

Significance
To elucidate their role in Behçet disease (BD), we used imputation to analyze MHC class I (MHC-I) alleles and amino acids in a large case-control collection. Several MHC-I alleles influenced BD susceptibility, some conferring risk and others affording protection. The association of MHC-I with BD was mapped to six positions around the peptide-binding groove and to one position in the signal peptide. These residues are critical in defining the peptide-binding specificity of MHC-I molecules, and they also affect the engagement of MHC-I molecules by killer immunoglobulin-like receptors on natural killer and T cells. These data implicate peptide binding by MHC-I in BD pathogenesis and suggest that altered regulation of cytotoxic cells by MHC-I may be pathogenic in BD.
Keywords: HLA imputation, autoinflammation, antigen presentation, killer immunoglobulin-like receptors, natural killer cells
Abstract
The HLA protein, HLA-B*51, encoded by HLA-B in MHC, is the strongest known genetic risk factor for Behçet disease (BD). Associations between BD and other factors within the MHC have been reported also, although strong regional linkage disequilibrium complicates their confident disentanglement from HLA-B*51. In the current study, we examined a combination of directly obtained and imputed MHC-region SNPs, directly obtained HLA-B locus types, and imputed classical HLA types with their corresponding polymorphic amino acid residues for association with BD in 1,190 cases and 1,257 controls. SNP mapping with logistic regression of the MHC identified the HLA-B/MICA region and the region between HLA-F and HLA-A as independently associated with BD (P < 1.7 × 10−8). HLA-B*51, -A*03, -B*15, -B*27, -B*49, -B*57, and -A*26 each contributed independently to BD risk. We directly examined rs116799036, a noncoding SNP upstream of HLA-B that was recently suggested to underlie the association of HLA-B*51 with BD, but we were unable to replicate that finding in our collection. Instead, we mapped the BD association to seven MHC class I (MHC-I) amino acid residues, including anchor residues that critically define the selection and binding of peptides to MHC-I molecules, residues known to influence MHC-I–killer immunoglobulin-like receptor interactions, and a residue located in the signal peptide of HLA-B. The locations of these variants collectively implicate MHC-I peptide binding in the pathophysiology of BD. Furthermore, several lines of evidence suggest a role for altered regulation of cellular cytotoxicity in BD pathogenesis.
Behçet disease (BD) is a multisystem inflammatory disease of complex inheritance with a clinical course marked by recurrent episodes of oral and genital ulceration, severe ocular inflammation often leading to visual impairment or blindness, and a range of inflammatory lesions of the skin and the gastrointestinal, neurologic, and circulatory systems (1). The predominant BD susceptibility locus is the MHC on chromosome 6 (2, 3), which contains the strongest known risk factor for BD, the MHC class I (MHC-I) allele HLA-B*51 (2–5). Several recent studies have expanded the list of genes or loci implicated in the pathophysiology of BD, which now includes HLA-B, IL10, IL23R, HLA-A, CCR1, STAT4, endoplasmic reticulum amino peptidase 1 (ERAP1), the killer lectin-like receptor cluster on chromosome 12, and, most recently, TLR4 and MEFV (2, 3, 6, 7). Although these genetic studies of BD have provided new clues and insights into the pathogenesis of BD, none has provided a thorough accounting of the individual risk factors within the MHC. The lack of such a study likely reflects the absence of a BD study population of adequate size to overcome the strong linkage disequilibrium (LD) and to disentangle from HLA-B*51 the additional risk factors within the MHC.
Multiple lines of evidence suggest that sources of BD risk, in addition to HLA-B*51, exist within the MHC. This evidence begins in the HLA-B locus, where associations between BD and several alleles in addition to HLA-B*51 have been reported (8–10). It also has been argued that variants in or around MHC class I polypeptide-related sequence A (MICA), the centromeric neighbor of HLA-B that encodes the MHC-I chain-related sequence A, contribute to BD susceptibility (11). However, efforts to parse the effects of MICA and HLA-B alleles definitively have been confounded by their particularly strong LD (11–14). Additionally, HLA-A has been identified as a BD susceptibility locus in numerous studies (2, 3, 14–17), and it has been suggested that HLA-C contributes to BD risk, as well (14).
To understand better the sources of BD risk within the MHC, we have analyzed directly ascertained and imputed SNP genotypes, together with HLA type and amino acid data from a very large and meticulously assembled collection of Turkish subjects with BD and geographically matched, healthy Turkish individuals. Using stepwise and multivariate logistic regression, conditional analysis, and haplotype analysis, we sought to characterize the range of genetic risk factors for BD contained within the MHC.
Results
Multiple HLA-B Alleles Independently Influence Susceptibility to BD.
We performed association testing of directly ascertained two-digit HLA-B locus types in 1,190 BD cases and 1,257 healthy subjects from Turkey. Our results affirmed that HLA-B*51 is the largest single risk factor for BD, acting in an additive fashion with an odds ratio (OR) of 3.0 per copy (P = 1.3 × 10−55) (Table 1 and Table S1). In addition to HLA-B*51, both the supervised conditional analysis (Table 1) and the unsupervised stepwise forward logistic regression analysis (Table S2) of HLA-B alleles in the full collection identified significant, independent effects of HLA-B*15 and HLA-B*27 on the risk of BD. HLA-B*15 also was significantly associated with BD in the HLA-B*51–negative subset of the collection, which included 487 cases and 889 controls (P = 3.4 × 10−5) (Table 1). Conditional analysis accounting for the effect of HLA-B*15 on BD risk in the HLA-B*51–negative subset unveiled a significant, protective effect of HLA-B*49 on BD (pregressor = 1.1 × 10−4) (Table 1), and, as is consistent with these findings, the model of BD risk generated by forward stepwise logistic regression included HLA-B*15 and HLA-B*49 (Table S2). Additionally, HLA-B*57 trended toward association with BD in analyses of the full collection and of the HLA-B*51–negative subset of the collection (Table 1).
Table 1.
Additive model association testing and stepwise conditional analysis of directly ascertained HLA-B antigens in BD
| Covariates | Risk allele | P value† | OR (95% CI) |
| Full collection | |||
| None | HLA-B*51 | 1.3 × 10−55 | 3.0 (2.6, 3.4) |
| HLA-B*51 | HLA-B*15 | 1.0 × 10−5 | 1.9 (1.4, 2.5) |
| HLA-B*51 | HLA-B*27 | 1.0 × 10−3 | 1.7 (1.2, 2.3) |
| + HLA-B*15 | |||
| HLA-B*51 | HLA-B*49 | 7.5 × 10−3 | 0.6 (0.4, 0.9) |
| + HLA-B*15 | HLA-B*57 | 9.4 × 10−3 | 1.7 (1.1, 2.6) |
| + HLA-B*27 | |||
| HLA-B*51-negative subset | |||
| None | HLA-B*15 | 3.4 × 10−5 | 2.0 (1.4, 2.7) |
| HLA-B*15 | HLA-B*49 | 1.1 × 10−4 | 0.4 (0.2, 0.7) |
| HLA-B*15 | HLA-B*57 | 5.5 × 10−3 | 2.0 (1.2, 3.2) |
| + HLA-B*49 | HLA-B*27 | 6.5 × 10−3 | 0.4 (0.2, 0.7) |
After correcting for 31 directly ascertained HLA-B types, significance was defined as P < 1.6 × 10−3.
HLA-B*51 Is the Primary BD Risk Factor in the HLA-B/MICA Region.
Association testing of 32,689 directly acquired and imputed SNPs from the MHC region revealed that the strongest peak of association included HLA-B and MICA and consisted of more than 775 BD-associated SNPs whose associations exceeded the threshold for genome-wide significance (P < 1.7 × 10−8), with the most significant associations observed under the additive model (Fig. 1). Among these was the most strongly BD-associated SNP in the study, rs79556279 [padditive = 2.2 × 10−50, OR 2.7 (95% confidence interval, CI, 2.3, 3.1)], which was located 4.9 kb 5′ of HLA-B. After controlling for the effect of rs79556279, we found that no other SNP in the HLA-B/MICA region was significantly associated with BD (Fig. 1A). Association testing of MHC-region SNPs conditioned on the effect of HLA-B*51 similarly identified no significant residual association in the HLA-B/MICA region (Fig. 1B), and, moreover, rs79556279 was in strong LD with HLA-B*51 [expectation–maximization r2 (r2EM) = 0.92; expectation-maximization pairwise linkage disequilibrium (D′EM) = 0.96], indicating that the effect of HLA-B*51 underlies the observed effect of rs79556279. In fact, the majority of BD-associated markers in the HLA-B/MICA region were in moderate to strong LD with HLA-B*51 (Fig. 1C).
Fig. 1.
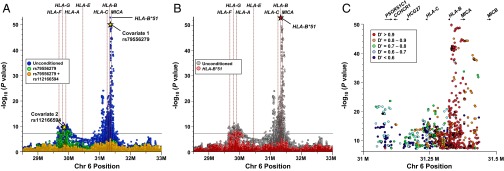
HLA-B*51 is the predominant risk allele, but variants between HLA-F and HLA-A are independently associated with BD. (A and B) The results of association testing and stepwise conditional analysis of imputed MHC region SNPs in 1,190 BD cases and 1,257 healthy control subjects from Turkey are displayed in A. Conditional analysis accounting for the effect of HLA-B*51 (B, red dots) produced a pattern of residual association virtually identical to that seen after conditioning for rs79556279 (A, green dots). (C) Association testing results of BD-associated SNPs in proximity to HLA-B/MICA were plotted, and data points were color-coded to demonstrate D′ of each SNP with HLA-B*51.
To examine the role of rs116799036, a noncoding SNP recently proposed to underlie the effect of HLA-B*51 on BD risk (14), we directly genotyped this marker in our collection. The association of rs116799036 with BD was seven orders of magnitude weaker than that of HLA-B*51 (Table S3). Furthermore, in contrast to a previous report (14), the association of HLA-B*51 with BD remained significant even after conditioning for the effect of rs116799036 (pregressor = 9.2 × 10−8; Table S3).
In an effort to localize further the source(s) of BD risk within the HLA-B/MICA region, we analyzed haplotypes of HLA-B/MICA region SNPs [minor allele frequency (MAF) >0.2] with HLA-B*51 using Haploview (18). We identified one haplotype, consisting of 48 HLA-B/MICA region SNPs and HLA-B*51, that was strongly associated with BD [P = 9.4 × 10−47; OR 2.81 (95% CI 2.44, 3.25)] (Fig. S1). Interestingly, a nearly identical form of this haplotype that bore the same 48 SNPs but lacked HLA-B*51 was present at equal frequencies in BD patients and healthy controls, thereby conferring no demonstrable effect on BD risk (P = 0.764).
The Region Between HLA-F and HLA-A Is an HLA-B*51-Independent BD Susceptibility Locus.
In addition to the HLA-B/MICA region, association testing of MHC-region SNPs under the additive model identified two other BD-associated regions, one located telomeric to HLA-C and a second that included HLA-F, -G, -H, and -A (Fig. 1). Conditional analysis correcting for the effect of rs79556279 revealed that variants in the HLA-F/HLA-A region were independently associated with BD, whereas those in the region telomeric to HLA-C were not (Fig. 1 A and B). After controlling for the effect of rs79556279, the HLA-F/HLA-A region included 113 SNPs whose associations with BD exceeded the genome-wide significance threshold, the strongest of which was rs112166594 [padditive = 8.3 × 10−10, OR 0.56 (95% CI 0.48, 0.67)]. The three regional SNPs most strongly associated with BD were located nearest to HLA-H, HLA-G, and HLA-A, respectively, whereas the majority of BD-associated SNPs in this region were located nearest to HLA-A. Finally, conditional analysis accounting for the effects of both rs79556279 and rs112166594 failed to identify any SNP associations whose significance met even a nominally significant threshold corrected for the 2,847 directly typed SNPs (P < 1.7 × 10−5) (Fig. 1A).
Multiple HLA-B and HLA-A Alleles Independently Influence the Risk of Developing BD.
Directly ascertained MHC SNPs were used to infer HLA types at the classical MHC-I and MHC-II loci using a large reference panel of European ancestry, as previously described (19). The concordance rate between imputed and directly ascertained two-digit HLA-B*51 alleles was 98.6%, the overall concordance rate among two-digit HLA-B alleles was 95.9%, and their allelic frequencies were highly correlated (r2 = 0.9913) (Fig. S2). Univariate logistic regression of imputed HLA-type dosage data affirmed that HLA-B*51 was the strongest genetic risk factor of BD [P = 3.4 × 10−58, OR 3.3 (95 % CI 2.8, 3.8)] (Table 2 and Table S1) with metrics of association nearly identical to those of imputed HLA-B*51:01 [P = 5.1 × 10−58, OR 3.3 (95 % CI 2.8, 3.8)] (Tables S1 and S4) and very similar to those of directly typed HLA-B*51 [P = 1.3 × 10−55, OR 3.0 (95 % CI 2.6, 3.4)] (Table 1 and Table S1). To identify additional MHC-I alleles that influence BD risk, we performed conditional analysis and stepwise forward logistic regression of imputed HLA types in the full collection and in the HLA-B*51–negative subset (Table 2). After controlling for the effect of imputed HLA-B*51 in the full collection, HLA-A*03 was significantly protective against BD (Table 2 and Table S2). We found that HLA-A*03 was in strong LD with the SNP most strongly BD-associated in the HLA-A region, rs112166594 (r2EM = 0.99, D′EM = 1.0). Additionally, HLA-B*15, -B*49, -A*26, and -B*27 each trended toward association with BD after correcting for the effects of HLA-B*51 and -A*03, although without reaching statistical significance (Table 2). In the HLA-B*51–negative subset of the population, stepwise forward logistic regression of imputed classical MHC alleles produced a model of BD risk that included HLA-A*03 and -B*49 (Table S2). In addition to HLA-A*03 and HLA-B*49, stepwise conditional analysis also demonstrated a trend toward association between BD and HLA-B*15 and HLA-A*26 (Table 2). Analogous analyses of imputed four-digit MHC alleles found that, upon controlling for the effect of imputed HLA-B*51:01 in the full study population, HLA-A*03:01 was significantly protective against BD [P = 2.4 × 10−6, OR 0.6 (95 % CI 0.4, 0.7)] (Table S2). Further, in the HLA-B*51–negative population, HLA-B*49:01 was significantly protective against BD [P = 1.2 × 10−5, OR 0.6 (95 % CI 0.4, 0.8)] (Table S2).
Table 2.
Additive model association testing and stepwise conditional analysis of imputed two-digit classic HLA alleles in BD
| Covariates | Risk allele | P value† | OR (95% CIl) |
| Full collection | |||
| None | HLA-B*51 | 3.4 × 10−58 | 3.3 (2.8, 3.8) |
| HLA-B*51 | HLA-A*03 | 4.0 × 10−8 | 0.6 (0.5, 0.7) |
| HLA-B*51 | HLA-B*15 | 8.7 × 10−4 | 1.6 (1.2, 2.1) |
| + HLA-A*03 | |||
| HLA-B*51 | HLA-B*49 | 2.3 × 10−3 | 0.6 (0.4, 0.8) |
| + HLA-A*03 | HLA-A*26 | 3.5 × 10−3 | 1.5 (1.1, 2.0) |
| + HLA-B*15 | HLA-B*27 | 5.8 × 10−3 | 1.6 (1.1, 2.2) |
| HLA-B*51–negative subset | |||
| None | HLA-B*49 | 1.1 × 10−5 | 0.3 (0.2, 0.6) |
| HLA-B*49 | HLA-A*03 | 1.1 × 10−4 | 0.6 (0.5, 0.8) |
| HLA-B*49 | HLA-B*15 | 4.5 × 10−3 | 1.6 (1.1, 2.1) |
| + HLA-A*03 | HLA-A*26 | 6.7 × 10−3 | 1.6 (1.1, 2.2) |
After correcting for 101 imputed two-digit classical HLA alleles, significance was defined as P < 5.0 × 10−4.
Haplotypes of imputed two-digit MHC-I alleles with frequencies >0.01 were assembled with the EM algorithm, as implemented in SVS7 (Table S5). With one exception, all HLA-B*51–containing haplotypes conferred risk of BD, and all HLA-A*03–containing haplotypes were protective against BD (Table S6). The sole exception to this rule was the haplotype bearing both HLA-A*03 and HLA-B*51, the only haplotype bearing either allele that did not influence BD risk, illustrating that the protective effect of HLA-A*03 and the risk effect of HLA-B*51 are independent and able to counteract one another (Table S6).
Five Amino Acid Residues of HLA-B and Two Residues of HLA-A Influence BD Risk.
In an effort to identify a motif that may explain the variety of protective and risk effects differentially conferred by HLA-B and HLA-A alleles in BD, polymorphic amino acid residues were derived from the imputed MHC type data, as previously described (19). Stepwise conditional analysis of the polymorphic amino acid positions of HLA-B in the full study population revealed that positions 97, 116, 152, and 67 each significantly and independently influenced the risk of developing BD (Table 3 and Fig. 2). Similar analyses of HLA-A amino acid positions identified significant and independent associations between residues 161 and 97 of HLA-A and BD risk (Table 3 and Fig. 2). When amino acids from HLA-B and HLA-A were evaluated together by stepwise conditional analysis, a model of BD risk was generated that included five of these six positions as independent influences on BD risk (Table 3). In further conditional analyses of the polymorphic amino acid positions of HLA-B and/or HLA-A in the HLA-B*51–negative subset (Table 3), residue −10 of HLA-B and residue 161 of HLA-A were significantly and independently associated with BD, and residue 149 of HLA-A also trended toward association with BD.
Table 3.
Stepwise conditional analysis of HLA-B and HLA-A amino acid positions in BD
| HLA locus | Covariates | Effect amino acids | Pregressor | OR (95% CI) |
| Full collection* | ||||
| HLA-B only† | None | Thr 97 | 1.5 × 10−38 | 2.3 (2.0, 2.7) |
| 97 | Leu 116 | 2.5 × 10−10 | 0.5 (0.4, 0.6) | |
| 97, 116 | Glu 152 | 6.2 × 10−8 | 1.6 (1.3, 1.9) | |
| 97, 116, 152 | Phe 67 | 2.0 × 10−4 | 1.4 (1.2, 1.6) | |
| HLA-A only‡ | None | Glu 161 | 2.0 × 10−10 | 0.5 (0.4, 0.7) |
| 161 | Arg 97 | 1.3 × 10−4 | 1.3 (1.1, 1.4) | |
| HLA-B and -A§ | None | Thr 97 (B) | 1.5 × 10−38 | 2.3 (2.0, 2.7) |
| B97 | Leu 116 (B) | 2.5 × 10−10 | 0.5 (0.4, 0.6) | |
| B97, B116 | Glu 161 (A) | 3.0 × 10−8 | 0.6 (0.5, 0.7) | |
| B97, B116 + A161 | Glu 152 (B) | 3.2 × 10−8 | 1.6 (1.4, 1.9) | |
| B97, B116, A161 + B152 | Phe 67 (B) | 2.2 × 10−4 | 1.4 (1.2, 1.6) | |
| HLA-B*51–negative subset* | ||||
| HLA-B only† | None | Ala −10 | 3.3 × 10−5 | 0.7 (0.6, 0.8) |
| HLA-A only‡ | None | Glu 161 | 6.1 × 10−5 | 0.6 (0.4, 0.8) |
| 161 | Glu 149 | 7.8 × 10−3 | 1.5 (1.1, 1.9) | |
| HLA-B and -A§ | None | Ala −10 (B) | 3.3 × 10−5 | 0.7 (0.6, 0.8) |
| B-10 | Glu 161 (A) | 2.5 × 10−10 | 0.5 (0.4, 0.6) | |
| B-10 + A161 | Glu 149 (A) | 1.5 × 10−3 | 1.6 (1.2, 2.1) | |
The inclusion of Bw4 as a potential covariate did not alter the results.
Cutoff P value adjusted for 69 HLA-B positions = 7.2 × 10−4.
Cutoff P value adjusted for 68 HLA-A positions = 7.4 × 10−4.
Cutoff P value adjusted for 137 HLA-A and HLA-B positions = 3.6 × 10−4.
Fig. 2.

Individual amino acids within HLA-A and HLA-B proteins influence the risk of BD. The allele frequencies of amino acid positions 97, 116, 152, 67, and −10 in HLA-B and positions 161 and 97 in HLA-A are plotted for cases (red) and controls (blue), and univariate ORs are displayed above the bars.
Residues 97, 116, 152, and 67 of the HLA-B protein are each located in the MHC-I antigen-binding groove (Fig. 3), where they act to define the sizes and shapes of individual peptide residues accommodated by specific MHC-I molecules (20, 21). Collectively these four MHC-I residues physically contact six of nine peptide residues (21), and residues 67 and 116 are critical anchor residues of particular importance in defining the peptide specificity of MHC-I antigen-binding grooves through their interactions with peptide positions P2 and P9, respectively (20, 21). Positions 67 and 116 also are critical determinants of the interactions of HLA-B molecules with the killer immunoglobulin-like receptors (KIR) KIR3DL1 and KIR3DS1, which regulate the activation of natural killer (NK) cells and CD8+ cytotoxic T lymphocytes (CTLs) (22). In addition, residue 67 is one of two residues at which HLA-B*51 differs from the nearly identical HLA-B protein HLA-B*52, which confers no significant effect on BD risk [P = 0.19, OR 0.77 (95 % CI 0.5, 1.1)], further emphasizing the importance of this position. One BD-associated residue was located distant from the antigen-binding groove at position −10 of the HLA-B signal peptide. Interestingly, the HLA-B signal peptide also regulates CTL and NK cell activation, although it does so by a mechanism independent of peptide binding and KIR engagement by HLA-B (23).
Fig. 3.

BD-associated positions within MHC-I molecules cluster around the antigen-binding groove. 3D modeling of HLA-B*5101 (A) and HLA-A*0301 (B) demonstrates the clustering of BD-associated amino acid positions (red balls) in and around the antigen-binding grooves of HLA-B and HLA-A. The peptide backbone and side chains of the epitope are displayed in green. This figure was prepared with PyMol, using Protein Data Bank entries 2XPG (40) and 1E27 (41).
MHC-I molecules bearing the Bw4 epitope of the α1-binding pocket, defined by leucine and arginine at positions 82 and 83, bind to a specific subset of KIRs, sometimes in an antigen-dependent fashion (24, 25). To investigate a potential role for Bw4 in BD, Bw4+ alleles of HLA-B and HLA-A were examined. Bw4 epitopes in HLA-B molecules were a weak risk factor for BD in univariate analysis of the full collection, but Bw4 epitopes present in HLA-A molecules conferred no risk of BD (Table S7). Upon conditioning for the effect of HLA-B*51 in the full collection, no Bw4 epitope was associated with BD; moreover, Bw4 had no effect on BD risk in the HLA-B*51–negative subset of the population (Table S7). Most importantly, addition of Bw4 variables to our stepwise analysis of MHC-I amino acid positions showed that Bw4 was not an independent risk factor for BD (see footnotes of Table 3).
In all, multivariate logistic regression analyses of classical MHC amino acid data identified seven MHC-I amino acid positions as risk factors for BD, implicating peptide binding by both HLA-B and HLA-A and the signal peptide of HLA-B in the pathophysiology of BD.
Discussion
By applying contemporary techniques to the examination of a large, well-matched collection of BD cases and healthy controls, this study has provided the most extensive interrogation of the HLA locus in BD to date. Our data have brought clarity to questions surrounding the relationship of HLA-B*51 with BD while also bringing to light additional MHC-I alleles that influence BD susceptibility. Moreover, this study has, for the first time to our knowledge, mapped the association of MHC-I molecules with BD to specific HLA-B and HLA-A amino acid residues, most of which cluster in the antigen-binding groove at positions known to influence strongly the selection and binding of peptide antigens by MHC-I molecules. These findings have important implications for the pathophysiology of BD, because peptide binding by MHC-I directly affects (i) the folding and stability of peptide–MHC-I complexes, which in turn determine their surface expression vs. retention in the endoplasmic reticulum; (ii) the recognition of peptide–MHC-I complexes by antigen-specific T-cell receptors on CTLs; and (iii) the engagement of KIRs on CTLs and NK cells by peptide–MHC-I complexes (26–30).
The relationship between HLA-B*51 and BD was first identified four decades ago; since then this association has been replicated in nearly every genetic study of BD. We have affirmed this relationship in our study of a very large collection of Turkish BD patients, defining the estimated effect size as OR 3.0 (95% CI 2.6, 3.4) per allele in an additive model. HLA-B*51 is in strong LD with genetic variants spanning the region that includes HLA-B and MICA (Fig. 1C), many of which are associated with BD. Using haplotype analysis of HLA-B*51 and SNPs in the HLA-B/MICA region, we demonstrated the importance of HLA-B*51 itself in BD risk. This observation is contrary to a recent report by Hughes et al. (14) that suggested that, instead of HLA-B*51, rs116799036, a noncoding variant between HLA-B and MICA, was the true source of BD risk in this region. In our study, HLA-B*51 was much more strongly associated with BD than was any SNP, including rs116799036 (Fig. 1B and Table S3); moreover, HLA-B*51 continued to confer significant risk of BD, even after controlling for the effect of rs116799036 (Table S3).
In comparing the results of our study with that of Hughes et al. (14), there are several important considerations. First, our study examined nearly twice as many BD cases as did the study by Hughes et al. and thus has greater statistical power to assess BD risk factors. Second, we assembled our study population prospectively with pairwise geographic matching of controls to cases to minimize population stratification (2). Third, we used a proven HLA imputation methodology that leverages a large reference panel of mixed European ancestry (19, 31), whereas Hughes et al. used a different algorithm that uses a reference panel of Northern European ancestry (32). Therefore, it is possible that differences in the imputation methodologies, differences in the ancestries of the imputation reference panels, or even different degrees of population stratification within the study populations may have produced the observed differences between our study and that of Hughes et al. The large sample size of our study, our approach to addressing population stratification, and the demonstrated accuracy of our HLA imputation strongly support the conclusion that coding variants account for a major part of the association of HLA-B*51 with BD.
Associations between other HLA-B types and BD have been reported (8–10), as have associations between BD and both HLA-A and HLA-C alleles (2, 3, 14–17). In our study, we observed that HLA-B*15, -B*27, and -B*57 were independent susceptibility alleles for BD, consistent with earlier reports. We have found suggestive evidence that HLA-A*26 is an independent risk factor for BD, but, contrary to earlier reports in other populations, neither HLA-A*02 nor -A*33 influenced BD risk in our study. Similarly, we found no HLA-C region SNP or allele with an HLA-B*51–independent effect on BD risk. Interestingly, we have found that HLA-B*49 and -A*03 are independent susceptibility alleles that are protective against the development of BD. Although the mechanism through which these alleles protect against BD is unclear, a closer examination of these alleles may provide insights into BD pathogenesis. HLA-A*03 and -B*51 are independent alleles that, when coinherited, produce neutral risk of BD (Table S6). HLA-A*03 also is unique among BD-associated molecules in its ability to engage the inhibitory receptor KIR3DL2, and it is possible that enhanced cytotoxic inhibition is protective against BD. The coinheritance of HLA-B*49 with HLA-B*51 did not reduce the risk of BD conferred by HLA-B*51. It also is worth noting that the HLA-B*49 and -B*51 proteins both contain the Bw4 motif, although differing at four of the five BD-associated amino acid positions.
Through the examination of amino acid sequences from MHC alleles, intramolecular mapping of disease-associated MHC amino acid positions has clarified the MHC associations in both rheumatoid arthritis (19) and host control of HIV infection (31). In BD, we have mapped the association of MHC-I molecules to positions that critically influence the peptide-binding specificity of the MHC molecule, particularly the anchor residues at positions 67 and 116, providing the most definitive data to date implicating peptide binding by MHC-I in BD. The importance of peptide binding is supported further by our recent observation that variants of ERAP1, an important enzyme in the processing of peptides for presentation by MHC-I, confers risk of BD through epistasis with HLA-B*51 (6). Collectively, these data support a role for one or more pathogenic peptides in BD, vindicating the investigation of infectious agents, exogenous peptides, endogenous autoantigens, and molecular mimicry in BD pathogenesis (33).
In addition to implicating peptide–MHC-I binding in BD, our results suggest that the regulation of CTL and NK activation by MHC-I is involved in the pathogenesis of BD. MHC-I molecules inherently regulate CTL activation by presenting peptide antigens to T-cell receptors; however, many BD-associated MHC-I variants in our study also regulate NK and CTL activation through interactions with the inhibitory KIR3DL1 or KIR3DL2 receptors or with the excitatory KIR3DS1 receptor (22, 24, 29). Furthermore, positions 67 and 116 (and to a lesser extent position 97) of the HLA-B antigen-binding groove are critical determinants of the binding specificity of some HLA-B alleles for KIR3DL1 or KIR3DS1 receptors (22). Beyond the MHC-I antigen-binding groove, we identified an association between BD and a variant of the HLA-B signal peptide that independently regulates CTL and NK-cell activation through HLA-E and the C-type lectin-like heterodimeric receptors, CD94/NKG2 (34–36). Coincidentally, we previously have identified an association between BD and KLCR4, which encodes the C-type lectin-like receptor, NKG2F (6).
Although the nature of BD is clearly inflammatory, it has been debated whether this inflammation is autoimmune, resulting from inappropriate adaptive immune activation and broken immune tolerance, or autoinflammatory, resulting from inappropriately activated innate immune pathways. Although the strongest risk factor for BD remains HLA-B*51, which encodes what usually is considered an adaptive immune molecule, several recent genetic studies have clearly linked innate immune mechanisms to BD risk, supporting an autoinflammatory contribution to its pathogenesis (6, 7). Our data bring additional insight to this discussion by offering a plausible mechanism through which MHC-I could act to confer risk of BD through the regulation of both innate NK cells and adaptive CTLs.
In summary, we have identified an array of MHC-I alleles that are associated with BD. Within MHC-I molecules, we have identified a group of amino acid positions that strongly influence the risk of developing BD, many of which are located at sites known to influence both peptide binding and the interactions between MHC-I molecules and receptors on CTL and NK cells. Further, our identification of a BD-associated residue in the signal peptide of HLA-B independently links BD pathogenesis to cytotoxicity. Taken together, these data further implicate MHC-I peptide binding in the pathophysiology of BD and provide several lines of evidence independently connecting the regulation of cell-mediated cytotoxicity to BD risk.
Materials and Methods
Patients.
BD patients fulfilling the International Study Group Criteria for BD (37) and geographically matched, healthy blood bank control subjects were enrolled in the study at the BD clinic of the Istanbul Faculty of Medicine, Division of Rheumatology, as previously described (2).
SNP Genotyping and Haplotype Analysis.
SNP genotype data were generated using Infinium Human CNV370 arrays (Illumina) and were processed with SVS7 (Golden Helix), as previously described (2). The subset of markers residing within the MHC (n = 2,832) were extracted for subsequent analyses. Haplotypes of SNPs from the HLA-B/MICA region with MAF >0.2 and HLA-B locus types were assembled manually, and haplotype association testing was performed by χ2 testing, as implemented in Haploview software (18). When necessary, validation genotyping was performed with custom MassArray assays (Sequenom).
Direct Typing of the HLA-B Locus.
Two-digit HLA-B locus typing of genomic DNA was performed as previously described (2). Associations between BD and HLA-B types were assessed using numeric regression in SVS7. Stepwise logistic regression with forward selection and conditional analysis of HLA-B types also were performed using SVS7. After correcting for 31 observed HLA-B types, significance was defined as P < 1.6 × 10−3.
SNP Genotype Imputation.
Using the 2,832 directly ascertained MHC region SNPs as a foundation, SNP imputation was performed using IMPUTE2 (38) software and using the 1,000 Genomes Project phase 1 integrated dataset as the reference dataset, as described (39). Imputation accuracy was internally assessed by independently masking and imputing every genotyped SNP, from which the overall concordance rate was determined to be 99.6%. Imputed SNP data were filtered to remove rare markers and markers that were imputed with poor quality using SVS7. Association testing and conditional analyses were performed using SNPTESTv2. Significance was determined using the genome-wide significance threshold (P < 5 × 10−8), adjusted for three models (P < 1.7 × 10−8).
Imputation of Classical HLA Types and Amino Acid Residues.
The directly ascertained MHC SNPs also were used to impute classical HLA alleles and their corresponding amino acid sequences using SNP2HLA and reference data collected by the Type I Diabetes Genetic Consortium, as described (19). Briefly, this dataset included a panel of 2,537 MHC SNPs that were selected to tag the entire MHC region, together with classical types for eight MHC-I and MHC-II loci at four-digit resolution in 2,767 unrelated individuals of European ancestry. All SNPs, amino acids, and HLA types in the reference panel were encoded as biallelic markers. Multivariate logistic regression analysis of classical HLA types was undertaken using PLINK-format dosage data in SVS7, and significance was determined using a Bonferroni correction for 101 imputed HLA types (P < 5 × 10−4). Haplotypes of two-digit MHC-I alleles were generated using the EM algorithm, and haplotype association testing was performed with SVS7.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Human Genome Research Institute of the National Institutes of Health (NIH) and the Istanbul University Research fund. P.I.W.d.B. received support from NIH Grant N1R01AR062886-01 and from the Netherlands Organization for Scientific Research Project Number 016.126.354.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 8706.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406575111/-/DCSupplemental.
References
- 1.Sakane T, Takeno M, Suzuki N, Inaba G. Behçet’s disease. N Engl J Med. 1999;341(17):1284–1291. doi: 10.1056/NEJM199910213411707. [DOI] [PubMed] [Google Scholar]
- 2.Remmers EF, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat Genet. 2010;42(8):698–702. doi: 10.1038/ng.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizuki N, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet’s disease susceptibility loci. Nat Genet. 2010;42(8):703–706. doi: 10.1038/ng.624. [DOI] [PubMed] [Google Scholar]
- 4.Ono S, Aoki K, Sugiura S, Nakayama E, Itakura K. Letter: HL-A5 and Behçet’s disease. Lancet. 1973;2(7842):1383–1384. doi: 10.1016/s0140-6736(73)93343-6. [DOI] [PubMed] [Google Scholar]
- 5.Ohno S, et al. Close association of HLA-Bw51 with Behçet’s disease. Arch Ophthalmol. 1982;100(9):1455–1458. doi: 10.1001/archopht.1982.01030040433013. [DOI] [PubMed] [Google Scholar]
- 6.Kirino Y, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013;45(2):202–207. doi: 10.1038/ng.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirino Y, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behçet disease. Proc Natl Acad Sci USA. 2013;110(20):8134–8139. doi: 10.1073/pnas.1306352110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choukri F, Chakib A, Himmich H, Hüe S, Caillat-Zucman S. HLA-B*51 and B*15 alleles confer predisposition to Behçet’s disease in Moroccan patients. Hum Immunol. 2001;62(2):180–185. doi: 10.1016/s0198-8859(00)00249-4. [DOI] [PubMed] [Google Scholar]
- 9.Gül A, et al. A weak association of HLA-B*2702 with Behçet’s disease. Genes Immun. 2002;3(6):368–372. doi: 10.1038/sj.gene.6363863. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad T, et al. Mapping the HLA association in Behçet’s disease: A role for tumor necrosis factor polymorphisms? Arthritis Rheum. 2003;48(3):807–813. doi: 10.1002/art.10815. [DOI] [PubMed] [Google Scholar]
- 11.Mizuki N, et al. Triplet repeat polymorphism in the transmembrane region of the MICA gene: A strong association of six GCT repetitions with Behçet disease. Proc Natl Acad Sci USA. 1997;94(4):1298–1303. doi: 10.1073/pnas.94.4.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ota M, et al. The critical region for Behçet disease in the human major histocompatibility complex is reduced to a 46-kb segment centromeric of HLA-B, by association analysis using refined microsatellite mapping. Am J Hum Genet. 1999;64(5):1406–1410. doi: 10.1086/302364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuki N, et al. Localization of the pathogenic gene of Behçet’s disease by microsatellite analysis of three different populations. Invest Ophthalmol Vis Sci. 2000;41(12):3702–3708. [PubMed] [Google Scholar]
- 14.Hughes T, et al. Identification of multiple independent susceptibility loci in the HLA region in Behçet’s disease. Nat Genet. 2013;45(3):319–324. doi: 10.1038/ng.2551. [DOI] [PubMed] [Google Scholar]
- 15.Mizuki N, et al. A strong association between HLA-B*5101 and Behçet’s disease in Greek patients. Tissue Antigens. 1997;50(1):57–60. doi: 10.1111/j.1399-0039.1997.tb02835.x. [DOI] [PubMed] [Google Scholar]
- 16.Meguro A, et al. Genetics of Behçet disease inside and outside the MHC. Ann Rheum Dis. 2010;69(4):747–754. doi: 10.1136/ard.2009.108571. [DOI] [PubMed] [Google Scholar]
- 17.Kang EH, et al. Associations between the HLA-A polymorphism and the clinical manifestations of Behcet’s disease. Arthritis Res Ther. 2011;13(2):R49. doi: 10.1186/ar3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 19.Raychaudhuri S, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44(3):291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: A revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huyton T, Ladas N, Schumacher H, Blasczyk R, Bade-Doeding C. Pocketcheck: Updating the HLA class I peptide specificity roadmap. Tissue Antigens. 2012;80(3):239–248. doi: 10.1111/j.1399-0039.2012.01928.x. [DOI] [PubMed] [Google Scholar]
- 22.Sanjanwala B, Draghi M, Norman PJ, Guethlein LA, Parham P. Polymorphic sites away from the Bw4 epitope that affect interaction of Bw4+ HLA-B with KIR3DL1. J Immunol. 2008;181(9):6293–6300. doi: 10.4049/jimmunol.181.9.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med. 1998;187(5):813–818. doi: 10.1084/jem.187.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gumperz JE, et al. Conserved and variable residues within the Bw4 motif of HLA-B make separable contributions to recognition by the NKB1 killer cell-inhibitory receptor. J Immunol. 1997;158(11):5237–5241. [PubMed] [Google Scholar]
- 25.Fadda L, et al. Common HIV-1 peptide variants mediate differential binding of KIR3DL1 to HLA-Bw4 molecules. J Virol. 2011;85(12):5970–5974. doi: 10.1128/JVI.00412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malnati MS, et al. Peptide specificity in the recognition of MHC class I by natural killer cell clones. Science. 1995;267(5200):1016–1018. doi: 10.1126/science.7863326. [DOI] [PubMed] [Google Scholar]
- 27.Mandelboim O, Wilson SB, Valés-Gómez M, Reyburn HT, Strominger JL. Self and viral peptides can initiate lysis by autologous natural killer cells. Proc Natl Acad Sci USA. 1997;94(9):4604–4609. doi: 10.1073/pnas.94.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405(6786):537–543. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- 29.Hansasuta P, et al. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur J Immunol. 2004;34(6):1673–1679. doi: 10.1002/eji.200425089. [DOI] [PubMed] [Google Scholar]
- 30.Vivian JP, et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature. 2011;479(7373):401–405. doi: 10.1038/nature10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereyra F, et al. International HIV Controllers Study The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330(6010):1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dilthey AT, Moutsianas L, Leslie S, McVean G. HLA*IMP—an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics. 2011;27(7):968–972. doi: 10.1093/bioinformatics/btr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Direskeneli H. Behçet’s disease: Infectious aetiology, new autoantigens, and HLA-B51. Ann Rheum Dis. 2001;60(11):996–1002. doi: 10.1136/ard.60.11.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braud V, Jones EY, McMichael A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol. 1997;27(5):1164–1169. doi: 10.1002/eji.1830270517. [DOI] [PubMed] [Google Scholar]
- 35.Rodgers JR, Cook RG. MHC class Ib molecules bridge innate and acquired immunity. Nat Rev Immunol. 2005;5(6):459–471. doi: 10.1038/nri1635. [DOI] [PubMed] [Google Scholar]
- 36.Merino AM, et al. Dimorphic HLA-B signal peptides differentially influence HLA-E- and natural killer cell-mediated cytolysis of HIV-1-infected target cells. Clin Exp Immunol. 2013;174(3):414–423. doi: 10.1111/cei.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.International Study Group for Behçet’s Disease Criteria for diagnosis of Behçet’s disease. Lancet. 1990;335(8697):1078–1080. [PubMed] [Google Scholar]
- 38.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 39.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011;1(6):457–70. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMahon RM, et al. Structure of HLA-A*0301 in complex with a peptide of proteolipid protein: Insights into the role of HLA-A alleles in susceptibility to multiple sclerosis. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 5):447–454. doi: 10.1107/S0907444911007888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maenaka K, et al. Nonstandard peptide binding revealed by crystal structures of HLA-B*5101 complexed with HIV immunodominant epitopes. J Immunol. 2000;165(6):3260–3267. doi: 10.4049/jimmunol.165.6.3260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.