

Abstract
Acetyl-CoA synthetase (ACS) catalyzes the formation of AcCoA from acetate, ATP and Coenzyme A, allowing the organism to grow on acetate as the sole carbon source. ACS was the first enzyme in Mycobacterium tuberculosis shown to be regulated by posttranslational acetylation by the cAMP-dependent protein acetyltransferase. This modification results in the inactivation of the enzyme and can be reversed in the presence of NAD+ and a mycobacterial sirtuin-like deacetylase. In this study we characterize the kinetic mechanism of MtACS, where the overall reaction can be divided into two half-reactions: the acetyl-adenylate forming reaction and the thiol-ligation reaction. We also provide evidence for the existence of the acetyl-adenylate intermediate via 31P-NMR spectroscopy. Furthermore, we dissect the regulatory role of K617 acetylation and show that acetylation inhibits only the first, adenylation half-reaction while leaving the second half reaction unchanged. Finally, we demonstrate that the chemical mechanism of the enzyme relies on a conformational change which is controlled by the protonation state of aspartate 525. Together with our earlier results, this suggests a degree of regulation of enzyme activity that is appropriate for the role of the enzyme in central carbon metabolism.
Keywords: Tuberculosis, Enzyme Kinetics, Enzyme Regulation, Central Carbon Metabolism
Acetyl-CoA (AcCoA) is an important intermediate in many pathways of intermediary metabolism, including fatty acid biosynthesis and oxidation, the TCA-cycle and the glyoxylate shunt (1). AcCoA can be synthesized from pyruvate by pyruvate dehydrogenase via the glycolytic sequence (2), or de-novo by acetyl-CoA synthetase (ACS) from acetate (Ac) and coenzyme A (CoA) in an ATP-dependent manner (Scheme 1) (3). The latter enables the bacterium to survive on a medium that contains acetate as a sole carbon source. ACS is a member of the adenylate-forming enzyme superfamily, among which are aminoacyl-tRNA synthetases, the nonribosomal peptide and polyketide synthetases and the fatty acyl CoA- and ACP-ligases. Members of this family are structurally characterized by a small C-terminal domain and a larger N-terminal domain that form a cleft that includes the active site of the enzyme (3). MtACS was recently shown to be the first enzyme in Mycobacterium tuberculosis to be regulated by reversible posttranslational acetylation (4). Mycobacteria contain a single cAMP-dependent protein acetyltransferase that catalyzes the acetylation of K617 of MtACS, resulting in inactivation of the enzyme. This acetylation is reversed in the presence of NAD+ and a mycobacterial sirtuin-like deacetylase. This regulation suggests an important role for ACS in the modulation of the energy pool in mycobacteria.
Scheme 1.
In E. coli, ACS is essential for growth in high-density cell culture. Under these conditions, glucose becomes limiting, and continued growth requires ACS to convert acetate into AcCoA which can be converted into hexoses via the glyoxylate shunt and gluconeogenesis. Acetate is a weakly lipophilic acid that can easily penetrate the membrane, acidify the cytoplasm, increase the osmotic pressure within the cell, and interfere with methionine biosynthesis (5). One of roles of ACS is thus to convert acetate into AcCoA thereby reducing the toxicity caused by acetate in the medium (5). It is possible that M. tuberculosis has to deal with similar environmental stresses when it resides within the macrophage phagosome, granuloma or tubercule, which are unique habitats for the organism (6). A recently published paper described an increase in the levels of different metabolites within the granuloma, one of which was acetate. This finding suggests a change in the selection of different metabolic pathways by M. tuberculosis at different stages of the disease (7). M. tuberculosis thus has to adapt to this changing external environment, probably in a similar manner to E. coli by upregulating ACS activity. However, in contrast to E. coli, M. tuberculosis can use glucose and acetate simultaneously as a carbon source and does not exhibit diauxonic growth (8). Thus it is very likely that ACS can both reduce the toxicity of Ac and serve as a gateway for Ac into central carbon metabolism.
Despite its obvious importance in allowing for growth on acetate alone, or the observations that ACS is posttranslationally acetylated in a number of microorganisms (9), including M. tuberculosis, there are no detailed mechanistic studies of the enzyme. As a member of the adenylate-forming acyl-CoA synthetases, the kinetic mechanism is predicted to be a bi-uni-uni-bi ping pong mechanism, where the overall reaction can be divided into two half reactions (Scheme 1): acetyl-adenylate formation and the thiol ligation reaction leading to AcCoA and AMP formation. In this study we provide evidence for the kinetic mechanism and the existence of the acetyl-adenylate intermediate via 31P-NMR spectroscopy. We show that acetylation prevents only the first adenylation half reaction while leaving the formation of AcCoA from the acetyladenylate and CoA unchanged. We also characterize the chemical mechanism of M. tuberculosis ACS by using pH rate profiles and show that a conformational change between the adenylate forming and thioester forming conformations is dependent on the ionization state of D525.
Materials and Methods
Materials
All chemicals were purchased from Sigma-Aldrich chemical company.
Expression and Purification of MtACS
The plasmid pET28a:MtACS was transformed into T7 express E. coli cells. A single colony was selected, grown in 25 ml of LB medium supplemented with 30μg/ml kanamycin, and these cells were used to inoculate 6L the same media. The cells were grown to mid-log phase at 37°C, and induced by the addition of 1mM IPTG for 4.5hr at 25°C. The cells were harvested by centrifugation and the pellet was resuspended in buffer containing 50 mM sodium phosphate buffer, pH 8.0 containing 300 mM NaCl (buffer A). The cells were lysed in an EmulsiFlex-C3 homogenizer (Avestin) and centrifuged for 1 hr at 38,000g. The supernatant was loaded onto Ni-NTA column (QIAGEN), that was pre-equilibrated with buffer A. The unbound proteins were eluted with 5 column volumes of 10 mM imidazole in buffer A, and eluted with a 20 column-volume linear imidazole gradient, from 10mM to 250mM imidazole. Fractions containing pure MtACS were determined using SDS-PAGE followed by Coomassie Blue staining. These fractions were pooled and dialyzed against 100 mM HEPES, pH 7.5 containing 100 mM KCl, followed by concentration with an Amicon concentrator with a 30Kd cutoff.
Site-Directed Mutagenesis
The D525N mutation was introduced into pET28a+/ ACS by using pfu ultra polymerase (from Stratagene) and the following primers: FW-GGTACTAGGCCGCATCAACGACGTGATGAACGTGTCCGGG, RV-CCCGGACACGTTCATCACGTCGTTGATGCGGCCTAGTACC, where the underlined residues are the site of the change.
Enzyme Activity Assay
Initial reaction velocities of MtACS were assayed spectrophotometrically by coupling the formation of AMP to the reactions of myokinase, pyruvate kinase and lactate dehydrogenate, and following the decrease in absorbance of NADH at 340 nm (ε340= 6220 M−1 s−1). All reactions were performed at 25°C using a Shimadzo UV-2450 spectrophotometer. A typical reaction mixture contained 100 mM HEPES, pH 7.5, 100 mM KCl, 10 mM MgCl2, 1 mM PEP, 0.1 mM NADH, 6 units of myokinase, 6 units of pyruvate kinase, 6 units of lactate dehydrogenase, MtACS at a final concentration of 100 nM and variable concentrations of the substrates: Ac, ATP and CoA. The reaction mixtures were incubated for 1 min at room temperature and reactions were initiated by the addition of Coenzyme A. Reactions were followed for 3–5 min and velocities were calculated assuming 2 molecules of NADH were oxidized for each AcCoA molecule formed.
Initial Velocity Experiments
Kinetic constants for ATP, Ac and CoA were determined by using at least five different concentrations of each substrate while saturating with the others (~1mM). The kinetic parameters for each substrate determined by fitting each substrate saturation curve to equation 1 with Sigma Plot 11.0:
| (1) |
where V is maximal velocity, A is substrate concentration, and K is the Michaelis-Menten constant for the variable substrate, A. Initial velocity patterns were determined at various concentrations of one substrate, fixed concentrations of the second substrate, and saturating concentrations of the third. The data were fit to equation 2 for patterns composed of parallel lines and equation 3 for patterns composed of intersecting lines:
| (2) |
| (3) |
where Ka and Kb are the Michaelis constants for the varied substrates A and B, and Kia is the dissociation constant for substrate A. Product and dead-end inhibition studies were performed using variable concentrations of one substrate, fixed concentrations of inhibitor or product, while the other substrate(s) were kept at saturating concentrations. The data were fit to equation 4 for a competitive inhibition pattern, equation 5 for an uncompetitive inhibition pattern and equation 6 for a noncompetitive pattern:
| (4) |
| (5) |
| (6) |
where I is the concentration of the product or inhibitor, and Kis and Kii are the inhibition constants for the slope and intercept terms.
18O Transfer Experiment
[18O2]-acetate was purchased from Sigma-Aldrich. 18O transfer from [18O2]-acetate to AMP was monitored by using 31P NMR spectroscopy with a Bruker AMX300 NMR spectrometer. A typical reaction contained 100 mM HEPES, pH 7.5, 100 mM KCl, 30 mM MgCl2, 22.5 mM ATP, 22.5 mM CoA, 45 mM 16O/18O-Ac (50% v/v) and 3.54 μM MtACS. The reaction was incubated for 2 hr at 25°C. The pH was adjusted to 8.6 and 20% D2O was added before spectroscopic analysis.
pH-rate profiles
The pH dependence of kcat and kcat/KATP were determined by using varying concentrations of ATP and saturating concentrations of Ac and CoA. For determination of kcat/KCoA, CoA was varied while Ac and ATP where held at saturating concentrations. MtACS activity was monitored over a pH ranging from 6.8 to 10.5 in Bis-Tris propane buffer. All reactions were repeated in the presence of 5x of the coupling enzymes, to ensure no changes were observed on the MtACS reaction rate at the pH extremes.
ATP-PPi Exchange Assay
PPi exchange assays were carried out in the presence of 100 mM HEPES pH 7.5, 100 mM KCl, 0.250 mM ATP, 0.250 mM Ac, 10 mM PPi , 20 μCi/μmol radiolabeled PPi (Perkin-Elmer), 5 mM MgCl2 and 100 nM MtbACS. The reaction was stopped at a given time points with the addition of 35% perchloric acid and 250 mM EDTA. Samples where boiled for 1min and centrifuged for 20 min at 13K rpm. 1μl from each sample was spotted onto a PEI-cellulose TLC plate. PPi was separated from ATP by using 0.9 M Guanidine-HCL as the mobile phase on chromatography sheets.
Synthesis of Acetyl-AMP
Acetyl-AMP was prepared as described at Berg et al. (10).
Results and Discussion
Initial velocity experiments
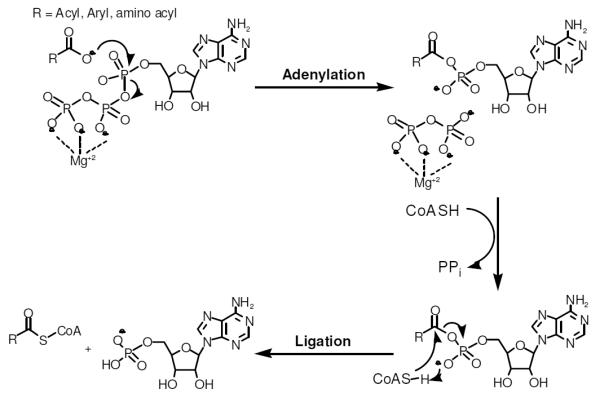
The kinetic mechanism of MtACS was determined using initial velocity studies and product and dead-end inhibition experiments. When acetate was varied against fixed concentrations of ATP, an intersecting initial velocity pattern was observed (Figure 1A). When acetate was varied against fixed concentrations of CoA, a parallel initial velocity pattern was observed (Figure 1B), indicating that there is a product released between the binding of Ac and CoA. This supports the predicted bi uni uni bi ping-pong kinetic mechanism. The Km values for ATP, Ac and CoA were calculated to be 79 ± 7, 55 ± 8 and 125 ± 5 μM, respectively, while the kcat value was determined to be 2.05 ± 0.03 sec−1.
Figure 1.
Initial velocity studies. (A) 1 mM CoA and 0.025 mM (circle), 0.05 mM (triangle), 0.100 mM (squares), 0.160 mM (diamond) and 0.500 mM Ac (inverted triangle) with 0.05–1 mM ATP. Data was fitted to equation 3 to obtain an intersecting pattern and the kinetic parameters were determined, kcat = 2.1 ± 0.1 sec−1, KAcm = 0.012 ± 0.001 mM, KATPm = 0.12 ± 0.01 mM. (B) 1 mM ATP and 0.04 mM (circles), 0.05 mM (triangle), 0.1 mM (square) and 1 mM CoA (diamond) with 0.025– 1 mM Ac. Data was fitted to equation 2 to obtain a parallel pattern and the kinetic parameters were determined, kcat = 2.4 ± 0.1 sec−1, KAcm = 0.030 ± 0.002 mM, KCoAm = 0.12 ± 0.05 mM. Dead-end inhibition studies with (C) 0 mM (circle), 4 mM (triangle) and 10 mM (square) AMPCPP in the presence of 1mM Ac and 0.05–0.4 mM ATP. Data was fitted to equation 4 to obtain a competitive inhibition pattern and Kis = 5.0 ± 0.8 mM was calculated. (D) 0 mM (circle), 4 mM (triangle), 10 mM (square) and 15 mM (diamond) AMPCPP in the presence of 1 mM ATP and 0.05–0.4 mM Ac. Data was fitted to equation 6 to obtain a noncompetitive inhibition pattern. Kis = 9 ± 3 mM, and Kii = 7.2 ± 0.6 mM were calculated.
To assign the order of ATP and acetate binding, the dead-end inhibitor AMPCPP, was used. When AMPCPP was held at several fixed concentrations and variable concentrations of ATP were used, a competitive pattern was observed (Figure 1C). This same experiment was performed using acetate as the variable substrate, and yielded a noncompetitive pattern (Figure 1D). Together, these two results support the ordered binding of ATP, followed by Ac. The release of the first product was probed using fixed concentrations of the product inhibitor PPi versus variable concentrations of ATP and resulted in noncompetitive inhibition (Figure S1A). When this experiment was repeated at saturating concentrations of acetate, an identical noncompetitive pattern was observed (Figure S1B), suggesting that at high concentrations of acetate, it can bind to the free enzyme prior to ATP binding, thus explaining the observed noncompetitive pattern rather than the uncompetitive pattern expected for an ordered mechanism.
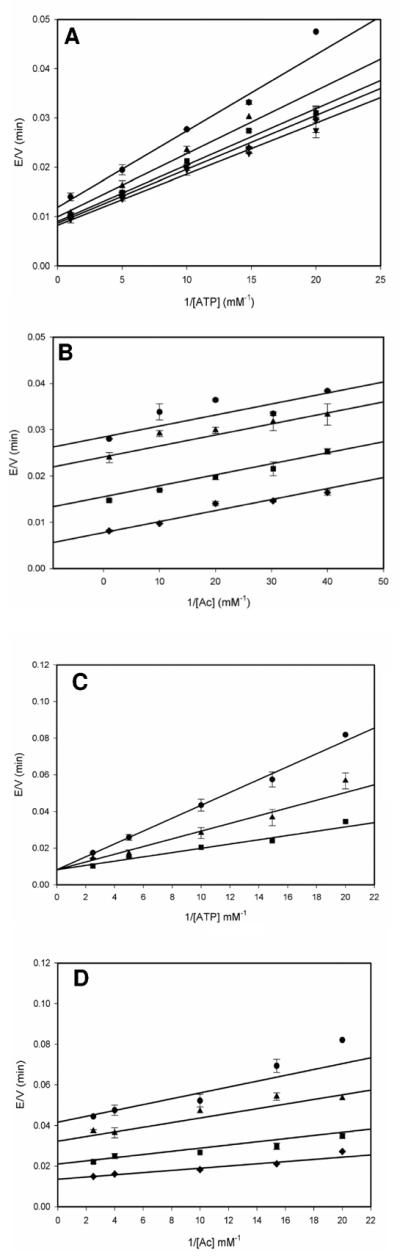
These results imply that the reaction is carried out by a bi-uni-uni-bi ping pong kinetic mechanism, where ATP binds first followed by acetate binding to form a ternary complex that allows for the adenylation chemistry to occur with the release of PPi and the generation of a tightly-bound acetyladenylate intermediate. Binding of CoA and nucleophilic attack of the thiol on the acetyladenylate in the thioester forming chemistry results in the formation of AcCoA and AMP, that are released randomly from the enzyme. At low concentrations of acetate, the kinetic mechanism is likely ordered due to the high intracellular concentration of ATP, but at higher acetate concentrations, the random binding of ATP and acetate cannot be excluded. We therefore propose a kinetic mechanism shown in Scheme 2 which allows for both possibilities under different acetate concentrations.
Scheme 2.
This kinetic mechanism is common to many ATP-dependent synthetases among which are panthothenate synthethase, involved in CoA biosynthesis, from Mycobacterium tuberculosis (11) and the Mycobacterium smegmatis mshC-encoded cysteine ligase, involved in mycothiol biosynthesis (12). These enzymes exhibit ordered binding of ATP and their adenylatable substrate, although the kinetic mechanism of E. coli tyrosyl-tRNA synthetase has been shown to involve the random binding of tyrosine and ATP (13).
Evidence for the existence of the acetyl-AMP intermediate
Our kinetic evidence suggested the existence of an acetyl-AMP intermediate; however, to confirm this we performed an experiment that would provide direct evidence for this intermediate. If [18O2]-acetate is used as the substrate, the acetyladenylate formed would carry an 18O atom in the phosphoric-acetic anhydride bond. Upon attack by CoA, this oxygen atom would leave with the product AMP. Figure 2 shows a stack plot of the 31P- NMR signals in reaction mixtures in which no enzyme has been added (A), where enzyme has been added using unlabeled Ac (B) and where enzyme has been added using a mixture of 50% [16O2]-acetate and 50% [18O2]-acetate (C). The region around 5 ppm has been expanded in the lower panel of the Figure. A single 31P resonance due to the α-P atom of AMP is observed at ~5 ppm in the control reaction containing only unlabeled [16O2]-acetate. However, when the reaction was carried out in the presence of a mixture of 50% [16O2]-acetate and 50% [18O2]-acetate, two 31P resonance peaks were observed, corresponding to the α-31P resonances of AMP and AMP labeled with one 18O atom which is shifted upfield by 0.024 ppm due to the presence of the heavy oxygen atom. These results require that an acetyl-AMP intermediate is formed as part of the reaction.
Figure 2.

31P-NMR spectrum of the reaction catalyzed by ACS. Incubation 22.5 mM ATP, 22.5 mM CoA, 45 mM 16O-Ac in the absence (1) or presence of 3.54 μM ACS (2). Incubation 22.5 mM ATP, 22.5 mM CoA, 45 mM 16O /18O-Ac (50% v/v) with 3.54 μM ACS (3).
The effect of acetylation on ACS activity
Having demonstrated the ping-pong nature of the kinetic mechanism, and demonstrated the formation of a bound acetyladenylate as a reaction intermediate, we sought evidence for the chemistry that is inhibited by the acetylation of K617 that we have previously reported (4). Our initial attempts were performed using a PPi exchange assay in which we measured a decrease in the amount of radioactive PPi as a function of time in the absence of CoA. We observed a constant, time-dependent decrease in radioactive PPi in the presence of native ACS. However, in the presence of acetylated ACS, no change in radioactive PPi concentrations was observed (Figure 3A), confirming that acetylated MtACS is not able to catalyze the pyrophosphate exchange reaction in the absence of CoA. The possibility that the exchange reaction could only be catalyzed in the presence of CoA could not be ruled out by this experiment, nor was the assay robust enough to provide quantitative data relevant to the relative rate-limiting nature of the adenylation or thioester ligation reactions.
Figure3.

The effects of acetylation on ACS activity. (A) PPi exchange assay in the presence of 1 mM Ac, 1 mM ATP, 0.1 mM PPi, radiolableled PPi and 200 nM ACS (triangles), or AcACS (circles). Representative of two experiments. (B) Activity of AcACS in the presence of 1 mM Ac, 1 mM ATP and 1 mM CoA (1) or 1 mM AcAMP and 1 mM CoA (3). Activity of ACS in the presence of 1 mM ATP, 1 mM Ac, and 1 mM CoA (2), or in the presence of 1 mM AcAMP and 1 mM CoA (4). Representative of two experiments.
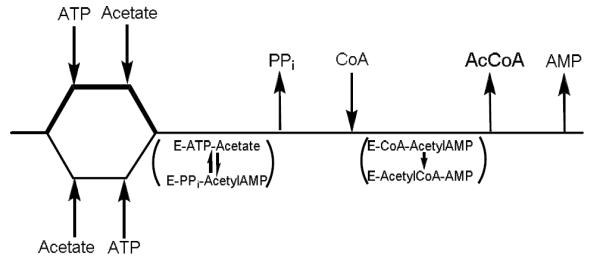
In order to gain more information on these issues, we synthesized AcAMP and used it as a substrate to monitor the second half reaction. Using either the complete reaction mixture components, or AcAMP and CoA, we could now compare, using the same AMP-forming assay, the rates of the overall reaction at saturating concentrations of ATP, Ac and CoA or the isolated second half reaction using AcAMP and CoA (Figure 3B). The formation of AMP is 2.3 times faster using AcAMP and CoA than the rate using ATP, Ac and CoA, suggesting that the adenylation half reaction is the slower of the two half reactions (Figure 4). Further, when acetylated ACS (AcACS) was substituted for WT ACS, the reaction requiring ATP and Ac was reduced to zero, while the reaction of AcAMP and CoA changed from 0.43 to 0.6 min−1. Previous X-ray crystallographic studies have shown that acetyl-CoA synthetases adopt two different conformations to catalyze each half reaction; the AcAMP-forming and the thiol-ligating conformations (14, 15). AcACS exhibited a small but significant increase in kcat when AcAMP was used to probe the second half reaction. If K617 acetylation specifically affected the first half reaction, then the rate of the second half reaction does not have to change when ACS is substituted with AcACS. However, the observed increase in AMP formation suggests that ACS may exist as an equilibrium mixture of the AcAMP-forming and the thiol-ligating conformations in the absence of substrates, and that K617 acetylation shifts the equilibrium towards the thiol-ligating conformation, resulting in an enhanced rate of the second half reaction for AcACS.
Figure 4.
Kinetics for ACS in the presence of 1 mM Ac, 1mM CoA and 0.01–0.50 mM ATP (triangles), data was fitted to equation 1 and yielded kcat = 0.184 ± 0.004 sec−1, ACS in the presence of 1 mM CoA and 0.01–0.50 mM AcAMP (squares). Data was fitted to equation 1 and yielded kcat = 0.43 ± 0.01 sec−1, and for AcACS in the presence of 1 mM CoA and 0.01–0.50 mM AcAMP (circles). Data was fitted to equation 1 and yielded kcat = 0.60 ± 0.03 sec−1.
These results demonstrate that monoacetylated ACS is incapable of catalyzing the overall reaction but is fully competent to catalyze the second half reaction. This supports a role for K617 in the adenylation reaction where the positive charge on the ε-nitrogen is used to stabilize the developing charge on the α-P atom of ATP during the adenylation reaction. Jogl et al. crystallized the S. cerevisiae ACS in the adenylate-forming conformation (14) in which it can be clearly seen that K675, which corresponds to K617 in MtACS, makes hydrogen bonds with the α-P of ATP. Acetylation of K617 would both prevent the electrostatic interaction with ATP, destabilizing the build-up of the negative charge in the adenylation transition state, as well as sterically prevent proper binding of ATP to allow adenylation chemistry to occur. Acetylation of the corresponding lysine residue in Salmonella enterica ACS has been shown to specifically affect the adenylation reaction, and further shown to be reversible by a bacterial NAD+-dependent Sir2 deacetylase (16). The corresponding lysine residue in Salmonella enterica propionyl-CoA synthetase has also been shown to be specifically involved in the adenylation chemistry (17).
pH-rate profiles
There have been no detailed studies of the chemical mechanism of any ACS, although structural studies have attempted to define potential groups involved in substrate binding and catalysis. In order to gain additional information about this, we determined the pH dependence of the kinetic parameters kcat, kcat/KATP and kcat/KCoA. The pH dependence of the maximum velocity, kcat, reports on the ionization states required for group(s) involved in catalysis. This parameter decreases at low pH values, and a fit of the experimental data to eq. 7 yielded a pK value of 7.5 ± 0.1 for a single group that must be deprotonated for efficient catalysis (Figure 4A). The kcat/KATP also decreased slightly as the pH decreased, and a fit of the experimental data to eq. 7 yielded a pK value of 6.5 ± 0.1 (Figure 4B). The pH dependence of kcat/KATP reports on either substrate or enzyme groups whose ionization affects ATP binding and also on chemistry occurring before the first product release step which in this reaction is inorganic pyrophosphate. In this case, the chemistry interrogated is the adenylation reaction: the nucleophilic attack of acetate on the α-phosphorous atom of bound ATP. It seems unlikely due to the low pK value of acetate (4.76) that the adenylation reaction requires an enzyme base to promote the reaction, and we ascribe the group responsible for the pH dependence of kcat/KATP to a group involved in ATP binding, either ATP itself or K617 that interacts with the oxygens on the α-P, and is the site of posttranslational acetylation. The pH independence of kcat/KCoA indicates that the thioester forming reaction does not depend on enzymatic groups for catalysis, and that the thiol of CoA is sufficiently nucleophilic to displace AMP from the acetyladenylate without the involvement of an active site base (Figure 4C).
The interpretation of the kcat pH profile is not straightforward. An examination of the ACS active site in the adenylate-forming conformation reveals no obvious or appropriate amino acid side chain that could act as a general base in this reaction (15). The involvement of K617 in adenylation chemistry was considered as a possible cause of the decrease in kcat at pH values below 7.5. However, if the ε-amino group of K617 is required to be protonated to polarize the α-phosphate, then its deprotonation at high pH values would prevent the adenylation chemistry from happening, which is not what is experimentally observed. Further, the pK value observed in the kcat profile is higher than the value observed in the kcat/KATP profile, and this is opposite to the behavior observed when the same groups are contributing to the pH dependence of two different kinetic parameters.
Alternatively, the protonation state of a group that influences the conformational change between the two conformations competent to perform the adenylation or thioester-forming reactions could influence the pH dependence of kcat. In the crystal structures of the S. cerevisia, and S. enterica ACSs, two conformations of the C-terminal domain relative to the N-terminal domain are observed which are interconverted by a 140° rotation of the C-terminal domain (14, 15). This conformational change has been proposed to be promoted by an aspartate residue (D517 in the S. enterica ACS) located in the hinge region between the C- and N-terminal domains (15). An alignment of the yeast, Salmonella and MtACS sequences reveals that this aspartic acid (D525) is also found in MtACS. We suggest that the pH-rate profiles report on the protonation of aspartate, which in the protonated state inhibits the conformational change between the adenylate- to the thiol-forming states of ACS, thus reducing the rate of the reaction at low pH values. Two previous mutants forms of the S. enterica ACS have been generated to probe the involvement of D517 in the conformational change associated with catalysis: D517G and D517P. Both of these mutant forms exhibit approximately 40% of the activity of the WT enzyme. We generated the isosteric substitution, D525N, and determined its overall activity with acetate, ATP and CoA, its pyrophosphate exchange capability and its activity with AcAMP and CoA. As seen in Figure 6A, the D525N mutant exhibited only 1% of the activity of the WT enzyme in the complete reaction (kcatWT = 1.9 sec−1; kcatD525N = 0.02 sec−1). In contrast, the ATP-pyrophosphate exchange reaction was significantly less affected by the loss of charge at position 525 (Figure 6B). The mutant catalyzed the exchange reaction only 3.3 times slower than the WT, an observation similar to the S. enterica ACS D517 mutants [14]. Finally, when only the thiol ligase chemistry was explored, the D525N mutant was 6.8 times slower than the WT enzyme (Figure 6C). Together, these results suggest that with the D525N mutant that both partial reactions occur at rates slower than, but comparable to that of the WT enzyme, but the mutation considerably inhibits the ability of the enzyme to switch from the conformation that permits adenylation to the one that permits thioester formation. We thus ascribe the pH dependence of kcat to the protonation of D525, preventing the conformational change required for the overall reaction.
Figure 6.
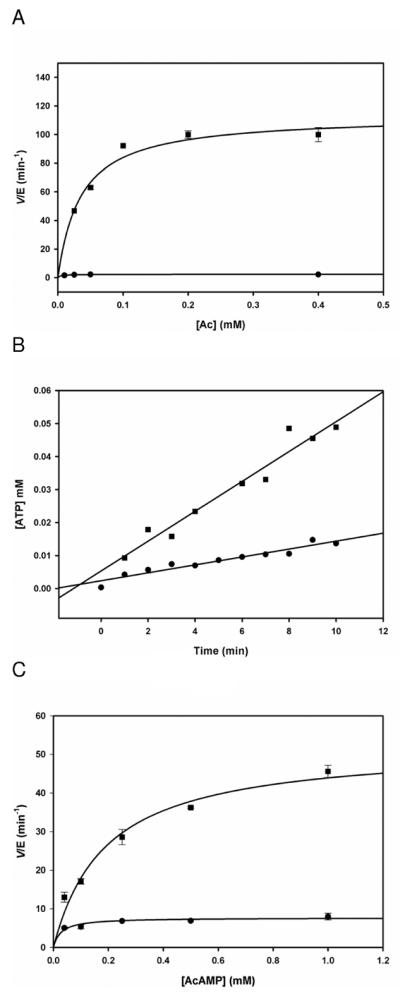
Kinetics of WT ACS (circles) and D525N ACS (squares). (A) In the presence of 1 mM ATP, 1 mM CoA and 0.025–0.4 mM Ac. Data was fitted equation 1 and yielded kWTcat = 1.9 ± 0.1 sec−1 and kD525Ncat = 0.02 ± 0.01 sec−1. (B) ATP/PPi exchange assay in the presence of 0.25 mM ATP, 0.25 mM Ac, 0.1 mM PPi, 0.04 μCi of P32 – labeled PPi. Representative of three experiments. (C) Kinetics of the second half reaction in the presence of 0.5 mM ATP, 1 mM CoA and 0.04 – 1 mM AcAMP.
Eubacterial acetyl-CoA synthetases were one of the first enzymes demonstrated to be post-translationally modified by acetylation (4, 18). In all cases studied to data, the single modification is on a lysine residue thought to interact with the α-P of bound ATP, and either electrostatically or sterically prevents the adenylation of acetate and thus inhibits the formation of AcCoA. In Mycobacterium tuberculosis, we previously demonstrated that the MtACS was similarly modified by the single protein acetyltransferase (PAT) in the genome (4), and that acetylation completely abolished the activity of the synthetase. In an additional regulatory wrinkle, the M. tuberculosis PAT contains an N-terminal cyclic nucleotide binding domain, and its activity is uniquely and strictly dependent on 3',5'-cyclic-AMP. We have also demonstrated that the geneome encodes a single NAD+-dependent sirtuin-like deacetylase, that can deacetylate acetylated-MtACS, with subsequent restoration of activity. The regulation of ACS in eubacteria is thought to prevent the unnecessary synthesis of AcCoA in an ATP-dependent process when organisms have access to carbon substrates that are easily converted into AcCoA. In M. tuberculosis, this situation is relevant after engulfment by human macrophages, where the organism maintains its metabolic equilibrium using host fatty acids and cholesterol (19). There are other fatty acyl synthetases that are similarly regulated in M. tuberculosis, including the fatty acyl synthetase implicated in mycobactin biosynthesis (20). Mycobacterium tuberculosis thus has evolved posttranslational modification, specifically enzyme acetylation, to adapt its metabolic activity to the various environments that it encounters and survives.
Supplementary Material
Acetyl-CoA synthetase is reversibly inactivated by acetylation of K617.
The kinetic mechanism was determined to be Bi Uni Uni Bi Ping Pong.
The adenylation reaction is specifically inhibited by K617 acetylation.
The reaction involves a conformational equilibrium dependent on D525.
Figure 5.
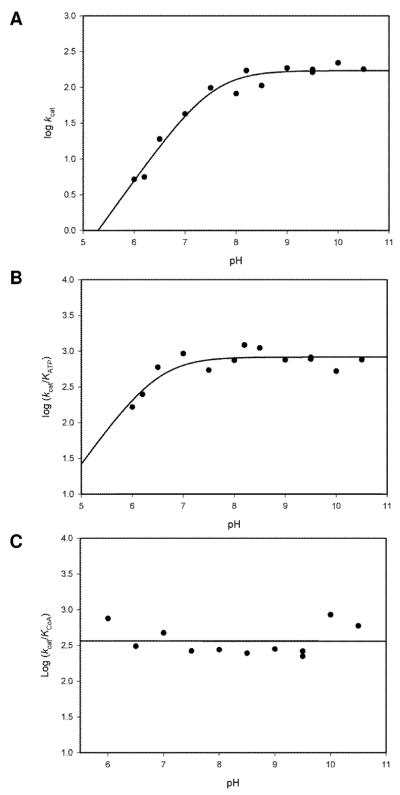
pH rate profiles. (A) pH dependence of kcat was assayed under saturating concentrations of ATP, Ac and CoA. Data was fitted to equation 7 and pKa = 7.5 ± 0.1 was obtained. (B) pH dependence of kcat /Km for ATP was assayed under saturating concentrations of Ac and CoA. Data was fitted to equation 7 and pKa = 6.5 ± 0.1 was obtained. (c) pH dependence of kcat /Km for CoA was assayed under saturating concentrations of ATP and Ac.
Acknowledgments
This work was supported by the National Institutes of Health Grant AI060899
Abbreviations
- MtACS
Mycobacterium tuberculosis Acetyl-CoA synthetase
- AcACS
acetylatedacetyl CoA synthetase
- Ac
acetate
- CoA
Coenzyme A
- AcCoA
acetyl Coenzyme A
- AcAMP
acetyl-adenylate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boshoff HI, Barry CE. A low-carb diet for a high-octane pathogen. Nat Med. 2005;11:599–600. doi: 10.1038/nm0605-599. [DOI] [PubMed] [Google Scholar]
- 2.Devlin TM. Textebook of Biochemistry with Clinical Correlation. 1986. pp. 211–225. [Google Scholar]
- 3.Schmelz S, Naismith JH. Adenylate-forming enzymes. Curr Opin Struct Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu H, Hegde SS, Blanchard JS. Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry. 2011;50:5883–5892. doi: 10.1021/bi200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 7.Somashekar BS, Amin AG, Rithner CD, Troudt J, Basaraba R, Izzo A, Crick DC, Chatterjee D. Metabolic profiling of lung granuloma in Mycobacterium tuberculosis infected guinea pigs: ex vivo 1H magic angle spinning NMR studies. J Proteome Res. 2011;10:4186–4195. doi: 10.1021/pr2003352. [DOI] [PubMed] [Google Scholar]
- 8.de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Crosby HA, Heiniger EK, Harwood CS, Escalante-Semerena JC. Reversible N epsilon-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol Microbiol. 2010;76:874–888. doi: 10.1111/j.1365-2958.2010.07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg P. Acyl adenylates; the synthesis and properties of adenyl acetate. J Biol Chem. 1956;222:1015–1023. [PubMed] [Google Scholar]
- 11.Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40:12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 12.Fan F, Luxenburger A, Painter GF, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium smegmatis cysteine ligase (MshC) Biochemistry. 2007;46:11421–11429. doi: 10.1021/bi7011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santi DV, Pena VA. Order of substrate binding to tyrosyl-tRNA synthetase of Escherichia coli B. FEBS Lett. 1971;13:157–160. doi: 10.1016/0014-5793(71)80224-7. [DOI] [PubMed] [Google Scholar]
- 14.Jogl G, Tong L. Crystal structure of yeast acetyl-coenzyme A synthetase in complex with AMP. Biochemistry. 2004;43:1425–1431. doi: 10.1021/bi035911a. [DOI] [PubMed] [Google Scholar]
- 15.Reger AS, Carney JM, Gulick AM. Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry. 2007;46:6536–6546. doi: 10.1021/bi6026506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 17.Horswill AR, Escalante-Semerena JC. Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: residue Lys592 is required for propionyl-AMP synthesis. Biochemistry. 2002;41:2379–2387. doi: 10.1021/bi015647q. [DOI] [PubMed] [Google Scholar]
- 18.Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vergnolle O, Xu H, Blanchard JS. Mechanism and regulation of mycobactin fatty acyl-AMP ligase FadD33. J Biol Chem. 2013;288:28116–28125. doi: 10.1074/jbc.M113.495549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.