

Significance
Synaptic transmission involves the release of neurotransmitters that activate receptors on postsynaptic cells. The results reveal that protons fulfill the criteria for a neurotransmitter and that they activate postsynaptic acid-sensing ion channels. This activity facilitates synaptic plasticity, a requirement for learning and memory in the amygdala.
Keywords: long-term potentiation, PcTX1, acid sensing ion channel
Abstract
Stimulating presynaptic terminals can increase the proton concentration in synapses. Potential receptors for protons are acid-sensing ion channels (ASICs), Na+- and Ca2+-permeable channels that are activated by extracellular acidosis. Those observations suggest that protons might be a neurotransmitter. We found that presynaptic stimulation transiently reduced extracellular pH in the amygdala. The protons activated ASICs in lateral amygdala pyramidal neurons, generating excitatory postsynaptic currents. Moreover, both protons and ASICs were required for synaptic plasticity in lateral amygdala neurons. The results identify protons as a neurotransmitter, and they establish ASICs as the postsynaptic receptor. They also indicate that protons and ASICs are a neurotransmitter/receptor pair critical for amygdala-dependent learning and memory.
Although homeostatic mechanisms generally maintain the brain’s extracellular pH within narrow limits, neural activity can induce transient and localized pH fluctuations. For example, acidification may occur when synaptic vesicles, which have a pH of ∼5.2–5.7 (1–3), release their contents into the synapse. Studies of mammalian cone photoreceptors showed that synaptic vesicle exocytosis rapidly reduced synaptic cleft pH by an estimated 0.2–0.6 units (4–6). Transient synaptic cleft acidification also occurred with GABAergic transmission (7). Some, but not all, studies also reported that high-frequency stimulation (HFS) transiently acidified hippocampal brain slices, likely as a result of the release of synaptic vesicle contents (8, 9). Neurotransmission also induces a slower, more prolonged alkalinization (10, 11). In addition to release of synaptic vesicle protons, neuronal and glial H+ and HCO3− transporters, channels, H+-ATPases, and metabolism might influence extracellular pH (10–12).
ASICs are potential targets of reduced extracellular pH. ASICs are Na+-permeable and, to a lesser extent, Ca2+-permeable channels that are activated by extracellular acidosis (13–19). In the brain, ASICs consist of homotrimeric and heterotrimeric complexes of ASIC1a, ASIC2a, and ASIC2b. The ASIC1a subunit is required for acid-activation in the physiological range (>pH 5.0) (20, 21). Several observations indicate that ASIC are located postsynaptically. ASICs are located on dendritic spines. Although similar to glutamate receptors, they are also present on dendrites and cell bodies (20, 22–24). ASIC subunits interact with postsynaptic scaffolding proteins, including postsynaptic density protein 95 and protein interacting with C-kinase-1 (20, 24–29). In addition, ASICs are enriched in synaptosome-containing brain fractions (20, 24, 30).
Although these observations raised the possibility that protons might be a neurotransmitter, postsynaptic ASIC currents have not been detected in cultured hippocampal neurons (31, 32), and whether localized pH transients might play a signaling role in neuronal communication remains unclear. In previous studies of hippocampal brain slices, extracellular field potential recordings suggested impaired hippocampal long-term potentiation (LTP) in ASIC1a−/− mice (20), although another study did not detect an effect of ASIC1a (33). Another study using microisland cultures of hippocampal neurons suggested that the probability of neurotransmitter release increased in ASIC1a−/− mice (32).
Here, we tested the hypothesis that protons are a neurotransmitter and that ASICs are the receptor. Criteria to identify substances as neurotransmitters have been proposed (34). Beg and colleagues (35) used these criteria to conclude that protons are a transmitter released from Caenorhabditis elegans intestine to cause muscle contraction. Key questions about whether protons meet criteria for a neurotransmitter are: Does presynaptic stimulation increase the extracellular proton concentration? Do protons activate currents in postsynaptic cells? Can exogenously applied protons reproduce effects of endogenous protons? What is the postsynaptic proton receptor? We studied lateral amygdala brain slices because amygdala-dependent fear-related behavior depends on a pH reduction (36). In addition, ASICs are abundantly expressed there, and ASIC1a−/− mice have impaired fear-like behavior (36–38).
Results and Discussion
Presynaptic Stimulation Induces ASIC Excitatory Postsynaptic Currents.
We found that an acidic pH stimulated currents in lateral amygdala pyramidal neurons, and ASIC1a−/− neurons lacked those currents (Fig. 1 A-C). We also stimulated cortical inputs and recorded excitatory postsynaptic currents (EPSCs). Under basal conditions, wild-type (WT) and ASIC1a−/− EPSCs had similar amplitudes (Fig. 1 E–G), and previous studies showed similar NMDA and AMPA receptor currents in cultured hippocampal neurons of both genotypes (20). After glutamate receptor (GluR) blockade with AMPA and NMDA receptor blockers, a small component of the EPSC remained (Fig. 1 E–G). HFS of cortical inputs also generated postsynaptic currents after GluR blockade (Fig. S1).
Fig. 1.
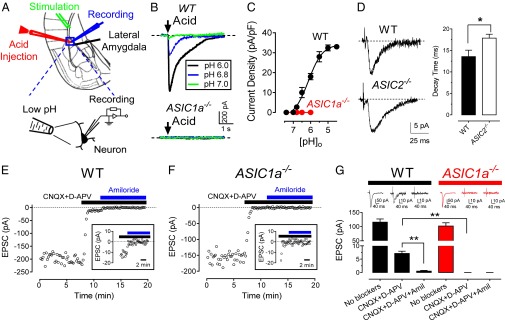
Stimulating presynaptic neurons elicits postsynaptic ASIC currents in lateral amygdala pyramidal neurons. (A) Schematic showing lateral amygdala, acid injection micropipette, stimulating electrode, and whole-cell patch-clamp recording electrode. (B) Representative ASIC currents elicited by acid injections in WT and ASIC1a−/− neurons. Holding potential, −70 mV. (C) pH-dependent activation of ASIC currents. Best-fit yielded EC50 pH of 6.1 ± 0.1 (n = 6–10 cells in 4 mice). (D) Left, representative traces of EPSCs in WT and ASIC2−/− neurons. Right, mean ± SEM of best-fit EPSC decay times. P < 0.05 (Student t test; n = 10 cells in 4 mice). (E and F) EPSC recordings in lateral amygdala brain slices from WT and ASIC1a−/− mice. To induce EPSCs, test pulses (100 µS, 0.05 or 0.1 Hz) were delivered through extracellular bipolar electrodes placed on cortical inputs. Slices were perfused with 25 µM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (an AMPA receptor antagonist) plus 50 µM (2R)-amino-5-phosphonovaleric acid (D-APV) (an NMDA receptor antagonist) and with 200 µM amiloride during times indicated. Data are from single slices. (Inset) EPSCs with expanded y axis. (G) EPSC amplitudes as recorded in D and E. **P < 0.01 (Student t test; n = 20 cells in 10 mice). Representative EPSC traces are shown at top.
ASIC1a−/− neurons lacked the current that was revealed in the presence of GluR blockers (Fig. 1 F and G and Fig. S1). Amiloride, which blocks ASICs (albeit a nonselective blocker) (39), inhibited the current (Fig. 1 E and G). The decay times of EPSCs were faster after glutamate receptor (GluR) blockade (Fig. S2), indicating that EPSCs observed in the presence of GluR blockers are likely not GluR-mediated. As an additional test of whether the GluR-independent EPSCs were caused by ASICs, we altered their subunit composition by eliminating ASIC2. Earlier work showed that ASIC current desensitization was slowed in cultured ASIC2−/− neurons (41). Consistent with that, we found prolonged GluR-independent EPSC decay times in ASIC2−/− lateral amygdala slices (Fig. 1D). Together, these results indicate that presynaptic stimulation activates postsynaptic ASICs. Thus, they suggested that protons were the neurotransmitter.
Presynaptic Stimulation Increases Extracellular Protons and Activates ASICs.
To test whether presynaptic stimulation increases extracellular protons, we fused the pH-sensitive superecliptic pHluorin (3) to the extracellular domain of syndecan 2, a postsynaptic membrane protein, and found that it reported changes in extracellular pH (Fig. 2A and Fig. S3A). In transfected pyramidal neurons, syndecan 2-pHluorin targeted spines, dendrites, and the cell membrane of soma (Fig. 2B). Stimulating cortical inputs transiently reduced pH at spines and the neighboring dendrites, followed by a slower alkalinization (Fig. 2 C and D and Fig. S3B). The extent of acidification and alkalinization depended on stimulus frequency. The alkalinization that followed the transient acidification has been reported in other preparations (11). Because ASICs inactivate in the continued presence of protons (39, 42–44), alkalinization between synaptic release stimulations might function to resensitize the receptor.
Fig. 2.

Presynaptic stimulation reduces extracellular pH. (A) Schematic of pHluorin linked to syndecan 2 to assay changes in extracellular pH. (B) Biolistic transfection of pyramidal neurons. (Left) Syndecan 2-pHluorin. (Middle) mCherry as a control fluorescent indicator. (Right) Merged image. (Bottom) Enlarged image of selected area from upper image. Note syndecan 2-pHlourin expression in spines. (C) Representative traces of fluorescence with stimulation at indicated frequencies for 1 s. (D) Ratio of change in fluorescence (acidic ∆F1, alkaline ∆F2) to basal fluorescence (F0) at indicated stimulation frequencies (n = 7).
To prevent synaptic vesicle fusion, we applied Ca2+ channel blockers and found that they eliminated both GluR-dependent and ASIC-dependent EPSCs (Fig. S4). Although Ca2+ channel blockers could have multiple effects, these results are consistent with proton release from synaptic vesicles as a source of the reduced pH. However, it is possible that other transport processes make important contributions to acidification (4–8).
If presynaptic stimulation increases extracellular protons that activate postsynaptic ASICs, we reasoned that decreasing the pH buffering capacity should augment ASIC EPSCs. Conversely, increasing pH buffering should attenuate ASIC EPSCs. A similar strategy has been used to attribute other responses to pH changes (4–7). In the presence of GluR blockers, reducing pH buffer capacity (10 mM HCO3−, 2% CO2 at pH 7.4) increased EPSC amplitude compared with control (25 mM HCO3−, 5% CO2 at pH 7.4) (Fig. 3A). In contrast, increasing buffer capacity (90 mM HCO3−, 15% CO2 at pH 7.4) decreased EPSCs. We obtained similar results with HFS (Fig. S5).
Fig. 3.

Changing pH buffer capacity alters ASIC-dependent EPSCs. (A, Left) Example of ASIC-dependent EPSCs in lateral amygdala neuron sequentially perfused with 25 mM HCO3−/5% CO2 ACSF, 10 mM HCO3−/2% CO2 ACSF, and 90 mM HCO3−/15% CO2 ACSF, all at pH 7.4. In addition, 25 µM CNQX and 50 µM D-APV were present throughout. Representative traces are shown at top. (Right) EPSC amplitude in indicated buffers, as described for left. Each set of connected points is from a different cell: 25 mM HCO3− group, −7.0 ± 0.6 pA; 10 mM HCO3−, −13.5 ± 1.5 pA; 90 mM HCO3−, −4.0 ± 0.4 pA (n = 10 cells in 4 mice); one-way ANOVA with Tukey’s post hoc multiple comparison test. *P < 0.05. (B, Left) ASIC-dependent EPSCs in lateral amygdala neuron in ASIC2−/− brain slice perfused with 25 mM HCO3−/5% CO2 ACSF and then 10 mM HCO3−/2% CO2 ACSF, both at pH 7.4. PcTX1 (100 nM) was present during time indicated. (Right) ASIC-dependent EPSC amplitude in buffers described for left (n = 8); one-way ANOVA with Tukey’s post hoc multiple comparison test. *P < 0.001.
To test further whether changing pH buffer capacity alters the ASIC component of EPSCs, we applied the ASIC-specific blocker Psalmotoxin-1 (PcTX1) (40). PcTX1 inhibits ASIC1a homomultimers, but not ASIC1/2 heteromultimers. In preliminary studies, PcTX1 had little effect on GluR-independent EPSCs in WT amygdala slices, consistent with the majority of ASIC currents arising from ASIC1/2 heteromultimers. However, in ASIC2−/− brain slices, reducing pH buffer capacity increased GluR-independent EPSC amplitude, and PcTX1 inhibited the current (Fig. 3B). These data indicate that reducing pH buffer capacity increased the ASIC component of EPSCs.
Protons and ASICs Are Required for Synaptic Plasticity.
Activation of postsynaptic ASICs by presynaptic stimulation suggested that this process might influence synaptic plasticity. To assess synaptic plasticity in lateral amygdala brain slices, we measured LTP. LTP is a prolonged increase in the strength of synaptic transmission after intense presynaptic stimulation and may be a correlate of the synaptic plasticity. We applied HFS to cortical inputs and assayed excitatory postsynaptic potentials (EPSPs), the depolarizations induced by test pulses delivered to cortical inputs. Immediately after HFS, EPSPs increased in slices from WT and ASIC1a−/− mice (Fig. 4A). However, LTP was strikingly reduced in ASIC1a−/− slices, decaying to baseline 15 min after HFS.
Fig. 4.

Protons and ASIC1a contribute to synaptic plasticity in the lateral amygdala. (A) EPSPs were induced by test pulses (100 µS, 0.05 or 0.1 Hz) delivered to cortical inputs. HFS (100 Hz, 1 s) was used to induce LTP. EPSPs were recorded before and after HFS in WT (blue, 135 ± 3% of baseline; P < 0.001; n = 12 cells in 6 mice) and ASIC1a−/− (red, 100 ± 2% of baseline, no significant difference; n = 12/6 mice) lateral amygdala pyramidal neurons. Solution was 25 mM HCO3−/5% CO2 at pH 7.4 ACSF. Representative EPSP traces at top were recorded under basal conditions and 50 min after HFS. WT differed from ASIC1a−/−. P < 0.01 (Student t test). (B) EPSPs before and after HFS (as in A) in WT lateral amygdala neurons. ACSF contained 25 mM HCO3−/5% CO2 at pH 7.4 (blue), 10 mM HCO3−/2% CO2 at pH 7.4 (green, 180 ± 7% of baseline; P < 0.01; n = 9/5 mice), or 90 mM HCO3−/15% CO2 at pH 7.4 (gray, 99 ± 5% of baseline; no significant difference; n = 7 cells in 4 mice). Data with 25 mM HCO3− differed from 10 mM HCO3− and 90 mM HCO3−. P < 0.01 (one-way ANOVA; Tukey’s post hoc multiple comparison). Representative EPSP traces at top were recorded before and 50 min after HFS.
Finding that ASICs were required for induction of amygdala LTP suggested that an increase in protons was involved. If that is the case, then reducing or increasing pH buffering capacity should enhance or minimize, respectively, a stimulus-induced fall in pH, and thereby increase or attenuate LTP. Although maintaining solution pH at 7.4, reducing pH buffer capacity strikingly enhanced LTP, and increasing buffer capacity diminished LTP (Fig. 4B). Attenuation of LTP by increased pH buffering was not a result of inhibition of EPSPs or irreversible changes in brain slices (Fig. S6). Together, these results suggest that increased protons and ASICs are required for normal synaptic plasticity.
Exogenous Application of Protons Induces LTP and Requires Glutamate.
To further test the hypothesis that protons are a neurotransmitter, we examined the effect of applying exogenous protons. Similar to in cultured neurons (45, 46), we found that a puff of acid (pH 7.0–6.0) elicited action potentials in lateral amygdala neurons, and pH 6.8 generated the greatest number of action potentials (Fig. 5A). Therefore, we gave three short puffs of pH 6.8 artificial cerebrospinal fluid (ACSF) and recorded EPSPs (Fig. 5B and Fig. S7). Acidic puffs induced LTP in WT, but not ASIC1a−/−, neurons.
Fig. 5.

Extracellular application of an acidic solution to lateral amygdala pyramidal neurons induces LTP. (A, Left) Representative traces of acid-generated action potentials. (Right) average number of action potentials generated at indicated pH (n = 10 for each pH; 4 mice). pH 6.8 induced the greatest number of action potentials. (B) EPSPs before and after three puffs of pH 6.8 ACSF were delivered to the target neuron in slices from WT (blue, 145 ± 6% of baseline; P < 0.001; n = 6 cells in 4 mice) and ASIC1a−/− (red, 101 ± 2% of baseline; no significant difference; n = 8 cells in 4 mice) mice. A test pulse was delivered 50 ms before the end of each puff of pH 6.8 ACSF. Representative EPSP traces from each group are at the top. WT differed from ASIC1a−/− (P < 0.01). Control data with test pulses are the same in B–D. Statistical analysis was one-way ANOVA; Tukey’s post hoc multiple comparison for B–D. (C) pH 6.8 application in slices treated with 50 µM D-APV (green, 116 ± 5% of baseline; P < 0.05; n = 6 cells in 4 mice) or vehicle control (blue). Control differed from 50 µM D-APV (P < 0.01). (D) Application of pH 6.8 ACSF to lateral amygdala pyramidal neurons in absence (gray, 99 ± 3% of baseline; no significant difference; n = 9 cells in 5 mice) or presence (blue) of test pulses during application of pH 6.8 solution. Control differed from slices without test pulses (P < 0.01).
To learn whether an acidic solution is sufficient to induce LTP or whether glutamate signaling is also required, we applied an NMDA receptor blocker. Inhibiting NMDA receptors substantially attenuated acid-induced LTP (Fig. 5C). Eliminating the presynaptic test pulses to decrease glutamate release also prevented acid-evoked LTP (Fig. 5D). Thus, both proton-ASIC and glutamate-GluR activities were required for LTP.
Protons Fulfill the Criteria for a Neurotransmitter.
Criteria for identifying substances as neurotransmitters have been proposed (34). We used the following criteria to gauge whether protons are neurotransmitters.
Chemical is present in the presynaptic cell.
Synaptic vesicles have a pH of 5.2–5.7 (3), and the presynaptic cell cytoplasm contains protons.
Stimulation of the cell releases the chemical.
Our data indicate that presynaptic stimulation causes transient extracellular acidification, and the greater the stimulation, the greater the increase in protons. Synaptic acidification, which has been estimated at 0.2–0.6 pH units at cone photoreceptor synapses (4, 5), might arise when synaptic vesicle exocytosis releases protons. Although calculations suggest that the number of free protons in a synaptic vesicle might be quite small, DeVries (4) has discussed that protons are buffered within vesicles and that buffers will be deprotonated on vesicle fusion. Alternatively, it has been proposed that sources of protons might be the H+-ATPase, which faces the synaptic cleft after vesicle fusion, and/or Na+/H+ exchange (4, 6, 7). Irrespective of the mechanism, our results are consistent with earlier studies indicating that presynaptic stimulation acutely increases the proton concentration (4–7).
There is a postsynaptic receptor.
We conclude that ASIC channels are the postsynaptic proton receptors because eliminating ASIC1a eliminated ASIC-dependent EPSCs, GluR EPSCs and ASIC EPSCs manifested different decay times, the biophysical properties of ASIC-dependent EPSCs changed as predicted when ASIC channel subunit composition was altered, PcTX1 and amiloride inhibited ASIC-dependent EPSCs, and ASICs localize to the postsynaptic membrane (20, 23, 24, 26).
Exogenous application of the chemical mimics the endogenous response.
We found that applying exogenous protons induced action potentials, as well as LTP.
A mechanism to terminate neurotransmitter action exists.
After acidification, we found that extracellular pH rapidly recovered and alkalinized. Several processes may restore and increase pH, including proton diffusion, pH buffering, and activity of membrane transporters (11). Increased interstitial pH after neurostimulation has also been reported in other preparations (11, 42, 47). Previous studies showed that raising extracellular pH before an acid stimulus reduced steady-state inactivation and increased the amplitude of ASIC currents (43, 44, 48). Thus, alkalinization might prepare postsynaptic ASICs for subsequent stimuli, and thereby maximize their current.
Blocking the receptor blocks the activity of the neurotransmitter.
Disrupting the ASIC1 gene eliminated the effect of exogenously applied protons, ASIC-dependent EPSCs, and acid-evoked LTP. Amiloride and PcTX1 also blocked ASIC EPSCs.
Additional Implications.
First, the results raise the question of whether protons function as a neurotransmitter entirely on their own. Although exogenous acid can activate action potentials, protons might not transmit information in isolation of other transmitters. We say that because when we puffed pH 6.8 solution into amygdala brain slices, LTP induction required delivery of a test pulse to the presynaptic neuron. In addition, compared with glutamate-dependent EPSCs, ASIC EPSCs are small.
Second, every time glutamate is released into a synapse, protons may be coreleased. Co-release has a parallel with other neurotransmitters (49): Co-release implies that proton exocytosis need not have a unique regulatory mechanism; acidification would be initiated by the same Ca2+-dependent exocytotic release mechanisms that regulate glutamate release.
Third, both the presence of ASICs in many brain areas (14–17, 22, 38) and a source for proton release at other sites position protons to function as a neurotransmitter in combination with other neurotransmitters and in many brain regions.
Fourth, an acidic pH and ASICs are associated with neuronal injury in models of cerebral ischemia (21, 50), multiple sclerosis (51), and traumatic brain injury (52). Function of protons as a neurotransmitter suggests involvement of excitotoxicity-like mechanisms.
Fifth, what is the physiological role of proton–ASIC signaling in neurotransmission? Compared with GluR currents, ASIC currents make a very small contribution to EPSCs induced by a single test pulse. Thus, we suspect that proton–ASIC signaling has little effect on neurotransmission under basal conditions. That conclusion is consistent with studies of ASIC1a−/− mice, which show little behavioral difference from WT mice in the absence of stress. However, we speculate that proton–ASIC signaling may be particularly important during intense presynaptic stimulation. Extracellular pH reductions are the greatest during HFS and could have at least two effects: The lower pH would generate larger ASIC currents, and in addition, the reduced pH may inhibit NMDA receptors (53), and thus concurrent activation of ASICs could sustain and enhance synaptic transmission. These effects could explain the involvement of protons and the requirement of ASICs for LTP induced by HFS. Consistent with these conclusions, both ASICs and GluRs are required for normal behavioral responses to stresses such as amygdala-dependent fear-related learning and memory.
Materials and Methods
Also see SI Materials and Methods.
Mice.
ASIC1a−/−, ASIC2−/−, and WT mice were maintained on a congenic C57BL/6 background. The University of Iowa Animal Care and Use Committee approved all procedures.
Brain Slices.
We used standard procedures to generate brain slices from 4–6-wk-old mice. For experiments with a varying HCO3− concentration, changes in HCO3− were balanced with gluconate, and Cl− concentration was maintained constant. Acidic solutions were delivered by direct injection into slices, using a microinjection system, or to the entire slice through the perfusion system.
Patch-Clamp Recording.
Standard procedures were used to patch-clamp lateral amygdala pyramidal neurons.
Measurement of Extracellular pH in Brain Slice Cultures.
Amygdala slice cultures were prepared from 3–4-d-old mice with procedures modified from our earlier work (23). A syndecan 2-pHluorin fusion was generated by inserting pHluorin sequence into mouse syndecan 2 cDNA immediately after the signal peptide sequence to generate a fusion with extracellular pHluorin. Biolistic transfection of slices was after 1–3 wk in culture, and slices were studied 48 h later. pHluorin signal was detected with a high-speed confocal microscope. Fluorescence of HeLa cells was measured 24–48 h after transfection.
Analysis.
Data are presented as means ± SEM. To statistically assess LTP, we analyzed the last 5 min of EPSPs of each LTP recording (total 30 EPSP points), averaged those 30 points, and then compared those data with average data before HFS. Statistical comparison of groups used one-way ANOVA and Tukey’s post hoc multiple comparison test. A Student t test was used to compare two groups.
Supplementary Material
Acknowledgments
We thank Jayasankar Jasti, Jacob Kundert, Rebecca Taugher, and Collin Kreple for assistance. We thank Dr. Christopher Benson, Dr. Peter Snyder, Dr. Amy Lee, Dr. Charles Harata, and Dr. Sodikdjon Kodirov for their comments. J.D. and M.J.W. are supported by the Howard Hughes Medical Institute. L.R.R. is supported by the Iowa Cardiovascular Interdisciplinary Research Fellowship (HL07121). J.A.W. is supported by the Department of Veterans Affairs and National Institutes of Mental Health (1R01MH085724-01).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407018111/-/DCSupplemental.
References
- 1.Füldner HH, Stadler H. 31P-NMR analysis of synaptic vesicles. Status of ATP and internal pH. Eur J Biochem. 1982;121(3):519–524. doi: 10.1111/j.1432-1033.1982.tb05817.x. [DOI] [PubMed] [Google Scholar]
- 2.Michaelson DM, Angel I. Determination of delta pH in cholinergic synaptic vesicles: Its effect on storage and release of acetylcholine. Life Sci. 1980;27(1):39–44. doi: 10.1016/0024-3205(80)90017-x. [DOI] [PubMed] [Google Scholar]
- 3.Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 4.DeVries SH. Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron. 2001;32(6):1107–1117. doi: 10.1016/s0896-6273(01)00535-9. [DOI] [PubMed] [Google Scholar]
- 5.Palmer MJ, Hull C, Vigh J, von Gersdorff H. Synaptic cleft acidification and modulation of short-term depression by exocytosed protons in retinal bipolar cells. J Neurosci. 2003;23(36):11332–11341. doi: 10.1523/JNEUROSCI.23-36-11332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vessey JP, et al. Proton-mediated feedback inhibition of presynaptic calcium channels at the cone photoreceptor synapse. J Neurosci. 2005;25(16):4108–4117. doi: 10.1523/JNEUROSCI.5253-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietrich CJ, Morad M. Synaptic acidification enhances GABAA signaling. J Neurosci. 2010;30(47):16044–16052. doi: 10.1523/JNEUROSCI.6364-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res. 1987;436(2):352–356. doi: 10.1016/0006-8993(87)91678-7. [DOI] [PubMed] [Google Scholar]
- 9.Gottfried JA, Chesler M. Temporal resolution of activity-dependent pH shifts in rat hippocampal slices. J Neurophysiol. 1996;76(4):2804–2807. doi: 10.1152/jn.1996.76.4.2804. [DOI] [PubMed] [Google Scholar]
- 10.Kaila K, Chesler M. Activity-evoked changes in extracellular pH. In: Kaila K, Ransom BR, editors. pH and Brain Function. New York: Wiley-Liss Inc; 1998. p 309. [Google Scholar]
- 11.Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83(4):1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- 12.Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15(10):396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- 13.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14(7):461–471. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherwood TW, Frey EN, Askwith CC. Structure and activity of the acid-sensing ion channels. Am J Physiol Cell Physiol. 2012;303(7):C699–C710. doi: 10.1152/ajpcell.00188.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gründer S, Chen X. Structure, function, and pharmacology of acid-sensing ion channels (ASICs): Focus on ASIC1a. Int J Physiol Pathophysiol Pharmacol. 2010;2(2):73–94. [PMC free article] [PubMed] [Google Scholar]
- 16.Deval E, et al. Acid-sensing ion channels (ASICs): Pharmacology and implication in pain. Pharmacol Ther. 2010;128(3):549–558. doi: 10.1016/j.pharmthera.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: Advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29(10):578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Chu XP, Xiong ZG. Physiological and pathological functions of acid-sensing ion channels in the central nervous system. Curr Drug Targets. 2012;13(2):263–271. doi: 10.2174/138945012799201685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zha XM. Acid-sensing ion channels: Trafficking and synaptic function. Mol Brain. 2013;6:1. doi: 10.1186/1756-6606-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wemmie JA, et al. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34(3):463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- 21.Xiong ZG, et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 22.García-Añoveros J, Samad TA, Zuvela-Jelaska L, Woolf CJ, Corey DP. Transport and localization of the DEG/ENaC ion channel BNaC1alpha to peripheral mechanosensory terminals of dorsal root ganglia neurons. J Neurosci. 2001;21(8):2678–2686. doi: 10.1523/JNEUROSCI.21-08-02678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zha X-M, Wemmie JA, Welsh MJ. Acid-sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci USA. 2006;103(44):16556–16561. doi: 10.1073/pnas.0608018103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zha XM, et al. ASIC2 subunits target acid-sensing ion channels to the synapse via an association with PSD-95. J Neurosci. 2009;29(26):8438–8446. doi: 10.1523/JNEUROSCI.1284-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hruska-Hageman AM, Benson CJ, Leonard AS, Price MP, Welsh MJ. PSD-95 and Lin-7b interact with acid-sensing ion channel-3 and have opposite effects on H+- gated current. J Biol Chem. 2004;279(45):46962–46968. doi: 10.1074/jbc.M405874200. [DOI] [PubMed] [Google Scholar]
- 26.Hruska-Hageman AM, Wemmie JA, Price MP, Welsh MJ. Interaction of the synaptic protein PICK1 (protein interacting with C kinase 1) with the non-voltage gated sodium channels BNC1 (brain Na+ channel 1) and ASIC (acid-sensing ion channel) Biochem J. 2002;361(Pt 3):443–450. doi: 10.1042/0264-6021:3610443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu H, et al. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21(24):9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duggan A, Garcia-Anoveros J, Corey DP. The PDZ domain protein PICK1 and the sodium channel BNaC1 interact and localize at mechanosensory terminals of dorsal root ganglion neurons and dendrites of central neurons. J Biol Chem. 2002;277(7):5203–5208. doi: 10.1074/jbc.M104748200. [DOI] [PubMed] [Google Scholar]
- 29.Baron A, et al. Protein kinase C stimulates the acid-sensing ion channel ASIC2a via the PDZ domain-containing protein PICK1. J Biol Chem. 2002;277(52):50463–50468. doi: 10.1074/jbc.M208848200. [DOI] [PubMed] [Google Scholar]
- 30.Jovov B, Tousson A, McMahon LL, Benos DJ. Immunolocalization of the acid-sensing ion channel 2a in the rat cerebellum. Histochem Cell Biol. 2003;119(6):437–446. doi: 10.1007/s00418-003-0525-4. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez de la Rosa D, et al. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol. 2003;546(Pt 1):77–87. doi: 10.1113/jphysiol.2002.030692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho JH, Askwith CC. Presynaptic release probability is increased in hippocampal neurons from ASIC1 knockout mice. J Neurophysiol. 2008;99(2):426–441. doi: 10.1152/jn.00940.2007. [DOI] [PubMed] [Google Scholar]
- 33.Wu PY, et al. Acid-sensing ion channel-1a is not required for normal hippocampal LTP and spatial memory. J Neurosci. 2013;33(5):1828–1832. doi: 10.1523/JNEUROSCI.4132-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowan WM, Kandel ER. A brief history of synapses and synaptic transmission. In: Cowan WM, Südhoff TC, Stevens CF, Davies K, editors. Synapses. Baltimore: Johns Hopkins University Press; 2001. pp. 1–87. [Google Scholar]
- 35.Beg AA, Ernstrom GG, Nix P, Davis MW, Jorgensen EM. Protons act as a transmitter for muscle contraction in C. elegans. Cell. 2008;132(1):149–160. doi: 10.1016/j.cell.2007.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziemann AE, et al. The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell. 2009;139(5):1012–1021. doi: 10.1016/j.cell.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wemmie JA, et al. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23(13):5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price MP, et al. Localization and behaviors in null mice suggest that ASIC1 and ASIC2 modulate responses to aversive stimuli. Genes Brain Behav. 2014;13(2):179–194. doi: 10.1111/gbb.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 40.Escoubas P, et al. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J Biol Chem. 2000;275(33):25116–25121. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- 41.Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem. 2004;279(18):18296–18305. doi: 10.1074/jbc.M312145200. [DOI] [PubMed] [Google Scholar]
- 42.Tong CK, Chen K, Chesler M. Kinetics of activity-evoked pH transients and extracellular pH buffering in rat hippocampal slices. J Neurophysiol. 2006;95(6):3686–3697. doi: 10.1152/jn.01312.2005. [DOI] [PubMed] [Google Scholar]
- 43.Babini E, Paukert M, Geisler HS, Gründer S. Alternative splicing and interaction with di- and polyvalent cations control the dynamic range of acid-sensing ion channel 1 (ASIC1) J Biol Chem. 2002;277(44):41597–41603. doi: 10.1074/jbc.M205877200. [DOI] [PubMed] [Google Scholar]
- 44.Sherwood TW, Lee KG, Gormley MG, Askwith CC. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J Neurosci. 2011;31(26):9723–9734. doi: 10.1523/JNEUROSCI.1665-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vukicevic M, Kellenberger S. Modulatory effects of acid-sensing ion channels on action potential generation in hippocampal neurons. Am J Physiol Cell Physiol. 2004;287(3):C682–C690. doi: 10.1152/ajpcell.00127.2004. [DOI] [PubMed] [Google Scholar]
- 46.Wu LJ, et al. Characterization of acid-sensing ion channels in dorsal horn neurons of rat spinal cord. J Biol Chem. 2004;279(42):43716–43724. doi: 10.1074/jbc.M403557200. [DOI] [PubMed] [Google Scholar]
- 47.Fedirko N, Svichar N, Chesler M. Fabrication and use of high-speed, concentric h+- and Ca2+-selective microelectrodes suitable for in vitro extracellular recording. J Neurophysiol. 2006;96(2):919–924. doi: 10.1152/jn.00258.2006. [DOI] [PubMed] [Google Scholar]
- 48.Benson CJ, Eckert SP, McCleskey EW. Acid-evoked currents in cardiac sensory neurons: A possible mediator of myocardial ischemic sensation. Circ Res. 1999;84(8):921–928. doi: 10.1161/01.res.84.8.921. [DOI] [PubMed] [Google Scholar]
- 49.Hnasko TS, Edwards RH. Neurotransmitter corelease: Mechanism and physiological role. Annu Rev Physiol. 2012;74:225–243. doi: 10.1146/annurev-physiol-020911-153315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA. 2004;101(17):6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friese MA, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13(12):1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 52.Yin T, et al. Loss of Acid sensing ion channel-1a and bicarbonate administration attenuate the severity of traumatic brain injury. PLoS ONE. 2013;8(8):e72379. doi: 10.1371/journal.pone.0072379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature. 1990;345(6273):347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.