

Significance
Mutations in the presenilin genes are the major cause of familial forms of Alzheimer’s disease. Nicastrin and presenilin are essential components of the γ-secretase complex, an intramembrane protease that cleaves type I membrane proteins such as Notch and the amyloid precursor protein. Presenilins are required for learning and memory, synaptic function, and age-related neuronal survival. In the current study we investigate whether nicastrin plays similar roles in hippocampal synapses by the generation and electrophysiological analysis of conditional knockout mice in which the nicastrin gene is deleted selectively in excitatory neurons of the adult cerebral cortex. Our data show that nicastrin is essential for both short-term and long-term synaptic plasticity, underscoring its importance in the regulation of synaptic function.
Abstract
Synaptic dysfunction is widely thought to play a key role in the pathogenesis of Alzheimer’s disease (AD). Presenilins, the major gene products involved in familial AD, are essential for short- and long-term synaptic plasticity in mature neurons as well as for the survival of cortical neurons during aging. Presenilin and nicastrin are both indispensable components of the γ-secretase complex, but it remains unknown whether presenilin regulates synaptic function in a γ-secretase–dependent or γ-secretase–independent manner and whether nicastrin plays similar roles in central synapses. In the current study, we address these questions using an electrophysiological approach to analyze nicastrin conditional knockout (cKO) mice in the hippocampal Schaffer collateral pathway. In these mice, we found that, even at 2 mo of age, deletion of nicastrin in excitatory neurons of the postnatal forebrain using Cre recombinase expressed under the control of the αCaMKII promoter led to deficits in presynaptic short-term plasticity including paired-pulse facilitation and frequency facilitation. Depletion of Ca2+ in the endoplasmic reticulum mimics and occludes the presynaptic facilitation deficits in nicastrin cKO mice, suggesting that disrupted intracellular Ca2+ homeostasis underlies the presynaptic deficits. In addition, NMDA receptor-mediated responses and long-term potentiation induced by theta-burst stimulation were decreased in nicastrin cKO mice at 3 mo but not at 2 mo of age. Together, these findings show that, similar to presenilins, nicastrin plays essential roles in the regulation of short- and long-term synaptic plasticity, highlighting the importance of γ-secretase in the function of mature synapses.
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder characterized by progressive memory loss and cognitive decline. The majority of familial AD cases are caused by missense mutations in genes encoding presenilin 1 (PS1) and presenilin 2, which are crucial components of the γ-secretase complex responsible for intramembrane cleavages of type I membrane proteins such as Notch. In addition to presenilin (PS), nicastrin (Nct), presenilin enhancer 2 (Pen-2), and anterior pharynx defective 1 (Aph-1) also are required to form the active γ-secretase complex. Nct is a type-1 transmembrane glycoprotein that originally was identified by its ability to form high molecular weight complexes with PS (1). Nct−/− mice die by embryonic day 10.5 and exhibit patterning defects similar to those in embryos lacking PS or Notch (2–7).
In the adult brain, genetic studies using conditional gene-targeting approaches demonstrated that both PS and Nct are essential for long-term memory and age-dependent neuronal survival (8–12). These findings highlight the importance of γ-secretase in memory and neuronal survival (13), even though γ-secretase–independent activities of PS have been reported also (14). However, Notch is unlikely to be the key mediator of γ-secretase in the adult brain, because Notch1 and Notch2 conditional knockout (cKO) mice using the same αCaMKII-Cre transgenic line had no major detectable phenotypes (15), whereas similar neurodevelopmental phenotypes were reported for mutant mice lacking PS or Notch in the developing brain (16–18).
Despite the importance of Nct in memory and neuronal survival, its role in the synapse is entirely unknown. In the current study, we performed electrophysiological analysis of Nct-deficient synapses in the Schaffer collateral pathway, using acute hippocampal slices of Nct cKO mice in which Nct was inactivated by a αCaMKII-Cre transgene (8). This transgene is known to recombine floxed alleles in excitatory neurons beginning at postnatal day 18 (8). In Nct cKO mice we found that long-term potentiation (LTP) induced by theta-burst stimulation (TBS) is normal at 2 mo but is impaired at age 3 mo, as is consistent with the progressive time course of Nct inactivation. NMDA receptor (NMDAR)-mediated responses similarly are normal at age 2 mo but are impaired at age 3 mo, suggesting that they likely contribute to the LTP deficits observed at this age. Presynaptic function, measured by paired-pulse facilitation (PPF) and frequency facilitation, is affected in Nct cKO mice at age 2 mo, before postsynaptic defects are apparent. Depletion of Ca2+ stores in the endoplasmic reticulum (ER) mimics and occludes the deficits in synaptic facilitation observed in Nct cKO mice. Our results demonstrate the importance of Nct in short- and long-term synaptic plasticity in mature hippocampal neurons.
Results
Time Course of Nct Inactivation in Nct cKO Mice.
We previously reported impairment of hippocampus-dependent spatial and associative memory in Nct cKO mice at 2–3 mo of age and an ∼50% reduction in Nct protein levels in the cerebral cortex of Nct cKO mice at age 2 mo (12). In the current study, we performed additional immunoblotting experiments to establish the time course of Nct inactivation using hippocampal lysates from Nct cKO and control mice at five time points, postnatal day 30, 45, 60, 75 and 90. Because Nct is modified posttranslationally by glycosylation, which makes the comparison of protein levels by immunoblotting difficult, we treated hippocampal lysates with peptide-N-glycosidase F (PNGase F) to remove saccharide groups from mature glycosylated forms of Nct. We found that the levels of Nct are reduced progressively in the hippocampus of Nct cKO mice with ∼50% remaining at 45–60 d of age and ∼25% remaining at 75–90 d of age (Fig. 1). The Nct protein still detected in the hippocampus of cKO mice is likely the result of Nct normally present in glia and/or interneurons, which are not targeted in this Nct cKO line, and of Nct remaining in excitatory neurons where Cre-mediated recombination and/or turnover of Nct mRNA and protein are not yet complete. Thus, the time course of Nct disappearance in Nct cKO mice is delayed relative to the disappearance of PS1 in PS conditional double knockout (cDKO) mice, in which we found hippocampal PSI reduced by ∼50% at age 4 wk (8, 9, 19).
Fig. 1.

Time course of Nct inactivation in the hippocampus of Nct cKO mice. (A) Western analysis of protein levels of Nct at five different time points. Hippocampal lysates were treated with PNGase-F to deglycosylate Nct. Nct levels are reduced progressively in the Nct cKO hippocampus. (B) Hippocampal lysates from Nct cKO and control mice at age 30, 45, 60, 75, and 90 d were analyzed by immunoblotting. Protein levels were normalized to β-actin and measured by LI-COR quantitative detection system. All data represent means ± SEM. The number of mice used in the experiment is indicated in parentheses.
Normal AMPA Receptor Responses but Progressive NMDAR Impairments in Nct cKO Mice.
To investigate whether Nct is involved in the modulation of synaptic function in the adult brain, we examined Nct cKO mice for deficits in synaptic transmission and plasticity in the Schaffer collateral pathway using acute hippocampal slices. We first evaluated basal synaptic transmission by quantifying the initial slope of evoked field excitatory postsynaptic potential (fEPSP) and the amplitude of the fiber volley (FV), which is a measure of the number of recruited axons, in acute hippocampal slices. Input/output (I/O) curves, which are primarily AMPA receptor (AMPAR)-mediated responses and were obtained by plotting the amplitude of FV versus the fEPSP slope in the presence of blockers of NMDAR and GABAA receptors (GABAAR) [50 µM APV (DL-2-amino-5-phosphonopentanoic acid) and 10 µM bicuculline, respectively] were similar in Nct cKO and control mice, indicating normal basal synaptic transmission (Fig. 2A). NMDAR-mediated synaptic responses were measured in the presence of blockers of AMPAR and GABAAR [10 µM 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) and 10 µM bicuculline, respectively]. We found that I/O curves of NMDAR-mediated synaptic responses were normal in Nct cKO mice at age 2 mo (Fig. 2B). To measure AMPAR and NMDAR responses more directly, we also performed whole-cell recording using voltage clamp in CA1 pyramidal neurons (Fig. S1). The AMPAR excitatory postsynaptic currents (EPSCs) were measured at a holding potential of −70 mV in the presence of Mg2+ to block the NMDAR-mediated component. The NMDAR-mediated synaptic current component was measured 60 ms after the peak of AMPAR EPSCs (recorded at −70 mV) at +40 mV, thus minimizing contamination by the AMPAR-mediated synaptic current (20). Again we found that postsynaptic AMPAR- and NMDAR-mediated responses and the ratio of NMDAR to AMPAR responses were normal in Nct cKO mice at age 2 mo (Fig. S1).
Fig. 2.

Age-dependent reduction of NMDAR-mediated responses in the hippocampal Schaffer collateral pathway of Nct cKO mice. (A and B) Normal AMPAR-mediated (A) and NMDAR-mediated (B) I/O curves of synaptic transmission in Nct cKO mice at age 2 mo. The FV amplitude is plotted against the initial slope of the evoked fEPSP for the Nct cKO and littermate control mice. Each point represents data averaged across all slices for a narrow bin of FV amplitude. (C and D) Normal AMPAR-mediated (C) but reduced NMDAR-mediated (D) I/O curves of synaptic transmission in Nct cKO mice at age 3 mo. The NMDAR I/O slope in Nct cKO mice (control: y = 0.429×, R2 = 0.980; cKO: y = 0.278×, R2 = 0.963) is significantly reduced (P < 0.05; Student t test). All data represent means ± SEM. The values in parentheses indicate the number of hippocampal slices (Left) and the number of mice (Right) used in each experiment.
To determine whether there is an age-dependent effect on synaptic function in the absence of Nct, we further evaluated AMPAR- and NMDAR-mediated responses in Nct cKO mice at age 3 mo. We found that basal synaptic transmission measured by AMPAR-mediated synaptic responses was normal in Nct cKO mice at age 3 mo (Fig. 2C). However, NMDAR-dependent responses were significantly reduced in Nct cKO mice (control: y = 0.429×, R2 = 0.980; cKO: y = 0.278×, R2 = 0.963; P < 0.05) (Fig. 2D). These results show that Nct cKO mice develop age-dependent specific deficits in NMDAR-mediated responses.
Progressive LTP Impairment in Nct cKO Mice.
Previous studies demonstrated that hippocampus-dependent spatial learning and memory are impaired in Nct cKO mice at 2–3 mo of age (12). We therefore examined the effect of Nct inactivation on LTP in the CA1 region of the hippocampus, which is the best-understood model of synaptic modification involved in learning and memory (21). LTP induced by five trains of TBS was unaffected in Nct cKO mice at age 2 mo (Fig. 3A) but was significantly impaired at age 3 mo (Fig. 3B). The magnitude of LTP measured during the last 10 min of the recording was significantly lower in Nct cKO mice (120.8 ± 2.7%) than in control mice (147.3 ± 3.5%) (P < 0.001). The age-dependent impairment of LTP in Nct cKO mice is consistent with the deficit in NMDAR-mediated responses and progressive loss of Nct protein in the hippocampus of these mutant mice.
Fig. 3.

Age-dependent impairment of long-term plasticity in the hippocampal Schaffer collateral pathway of Nct cKO mice. (A) Normal LTP induced by 5 TBS in Nct cKO mice (closed circles) compared with controls (open circles) at age 2 mo. Superimposed traces are averages of four consecutive responses 1 min before (thin line) and 50 min after (thick line) TBS induction. (B) Impaired TBS-induced LTP in Nct cKO mice at age 3 mo. Superimposed traces are averages of four consecutive responses 1 min before (thin line) and 50 min after (thick line) TBS induction. The magnitude of LTP during the last 10 min of the recording is significantly reduced in Nct cKO mice (120.8 ± 2.7%) relative to the control (147.3 ± 3.5%) (P < 0.001; Student t test). All data are means ± SEM. The values in parentheses indicate the number of hippocampal slices (Left) and the number of mice (Right) used in each experiment. [Scale bar: 10 ms (x axis) or 1 mV (y axis).]
Impaired Short-Term Plasticity in Nct cKO Mice.
Short-term plasticity also has been implicated in learning and memory (22). PPF and frequency facilitation are measures of presynaptic short-term plasticity, reflecting the ability of synapses to modulate neurotransmitter release induced by two closely spaced stimuli or repetitive stimulation, respectively. To examine whether PPF and synaptic frequency facilitation are affected in the absence of Nct, we recorded fEPSPs in the hippocampal Schaffer collateral pathway of Nct cKO mice. Stimulus intervals between 20 and 2,000 ms were used. Compared with control mice, PPF was reduced significantly in Nct cKO mice at age 2 mo, indicating impairment of short-term plasticity (Fig. 4).
Fig. 4.

Impaired short-term synaptic plasticity in the hippocampal Schaffer collateral pathway of Nct cKO mice at age 2 mo. (A) Representative traces from control and Nct cKO mice of fEPSPs evoked by two consecutive stimuli with a 60-ms interpulse interval. (B) Average paired-pulse ratios plotted as a function of the interstimulus interval. All data represent means ± SEM (□, P < 0.05; ■, P < 0.01; Student t test). The values in parentheses indicate the number of hippocampal slices (Left) and the number of mice (Right) used in each experiment. [Scale bars: 30 ms (x axis) or 1 mV (y axis).]
Moreover, frequency facilitation induced by 10 stimuli applied at frequencies ranging from 1 to 20 Hz also was reduced substantially (Fig. 5A), providing further evidence for presynaptic deficits in short-term plasticity. Thus, Nct is required for normal presynaptic short-term plasticity. Furthermore, the presynaptic defects occurred before the LTP and NMDAR deficits in Nct cKO mice.
Fig. 5.

Depletion of ER calcium mimics and occludes the impaired presynaptic facilitation in Nct cKO mice. (A) Synaptic facilitation elicited by stimulus trains is impaired in a frequency-dependent manner in hippocampal area CA1 of Nct cKO mice at age 2 mo. fEPSP slopes shown are normalized to the slope of the first fEPSP of the stimulus train. (B and C) Effects of TG treatment (2 µM for 1 h) on synaptic facilitation induced by high-frequency stimulus trains in the hippocampal CA1 region of control (B) and Nct cKO (C) mice at age 2 mo. All data are means ± SEM (□, P < 0.05; ■, P < 0.01; Student t test). The values in parentheses indicate the number of hippocampal slices (Left) and the number of mice (Right) used in each experiment.
ER Calcium Dependency of Synaptic Facilitation in Nct cKO Mice.
Synaptic facilitation is caused by local increases of presynaptic Ca2+ concentration, leading to increased release of neurotransmitter. PS has been reported to be involved in the regulation of Ca2+ release from intracellular stores (13, 23, 24). Therefore we tested whether the deficits in synaptic facilitation observed in Nct cKO mice were caused by disrupted ER Ca2+ homeostasis. We assessed synaptic facilitation in acute hippocampal slices from Nct cKO and control mice in the presence or absence of thapsigargin (TG), which irreversibly blocks sarco-endoplasmic reticulum Ca2+-ATPase on the ER and depletes Ca2+ in the ER (25). After treatment with 2 µM of TG for 1 h, synaptic facilitation during high-frequency stimulation (10 and 20 Hz) was markedly suppressed in control synapses, so that synaptic facilitation in control synapses in the presence of TG was similar to that in Nct cKO synapses in the absence of TG (Fig. 5B). Moreover, in Nct cKO synapses TG treatment had no discernible effect on synaptic facilitation at any of the frequencies examined (1, 5, 10–20 Hz; Fig. 5C). Thus, TG treatment mimics and occludes the effect of Nct inactivation on synaptic facilitation, suggesting that Nct inactivation may affect the regulation of presynaptic facilitation by disrupting intracellular Ca2+ stores.
Normal Levels of Neuronal and Synaptic Proteins in Nct cKO Mice.
The deficits in short-term and long-term synaptic plasticity in Nct cKO mice prompted us to examine whether levels of neuronal, presynaptic, and postsynaptic markers are altered in the absence of Nct. Immunoblotting analysis of cortical lysates from Nct cKO and control mice at 2 and 3 mo of age showed that levels of axonal and dendritic proteins (Tau, MAP2), receptors, and postsynaptic markers (GABAAR, GluR1, NMDAR1, PSD95) were normal (Fig. 6). Levels of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins Syntaxin-1, SNAP-25, and Synaptobrevin-2, the SM protein Munc18-1, complexin, and the chaperones α-synuclein, CSPα, SGT, and Hsc70 also were unaltered in the cortical lysates of Nct cKO mice at 2 and 3 mo of age (Fig. 6). Thus, the impairment in synaptic plasticity observed in Nct cKO mice is unlikely to be caused by alterations of receptor levels or deficits in the assembly of the SNARE complex. Similar to our earlier report (12), GFAP levels are increased in cortical lysates of Nct cKO mice (Fig. 6), indicating astrogliosis. This result is consistent with our prior findings showing ongoing apoptotic cell death in a small percentage (∼0.1%) of cortical neurons in PS cDKO mice beginning at age 2 mo and 10-fold increases of GFAP levels in cortical lysates of PS cDKO mice at age 6 mo (10, 11).
Fig. 6.
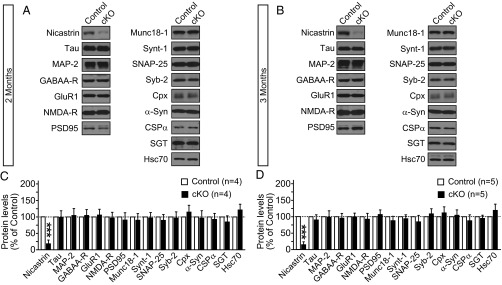
Normal levels of neuronal and synaptic markers in Nct cKO mice. Levels of indicated proteins in control and Nct cKO mice at 2 and 3 mo of age were analyzed by quantitative immunoblotting of cortical lysates using iodinated secondary antibodies and were normalized to GDP dissociation inhibitor as a loading control. Immunoblotting for Nct was performed after treatment with PNGase F to remove N-glycosylation. Data shown are means ± SEM (***P < 0.001; Student t test). The number of mice used in the experiment is indicated in parentheses.
Discussion
Role of Nct at the Synapse.
Through the generation and analysis of Nct cKO mice, which circumvent the embryonic lethality of Nct −/− mice, we uncovered essential roles of Nct in the synapse of the adult hippocampus. Nct is required for presynaptic short-term plasticity, such as PPF and frequency facilitation, and long-term plasticity, such as LTP (Figs. 3–5). These synaptic deficits are specific, because basal synaptic transmission is normal in Nct cKO mice (Fig. 2), and are not caused by alterations in receptor expression levels or SNARE complex assembly (Fig. 6). Moreover, the development of synaptic deficits in Nct cKO mice is age dependent, with the earliest synaptic changes occurring at age 2 mo. Presynaptic deficits in PPF and frequency facilitation during repetitive 10-stimulus trains (Figs. 4 and 5) are followed by reduced induction of LTP and NMDAR-mediated responses (Figs. 2 and 3). These results are consistent with our earlier findings showing that Nct cKO mice exhibit impairment of hippocampal-dependent learning and memory at 2–3 mo of age using Morris water maze and contextual fear-conditioning paradigms (12). Although basal synaptic transmission is normal, depletion of ER Ca2+ stores by TG in wild-type hippocampal slices mimics the defects in synaptic facilitation observed in Nct cKO slices, and TG treatment of Nct cKO slices does not worsen the presynaptic defects (Fig. 5). These results suggest that disruption of ER Ca2+ stores underlies the impairment in short-term plasticity in the absence of Nct. Furthermore, the deficits in NMDAR-mediated responses likely contribute to the LTP impairment in Nct cKO mice. The lack of alterations in receptor and SNARE complex proteins in Nct cKO mice (Fig. 6) are consistent with this interpretation. Thus, Nct plays crucial roles in short- and long-term synaptic plasticity, highlighting the importance of γ-secretase in the regulation of synaptic function.
γ-Secretase, PS, and Nct.
PS and Nct are both essential components of the γ-secretase complex. However, in principle, both PS and Nct may have both γ-secretase–dependent and –independent activities. In fact, γ-secretase–independent activities of PS have been suggested, including targeting the v-ATPase V0a1 subunit to lysosomes (14). We previously reported that PS is required for neurotransmitter release as well as for short- and long-term synaptic plasticity in the hippocampal Schaffer collateral pathway (9, 19, 23). Specifically, the presynaptic deficits, such as reduced synaptic facilitation and release probability, are first detected in PS cDKO mice at age 5 wk, followed by impairment in LTP and NMDAR-mediated responses at age 6 wk (19). Both reduced NMDAR responses and decreased neurotransmitter release probability are thought to contribute to the LTP deficits, because presynaptic inactivation of PS is sufficient to cause LTP impairment in the absence of reduction in NMDAR responses (13, 23).
The difference in the age-dependent development of LTP impairment at 3 mo in Nct cKO mice and at 2 mo in PS cDKO mice initially raised the possibility that Nct and PS may have differential roles in the synapse. However, detailed quantitative analysis revealed that there is a delay in the inactivation of Nct relative to PS (Fig. 1), even though the same αCaMKII-Cre line is used (8, 9, 12, 19); this delay may be caused by differences in the genetic loci and/or the half life of the mRNA and/or protein. Specifically, levels of Nct are reduced by ∼50% in the hippocampus of Nct cKO mice at age 2 mo (Fig. 1) (12), whereas levels of PS1 are reduced by ∼50% in the cortex of PS cDKO mice at age 4 wk (19). Moreover, that conditional inactivation of Nct or PS in excitatory neurons of the postnatal hippocampus results in the same pre- and postsynaptic changes suggests that the γ-secretase–dependent activity of Nct or PS is essential for their role in regulating synaptic function and cannot be explained by the role of PS as an ER Ca2+ leak channel (26).
γ-Secretase in Brain and Skin Diseases.
Although more than 200 mutations in PS genes have been identified in familial AD, no mutations in genes encoding other γ-secretase components have been reported in AD, suggesting that PS has a unique role in AD. This unique role may be caused by γ-secretase–independent activities of PS that are more relevant to AD pathogenesis, by a higher intrinsic mutation rate of the human PSEN genes than of the other γ-secretase subunit genes, or by PS forming the catalytic core of the γ-secretase complex. Interestingly, recent human genetic studies identified large numbers of loss-of-function mutations in the Nct (17) and Pen-2 (3) genes that are associated with familial acne inversa or hidradenitis suppurativa (27–31). The lack of mutations identified in the Aph-1A and Aph-1B genes may reflect the genetic redundancy of the Aph-1 family; however, one mutation was reported in the PSEN1 gene despite the presence of its family member PSEN2 (27). Because the identified mutations are mostly dominantly inherited loss-of-function mutations (nonsense or frame-shift), these findings indicate that partial loss of γ-secretase activity because of haploinsufficiency of these genes leads to acne inversa. This notion is consistent with findings from mouse studies showing that γ-secretase or Notch deficiency results in follicular hyperkeratosis, which is the initiating event in acne inversa (32–34).
Thus, haploinsufficiency of nonredundant genes encoding Nct and Pen-2 causes acne inversa, likely through the Notch pathway, without accompanying AD. Likewise, acne inversa is not associated with AD in AD patients carrying dominantly inherited mutations in the PSEN1 gene, even those exhibiting complete loss of PS1 function such as the L435F mutation (35). Possible explanations for the difference in disease manifestation are as follows. First, the molecular targets or pathways regulated by γ-secretase in mediating acne inversa or AD are distinct. Although acne inversa is mediated through a γ-secretase–dependent Notch pathway, Notch1 and Notch2 clearly are not the targets of PS or γ-secretase mediating its function in the adult brain (15). Second, FAD-linked dominantly inherited missense mutations in PS not only have cis-acting effects, reducing its γ-secretase activity, but also have trans-acting effects, inhibiting the γ-secretase activity of the wild-type PS protein (36). Further studies will be needed to elucidate the mechanisms underlying AD and acne inversa, and identification of the γ-secretase substrates responsible for synaptic function and neuronal survival will provide additional mechanistic insight into the role of γ-secretase in the aging brain.
Materials and Methods
Nct cKO Mice.
The generation of Nct cKO mice has been described previously (12). Briefly, to obtain forebrain-specific Nct cKO (fNct/fNct;CaM-Cre) mice, we crossed floxed Nct (fNct/fNct) mice with αCaMKII-Cre transgenic mice (8). Homozygous fNct/fNct mice were generated in a C57BL/6129 hybrid background, whereas αCaMKII-Cre transgenic mice were generated in a C57BL/6CBA hybrid strain and then were backcrossed to B6 for more than 20 generations. The genetic background of all the mice used in this study was C57BL/6 and 129 hybrid, and only littermates were used. All procedures relating to animal care and treatment conformed to the Harvard Institutional Animal Care and Use Committee and National Institutes of Health (37) guidelines.
Western Analysis.
Mouse hippocampi were dissected and homogenized in cold radioimmunoprecipitation assay lysis buffer [consisting of the following (in mM): 50 mM Tris⋅HCl (pH 8), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS] containing protease and phosphatase inhibitors (Sigma-Aldrich). Standard Western blotting was performed using anti-nicastrin (N1660; 1:1,000; Sigma-Aldrich) and anti–β-actin (1:20,000; Abcam) followed by infrared dye-coupled secondary antibodies (goat anti-mouse IRdye800 and goat anti-rabbit IRdye680 from Li-Cor). Image acquisition was performed using the Odyssey Infrared Imaging System (Li-Cor). Mouse neocortices were homogenized in 2 mL of PBS (pH 7.4) containing protease inhibitor mixture (Roche). Homogenates were dissolved in 2× Laemmli sample buffer and were passed through an insulin syringe 20 times before loading. Protein (8 μg) from each sample was separated by SDS/PAGE, transferred onto nitrocellulose membranes, and incubated with the primary antibodies listed below and secondary antibody (1:5,000; MP Biomedicals). HRP immunoblots were developed using enhanced chemiluminescence (GE Healthcare). All quantitative immunoblotting experiments were performed with iodinated secondary antibodies (1:1,000; Perkin-Elmer) overnight at room temperature, as described (38). The 125I blots were exposed to phosphorimager screens (GE Healthcare) overnight and were scanned using a Typhoon scanner (GE Healthcare), followed by quantification with ImageQuant software (GE Healthcare). Primary antibodies used were as follows: CSPα (R807), complexin 1, 2 (122002; SYSY), GABAAR (06-868; Upstate), GluR1 (160-E5; SYSY), Hsc70 (clone 3C5; SYSY), MAP2 (AB5622; Millipore), Munc18 (610337; BD Transduction), Nct (N1660 and T3749; Sigma), NMDAR1 (M68; SYSY), PSD-95 (MA1046; Thermo), SGT (CHAT33), SNAP-25 (SMI81; Sternberger Monoclonals), synaptobrevin-2 (cl. 69.1; SYSY), syntaxin-1 (HPC1; SYSY), and tau (MAB361; Millipore).
Preparation of Brain Slices.
Brain slices were prepared from 2- to 3-mo-old Nct cKO mice and littermate control mice. Transverse hippocampal slices (400 μm thick) were prepared using a vibratome (VT1200S; Leica). For functional studies, slices were incubated at 35 °C for 1 h and thereafter were maintained at 32 °C until in situ slice recordings were made. Hippocampal slices were visualized using an upright microscope equipped with differential interference contrast optics (BX51WI; Olympus). All experiments procedures were conducted in accordance with institutional and National Institutes of Health (37) guidelines.
Field and Whole-Cell Electrophysiological Analysis of Acute Hippocampal Slices.
All electrophysiological analyses were performed in a genotype-blind manner. The slices were maintained in a storage chamber containing artificial CSF (aCSF) [125 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, 25 mM NaHCO3, 10 mM dextrose, 1.2 mM pyruvate, and 0.4 mM Na-ascorbate, pH 7.4 (300 ± 5 mOsm)] when saturated with carbogen (95% O2 and 5% CO2) at 30 °C. Stimulation by 200-μs pulses was delivered with a bipolar concentric metal electrode at the Schaffer collateral pathway. Synaptic strength was quantified as the initial slope of field potentials recorded with aCSF-filled microelectrodes (1–2 MΩ). In LTP recordings, baseline responses were collected every 15 s with a stimulation intensity that yielded 60% of maximal response. LTP was induced by five episodes of TBS delivered at 0.1 Hz. Each episode contains 10 stimulus trains (five pulses at 100 Hz) delivered at 5 Hz. Average responses (mean ± SEM.) are expressed as percentage of pre-TBS baseline response. Synaptic facilitations were measured as the percentage of the fEPSP slope versus the first fEPSP slope at a given stimulus train in individual slices.
Intracellular (whole-cell) recordings were performed using Multiclamp 700B (Molecular Devices) in CA1 pyramidal neurons. Patch pipettes (3–5 MΩ) were filled with internal solution consisting of (in mM) 110 Cs-methanesulfonate, 20 tetraethylammonium-chloride, 8 KCl, 10 EGTA, 10 Hepes, 5 QX-314 (a derivative of lidocaine), 3 Mg-ATP, and 0.3 Na-GTP (pH 7.3); 275–285 mOsm. The AMPAR EPSC amplitude was measured at a holding potential of −70 mV. The NMDAR-mediated component of the EPSC at +40 mV was measured 60 ms after the peak of the AMPAR EPSCs. Data were analyzed using Igor (version 6.3; Wave-Metrics) and Clampfit (version 10.3; Molecular Devices).
Statistics.
All data are presented as mean ± SEM. The statistical significances were evaluated using Student t test.
Supplementary Material
Acknowledgments
We thank H. Zhao for breeding and genotyping the mice used in the study and members of the J.S. laboratory for discussions and comments. This work was supported by National Institutes of Health Grants R01NS041779 and R01NS042818.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408554111/-/DCSupplemental.
References
- 1.Yu G, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407(6800):48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 2.Li T, Ma G, Cai H, Price DL, Wong PC. Nicastrin is required for assembly of presenilin/gamma-secretase complexes to mediate Notch signaling and for processing and trafficking of beta-amyloid precursor protein in mammals. J Neurosci. 2003;23(8):3272–3277. doi: 10.1523/JNEUROSCI.23-08-03272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, et al. Positive and negative regulation of the gamma-secretase activity by nicastrin in a murine model. J Biol Chem. 2003;278(35):33445–33449. doi: 10.1074/jbc.M301288200. [DOI] [PubMed] [Google Scholar]
- 4.Shen J, et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89(4):629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 5.Wong PC, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387(6630):288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 6.Donoviel DB, et al. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13(21):2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8(6):707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, et al. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31(5):713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 9.Saura CA, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 10.Beglopoulos V, et al. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem. 2004;279(45):46907–46914. doi: 10.1074/jbc.M409544200. [DOI] [PubMed] [Google Scholar]
- 11.Wines-Samuelson M, et al. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS ONE. 2010;5(4):e10195. doi: 10.1371/journal.pone.0010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabuchi K, Chen G, Südhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J Neurosci. 2009;29(22):7290–7301. doi: 10.1523/JNEUROSCI.1320-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho A, Shen J. Presenilins in synaptic function and disease. Trends Mol Med. 2011;17(11):617–624. doi: 10.1016/j.molmed.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JH, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng J, et al. Conditional deletion of Notch1 and Notch2 genes in excitatory neurons of postnatal forebrain does not cause neurodegeneration or reduction of Notch mRNAs and proteins. J Biol Chem. 2012;287(24):20356–20368. doi: 10.1074/jbc.M112.349738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127(12):2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- 17.Wines-Samuelson M, Handler M, Shen J. Role of presenilin-1 in cortical lamination and survival of Cajal-Retzius neurons. Dev Biol. 2005;277(2):332–346. doi: 10.1016/j.ydbio.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 18.Wines-Samuelson M, Shen J. Presenilins in the developing, adult, and aging cerebral cortex. Neuroscientist. 2005;11(5):441–451. doi: 10.1177/1073858405278922. [DOI] [PubMed] [Google Scholar]
- 19.Zhang D, et al. Inactivation of presenilins causes pre-synaptic impairment prior to post-synaptic dysfunction. J Neurochem. 2010;115(5):1215–1221. doi: 10.1111/j.1471-4159.2010.07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myme CI, Sugino K, Turrigiano GG, Nelson SB. The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. J Neurophysiol. 2003;90(2):771–779. doi: 10.1152/jn.00070.2003. [DOI] [PubMed] [Google Scholar]
- 21.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 22.Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22(1):139–146. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- 23.Zhang C, et al. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460(7255):632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mattson MP. ER calcium and Alzheimer’s disease: In a state of flux. Sci Signal. 2010;3(114):pe10. doi: 10.1126/scisignal.3114pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Treiman M, Caspersen C, Christensen SB. A tool coming of age: Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19(4):131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 26.Tu H, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126(5):981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang B, et al. Gamma-secretase gene mutations in familial acne inversa. Science. 2010;330(6007):1065. doi: 10.1126/science.1196284. [DOI] [PubMed] [Google Scholar]
- 28.Kelleher RJ, 3rd, Shen J. Genetics. Gamma-secretase and human disease. Science. 2010;330(6007):1055–1056. doi: 10.1126/science.1198668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li CR, et al. Two novel mutations of the nicastrin gene in Chinese patients with acne inversa. Br J Dermatol. 2011;165(2):415–418. doi: 10.1111/j.1365-2133.2011.10372.x. [DOI] [PubMed] [Google Scholar]
- 30.Nomura Y, et al. A novel splice site mutation in NCSTN underlies a Japanese family with hidradenitis suppurativa. Br J Dermatol. 2013;168(1):206–209. doi: 10.1111/j.1365-2133.2012.11174.x. [DOI] [PubMed] [Google Scholar]
- 31.Pink AE, Simpson MA, Desai N, Trembath RC, Barker JN. γ-Secretase mutations in hidradenitis suppurativa: New insights into disease pathogenesis. J Invest Dermatol. 2013;133(3):601–607. doi: 10.1038/jid.2012.372. [DOI] [PubMed] [Google Scholar]
- 32.Xia X, et al. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci USA. 2001;98(19):10863–10868. doi: 10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li T, et al. Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma-secretase. J Biol Chem. 2007;282(44):32264–32273. doi: 10.1074/jbc.M703649200. [DOI] [PubMed] [Google Scholar]
- 34.Nicolas M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33(3):416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 35.Heilig EA, Xia W, Shen J, Kelleher RJ., 3rd A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J Biol Chem. 2010;285(29):22350–22359. doi: 10.1074/jbc.M110.116962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ., 3rd Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Aβ production. J Neurosci. 2013;33(28):11606–11617. doi: 10.1523/JNEUROSCI.0954-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Committee on Care and Use of Laboratory Animals (1985) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85-23.
- 38.Rosahl TW, et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375(6531):488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.