHR to methacholine was also enhanced in saline-treated PKCε-deficient mice as compared to saline-treated WT mice. This observation suggests that PKCε-deficient mice are primed for a hypersensitive response. Because airway smooth muscle relaxation and thus decreased AHR would be a product of NO production, it is possible that PKCε-deficient mice with a dampened NO response have an enhanced ODE-induced AHR following challenge [38]. Finally, we investigated whether other PKC isoforms (i.e. PKCε, PKCε, and PKCε) were alternatively activated (or repressed) in a potential compensation for the loss of PKCε, but we did not observe any abnormal responses in these other PKC isoforms (data not shown).
In conclusion, PKCε-deficient animals demonstrated a hypersensitive response to acute and repetitive ODE treatment marked by increased neutrophil influx, lung pathology, and AHR to methacholine. These findings are not explained by modulation in ODE-induced pro-inflammatory cytokines/chemokines, but may be due to derangements in the NO pathway. Because emerging studies suggest that PKCε communicates with various intracellular TLR signaling proteins including the common adaptor protein MyD88 [39, 40], future studies might be warranted to explore the relationship of PKCε with the classic TLR signaling proteins (i.e. Mal, MyD88, IRAK4, IRAK1, TRAF) in the context of organic dust exposures. Finally, it may also be important to investigate PKCε gene polymorphisms in exposed agriculture workers.
Supplementary Material
{kind=link}
Acknowledgements
The authors wish to thank Ashley Bauer, Jane DeVasure, and Jackie Pavlik for technical assistance, and Dr. Geoffrey Talmon, MD, David Muirhead, and particularly David Wert, Facility Supervisor, Pathology and Microbiology Department in the UNMC Tissue Science Facility for assistance with lung tissue processing, sectioning, H&E staining, and assistance with digital microscopy images prepared for the manuscript. We also thank Lisa Chudomelka for manuscript preparation assistance.
Disclosure footnote: Study supported by grants from the National Institute of Environmental Health Sciences (R01: ES019325; K08: ES015522-01 and [ARRA] ES015522-03S1 to JAP), National Institute of Occupational Safety Health (R01OH008539 to DJR and P01OH010162 to TAW), National Institute on Alcoholism and Alcohol Abuse (R01AA017993 to TAW) and Department of Veterans Affairs (I01BX000728 to TAW).
Footnotes
Declaration of Interest
The authors report no conflicts of interest.
References
- 1.Poole JA, Romberger DJ. Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol. 2012;12:126–32. doi: 10.1097/ACI.0b013e3283511d0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz DA, Landas SK, Lassise DL, Burmeister LF, Hunninghake GW, Merchant JA. Airway injury in swine confinement workers. Ann Intern Med. 1992;116:630–5. doi: 10.7326/0003-4819-116-8-630. [DOI] [PubMed] [Google Scholar]
- 3.Sundblad BM, Larsson BM, Palmberg L, Larsson K. Exhaled nitric oxide and bronchial responsiveness in healthy subjects exposed to organic dust. Eur Respir J. 2002;20:426–31. doi: 10.1183/09031936.02.00257402. [DOI] [PubMed] [Google Scholar]
- 4.Zhiping W, Malmberg P, Larsson BM, Larsson K, Larsson L, Saraf A. Exposure to bacteria in swine-house dust and acute inflammatory reactions in humans. Am J Respir Crit Care Med. 1996;154:1261–6. doi: 10.1164/ajrccm.154.5.8912733. [DOI] [PubMed] [Google Scholar]
- 5.Palmberg L, Larssson BM, Malmberg P, Larsson K. Airway responses of healthy farmers and nonfarmers to exposure in a swine confinement building. Scand J Work Environ Health. 2002;28:256–63. doi: 10.5271/sjweh.673. [DOI] [PubMed] [Google Scholar]
- 6.Sundblad BM, von Scheele I, Palmberg L, Olsson M, Larsson K. Repeated exposure to organic material alters inflammatory and physiological airway responses. Eur Respir J. 2009;34:80–8. doi: 10.1183/09031936.00105308. [DOI] [PubMed] [Google Scholar]
- 7.Poole JA, Wyatt TA, Oldenburg PJ, Elliott MK, West WW, Sisson JH, Von Essen SG, Romberger DJ. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1085–95. doi: 10.1152/ajplung.90622.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charavaryamath C, Janardhan KS, Townsend HG, Willson P, Singh B. Multiple exposures to swine barn air induce lung inflammation and airway hyper-responsiveness. Respir Res. 2005;6:50–63. doi: 10.1186/1465-9921-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wyatt TA, Slager RE, Heires AJ, Devasure JM, Vonessen SG, Poole JA, Romberger DJ. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am J Respir Cell Mol Biol. 2010;42:706–15. doi: 10.1165/rcmb.2009-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyatt TA, Slager RE, Devasure J, Auvermann BW, Mulhern ML, Von Essen S, Mathisen T, Floreani AA, Romberger DJ. Feedlot dust stimulation of Interleukin-6 and -8 requires protein kinase C epsilon in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1163–70. doi: 10.1152/ajplung.00103.2007. [DOI] [PubMed] [Google Scholar]
- 11.Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol. 2002;93:289–96. doi: 10.1152/japplphysiol.00815.2001. [DOI] [PubMed] [Google Scholar]
- 12.Poole JA, Wyatt TA, Von Essen SG, Hervert J, Parks C, Mathisen T, Romberger DJ. Repeat organic dust exposure-induced monocyte inflammation is associated with protein kinase C activity. J Allergy Clin Immunol. 2007;120:366–73. doi: 10.1016/j.jaci.2007.04.033. [DOI] [PubMed] [Google Scholar]
- 13.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. Protein kinase C epsilon is required for macrophage activation and defense against bacterial infection. J Exp Med. 2001;194:1231–42. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Littler CM, Wehling CA, Wick MJ, Fagan KA, Cool CD, Messing RO, Dempsey EC. Divergent contractile and structural responses of the murine PKC-epsilon null pulmonary circulation to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2005;289:L1083–93. doi: 10.1152/ajplung.00472.2004. [DOI] [PubMed] [Google Scholar]
- 15.Poole JA, Wyatt TA, Kielian T, Oldenburg P, Gleason AM, Bauer A, Golden G, West WW, Sisson JH, Romberger DJ. Toll-like receptor 2 (TLR2) regulates organic dust-induced airway inflammation. Am J Respir Cell Mol Biol. 2011;45:711–9. doi: 10.1165/rcmb.2010-0427OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyatt TA, Kharbanda KK, McCaskill ML, Tuma DJ, Yanov D, Devasure J, Sisson JH. Malondialdehyde-acetaldehyde-adducted protein inhalation causes lung injury. Alcohol. 2012;46:51–9. doi: 10.1016/j.alcohol.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanderson MJ. Exploring lung physiology in health and disease with lung slices. Pulm Pharmacol Ther. 2011;24:452–65. doi: 10.1016/j.pupt.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole JA, Kielian T, Wyatt TA, Gleason AM, Stone J, Palm K, West WW, Romberger DJ. Organic dust augments nucleotide-binding oligomerization domain (NOD2) expression via an NF-kappa B pathway to negatively regulate inflammatory responses. Am J Physiol Lung Cell Mol Physiol. 2011;301:L296–306. doi: 10.1152/ajplung.00086.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giemsa G. Farbemethoden fur malariaparasiten. Zentrallbl Bakteriol. 1902;31:429–30. [Google Scholar]
- 20.Ward JM, Erexson CR, Faucette LJ, Foley JF, Dijkstra C, Cattoretti G. Immunohistochemical markers for the rodent immune system. Toxicol Pathol. 2006;34:616–30. doi: 10.1080/01926230600941340. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Larsson K, Palmberg L, Malmberg P, Larsson P, Larsson L. Inhalation of swine dust induces cytokine release in the upper and lower airways. Eur Respir J. 1997;10:381–7. doi: 10.1183/09031936.97.10020381. [DOI] [PubMed] [Google Scholar]
- 22.Von Essen SG, Scheppers LA, Robbins RA, Donham KJ. Respiratory tract inflammation in swine confinement workers studied using induced sputum and exhaled nitric oxide. J Toxicol Clin Toxicol. 1998;36:557–65. doi: 10.3109/15563659809028049. [DOI] [PubMed] [Google Scholar]
- 23.Wyatt TA, Sisson JH, Von Essen SG, Poole JA, Romberger DJ. Exposure to hog barn dust alters airway epithelial ciliary beating. Eur Respir J. 2008;31:1249–55. doi: 10.1183/09031936.00015007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, Schiffer M, Mengel M, Lindschau C, Leitges M, Haller H. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol. 2007;18:1190–8. doi: 10.1681/ASN.2005070694. [DOI] [PubMed] [Google Scholar]
- 25.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poole JA, Dooley GP, Saito R, Burrell AM, Bailey KL, Romberger DJ, Mehaffy J, Reynolds SJ. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J Toxicol Environ Health A. 2010;73:684–700. doi: 10.1080/15287390903578539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nehme B, Gilbert Y, Letourneau V, Forster RJ, Veillette M, Villemur R, Duchaine C. Culture-independent characterization of archaeal biodiversity in swine confinement building bioaerosols. Appl Environ Microbiol. 2009;75:5445–50. doi: 10.1128/AEM.00726-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner JG, Roth RA. Neutrophil migration during endotoxemia. J Leukoc Biol. 1999;66:10–24. doi: 10.1002/jlb.66.1.10. [DOI] [PubMed] [Google Scholar]
- 29.Sperry JF, Burns JM. Polymorphonuclear neutrophil chemotaxis modulated by bacteroides fragilis peptidoglycan. Infect Immun. 1987;55:1725–7. doi: 10.1128/iai.55.7.1725-1727.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buck MG, Schachter EN, Fick RB, Merrill WW, A CJ, Jr, Keirns JJ, Oliver J, Wall JH. Biologic activity of purified cotton bract extracts in man and guinea pig. Environ Health Perspect. 1986;66:37–44. doi: 10.1289/ehp.866637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Von Essen SG, O'Neill DP, Robbins RA, Rennard SI. Neutrophil chemotaxis to extracts of grain plant components. Am J Ind Med. 1994;25:85–8. doi: 10.1002/ajim.4700250122. [DOI] [PubMed] [Google Scholar]
- 32.Von Essen SG, Robbins RA, Thompson AB, Ertl RF, Linder J, Rennard S. Mechanisms of neutrophil recruitment to the lung by grain dust exposure. Am Rev Respir Dis. 1988;138:921–7. doi: 10.1164/ajrccm/138.4.921. [DOI] [PubMed] [Google Scholar]
- 33.Fagan KA, Tyler RC, Sato K, Fouty BW, Morris KG, Jr, Huang PL, McMurtry IF, Rodman DM. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am J Physiol. 1999;277:L472–8. doi: 10.1152/ajplung.1999.277.3.L472. [DOI] [PubMed] [Google Scholar]
- 34.Charavaryamath C, Juneau V, Suri SS, Janardhan KS, Townsend H, Singh B. Role of toll-like receptor 4 in lung inflammation following exposure to swine barn air. Exp Lung Res. 2008;34:19–35. doi: 10.1080/01902140701807779. [DOI] [PubMed] [Google Scholar]
- 35.Pennings HJ, Kramer K, Bast A, Buurman WA, Wouters EF. Tumour necrosis factor-alpha induces hyperreactivity in tracheal smooth muscle of the guinea-pig in vitro. Eur Respir J. 1998;12:45–9. doi: 10.1183/09031936.98.12010045. [DOI] [PubMed] [Google Scholar]
- 36.Savov JD, Gavett SH, Brass DM, Costa DL, Schwartz DA. Neutrophils play a critical role in development of LPS-induced airway disease. Am J Physiol Lung Cell Mol Physiol. 2002;283:L952–62. doi: 10.1152/ajplung.00420.2001. [DOI] [PubMed] [Google Scholar]
- 37.Lefort J, Singer M, Leduc D, Renesto P, Nahori MA, Huerre M, Creminon C, Chignard M, Vargaftig BB. Systemic administration of endotoxin induces bronchopulmonary hyperreactivity dissociated from TNF-alpha formation and neutrophil sequestration into the murine lungs. J Immunol. 1998;161:474–80. [PubMed] [Google Scholar]
- 38.Oldenburg PJ, Poole JA, Sisson JH. Alcohol reduces airway hyperresponsiveness (AHR) and allergic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2012;302:L308–15. doi: 10.1152/ajplung.00077.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loegering DJ, Lennartz MR. Protein kinase C and toll-like receptor signaling. Enzyme Res. 2011;2011:537821. doi: 10.4061/2011/537821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faisal A, Saurin A, Gregory B, Foxwell B, Parker PJ. The scaffold MyD88 acts to couple protein kinase c epsilon to toll-like receptors. J Biol Chem. 2008;283:18591–600. doi: 10.1074/jbc.M710330200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.