

Abstract
Diabetes is associated with an increased risk of sudden cardiac death, but the underlying mechanisms remain unclear. Our goal was to investigate changes occurring in the action potential duration (APD) and conduction velocity (CV) in the diabetic rabbit ventricle, and delineate the principal ionic determinants. A rabbit model of alloxan-induced diabetes was utilized. Optical imaging was used to record electrical activity in isolated Langendorff-perfused hearts in normo-, hypo- and hyper-kalemia ([K+]o=4, 2, 12 mM respectively). Patch clamp experiments were conducted to record Na+ current (INa) in isolated ventricular myocytes. The mRNA/protein expression levels for Nav1.5 (the α-subunit of INa) and connexin-43 (Cx43), as well as fibrosis levels were examined. Computer simulations were performed to interpret experimental data. We found that the APD was not different, but that the CV was significantly reduced in diabetic hearts in normo-, hypo-, and, in hyper-kalemic conditions (13%, 17% and 33% reduction in diabetic vs. control, respectively). The cell capacitance (Cm) was increased (by ~14%), and the density of INa was reduced by ~32% in diabetes compared to controls, but the other biophysical properties of INa were unaltered. The mRNA/protein expression levels for Cx43 were unaltered. For Nav1.5, the mRNA expression was not changed, and though the protein level tended to be less in diabetic hearts, this reduction was not statistically significant. Staining showed no difference in fibrosis levels between the control and diabetic ventricles. Computer simulations showed that the reduced magnitude of INa was a key determinant of impaired propagation in the diabetic ventricle, which may have important implications for arrhythmogenesis.
Keywords: Diabetes Rabbit Cardiac Electrophysiology Na+-current
1. Introduction
It is estimated that ~26 million people have diabetes in the US alone [1]. The majority have type-2, while 5–10% have the type-1 form [2]. Cardiovascular complications are a significant cause of mortality and morbidity in diabetic patients [3]. Diabetes has also been associated with an increased risk of arrhythmias and sudden cardiac death [4,5]. However, the underlying mechanisms remain poorly understood.
It has been shown that diabetes (both type 1 and 2) is associated with ECG abnormalities such as QT prolongation and increased QT dispersion in patients [6,7], suggesting irregularities in cardiac repolarization. To examine the underlying mechanisms, studies have been conducted in animal models of diabetes [8,9]. In rats with streptozotocin (STZ) induced diabetes, the ventricular action potential duration (APD) is prolonged, which has been mainly attributed to the downregulation of the transient outward K+ current (Ito) [8,9]. Although studies using the rodent models of diabetes have yielded many useful insights, the cardiac ventricular action potential profile (shape and duration) and the underlying ionic currents in rodents are very different from those observed in humans [10]. Rodents have a short, triangular action potential (AP) profile, and the key repolarizing delayed rectifier K+ currents in the human ventricle, i.e. the fast and the slow delayed rectifiers, namely IKr, and IKs respectively, are functionally not active in rodents [10]. In contrast, rabbit, guinea pig and canine hearts present a longer AP with a characteristic plateau that is typical of the human ventricular AP. Furthermore, in rabbits, IKr and IKs constitute the key repolarizing currents, and these currents show similar biophysical characteristics to those found in humans [10,11]. As a result, in the last 6–7 years, several studies have utilized rabbit/canine/guinea pig models to study the electrophysiological/ionic alterations in diabetes [12–16].
In a rabbit model of alloxan-induced diabetes, Zhang et al [14] showed considerable prolongation of heart-rate-corrected QT interval (QTc) and APD, and found that this was in part due to a substantial reduction in the density of IKr. In contrast, Lengyel et al [12] showed a small increase in QTc, and a reduced density of IKs in the diabetic rabbit hearts, but observed no alterations in the density/properties of IKr. In the canine model of diabetes, only little to moderate QTc and APD prolongation were shown, with decreases in Ito and IKs, but no change in IKr was observed [15]. A recent study showed that the ventricular APD was not altered in the diabetic guinea pig ventricle [16]. Thus, the reports regarding the APD changes in diabetes in higher animal models show varied and conflicting results.
An alternative explanation for enhanced arrhythmia risk in diabetic hearts may be impaired cardiac conduction. Nygren et al [17] used optical mapping in hearts from streptozotocin (STZ) induced diabetic rats (7–14 days post-injection) to show that, while there was no difference between diabetic and control at lower extracellular K+ levels ([K+]o=5.9mM), elevated potassium ([K+]o=9mM) caused significantly slowing of conduction velocity (CV) in the diabetic hearts. They were also able to demonstrate that the CV was slower in diabetes compared to control hearts, when challenged with experimental conditions mimicking ischemia/low pH [18]. Studies in a mouse model with cardiomyocyte-specific knock out of insulin receptors (CIRKO) showed similar results [19]. Recent results from optical mapping studies in the diabetic guinea pig ventricle showed that the CV was reduced by ~14% [16]. However, the underlying ionic mechanisms of the slower CV in diabetes remain unclear.
The objective of our study was to study the cardiac electrophysiology alterations and also determine their underlying mechanisms by utilizing a rabbit model of diabetes. Diabetes in this model was induced by injecting alloxan monohydrate, which destroys pancreatic-β cells, and is thus more representative of type 1- diabetes. Our results suggest that the APD is not altered, but CV is slower in the diabetic rabbit ventricle, compared to healthy controls. A reduced density of the Na+ current, INa, is a key determinant of this impaired impulse propagation.
2. Materials and Methods
Male New Zealand White rabbits were obtained from Harlan Laboratories. The investigation conformed to the United States National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health publication no. 85–23, revised 1996), and protocols approved by the local University Committee on Use and Care of Animals at the University of Michigan, Ann Arbor.
2.1 Induction of Diabetes
Diabetes was induced using techniques adapted from published studies [13–15, 20]. A single injection of alloxan monohydrate (140–160 mg/kg body weight) was administered, via the ear vein during brief sedation (via a combination of ketamine/xylazine). To reduce risk of nephrotoxicity from hyperuricemia, a 7 ml/kg body weight intravenous injection of 0.9% saline was given immediately after the injection of alloxan. To counteract initial hypoglycemia, 5% glucose was provided in the drinking bottle ad libitum for 24 h. Blood collected via the ear vein was used for determination of the plasma level of glucose with a glucometer (Abbott MediSense Precision Xtra). After alloxan injection, rabbits in the diabetic state were maintained for 146±16 days, after which non-survival surgery was performed and the heart removed. Characteristics of the rabbits that were successfully made diabetic and used in our experiments are shown in Table 1 in the online data supplement. Diabetic rabbits were hyperglycemic, compared to controls (399±26 mg/dL vs. 148±10 mg/dL; p<0.05). In some rabbits, the ECG parameters (Table 2), and the serum lipid levels were measured (Fig. S2), and these values are reported in the online supplement.
2.2 Optical mapping in Langendorff-perfused isolated hearts
Optical mapping was performed as described in our previous studies [21,22]. Briefly, following sedation (ketamine and xylazine; 10–40 mg/kg, and 3–5 mg/kg respectively, IM), rabbits were treated with heparin and anesthetized with sodium pentobarbital (50–100 mg/kg intravenously via ear vein). Upon reaching surgical-level anesthesia, hearts were quickly removed and placed in cold cardioplegic solution. Hearts were cannulated and perfused under constant pressure (70 mm Hg) with standard Tyrode’s solution (composition as in previous studies [21]), pH 7.4, and continuously gassed with 95% O2, 5% CO2. The heart was immersed in the same Tyrode’s solution, in a custom-made plastic chamber, and the temperature of both the perfusate leaving the cannula and that in the superfusion chamber was maintained at 35.5±1.5°C. Blebbistatin (5–10μM) was added to the perfusate to uncouple electrical impulses from contraction, thus minimizing motion artifact. Blebbistatin has been shown to have minimal effect on rabbit ventricular electrophysiology [23]. A 1–2 ml bolus of the voltage-sensitive dye Di-4-ANEPPS (10 μM) was added to the perfusion line. Green light from a 532nm, 1 Watt laser was directed at the anterior surface of the heart and emitted light passed through a 650±50nm filter before being captured by a high-resolution 80×80 pixel Little Joe CCD camera, at 1kHz sampling frequency [22]. Typical duration of a recorded optical movie was 5 seconds. The hearts were paced from the RV epicardium, using a bipolar electrode, at 4Hz (250ms cycle length). This allowed us to capture and pace the ventricle over and above the intrinsic pacing cycle length (~2–3 Hz), without crushing the SA node. APD and CV measurements were constructed at different extracellular potassium concentrations, in normo-, hypo- and hyper-kalemia (4, 2 and 12mM [K+]o respectively). The choice of high [K+]o (12 mM) in our studies was dictated by the goal to avoid the “biphasic response phase” of the CV in cardiac tissue, i.e. CV is increased, when [K+]o is increased from 4 to 8 mM, but goes down above ~10 mM, as was shown in previous experiments [24] and simulation studies [25]. Analytical tools developed by members of our laboratory (Dr. Sergey Mironov: SCROLL) were used to calculate APDs/CV in these hearts, as in previous studies [21,22]. APD at 70% repolarization (APD70) was quantified.
2.3 Patch Clamp Experiments
Hearts were enzymatically digested as previously described [21,25]. Tissue from the ventricular epicardium was separated following enzymatic digestion, and myocytes were isolated by mincing and pulsatile agitation. Whole-cell voltage-clamp experiments were then performed. Voltage-dependent steady-state inactivation, recovery from inactivation, as well as current density of INa was measured using standard protocols, as in previous studies [27,28]. A low extracellular Na+ concentration (5mM) was used to ensure adequate voltage control. Recordings were performed at 22°C.
2.4 Quantitative PCR (qPCR)
Tissue samples for qPCR were obtained from hearts prior to myocyte isolation (for patch clamp experiments above). Following clearance of the blood by perfusion, but prior to enzymatic digestion, LV epicardial tissue samples were taken and placed into RNAlater (Qiagen). RNAlater samples were processed using the RNeasy mini kit (Qiagen) to obtain RNA, and SuperScript™ III First-Strand Synthesis System for RT-PCR (Invitrogen) was used to obtain cDNA. qPCR was performed using SYBR Green PCR Master Mix (Applied Biosystems), StepOnePlus Real-Time PCR machine (Applied Biosystems) and primers for cardiac myosin heavy chain (MHC), Nav1.5, Kir2.1 and Cx43, sequences for which were taken from Tellez et al [29]. Expression of each gene was calculated based on a cDNA titration within each plate.
2.5 Western blots
Tissue samples for western blots were taken from hearts prior to myocyte isolation (for patch clamp experiments above) and flash frozen in liquid nitrogen. Samples were stored at −80°C until being homogenized with a polytron in cold ice homogenization buffer (25 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 2mM Na3VO4, 4 mM NaF, 1 % Triton 100X, phosphatase inhibitor cocktail (Roche)). Homogenates were centrifuged at 4°C for 10 minutes at 10,000 rpm. Supernatants were aliquoted, and protein concentrations were determined using a modified Lowry assay (Pierce). SDS/PAGE was carried out according to the method in the work by Laemmli [30]. Briefly, 25 μg of supernatant protein samples were run on 4–20% precast acrylamide gels (Invitrogen) and transferred to nitrocellulose membranes (Bio-Rad). Nonspecific binding sites were blocked with 5% nonfat dry milk in PBS with Tween-20 (0.05%). Membranes were then incubated with Cx43 (Millipore), Nav1.5 (Alomone), and GAPDH (Sigma) antibodies diluted in 5% nonfat dry milk overnight at 4 °C. After washing, membranes were incubated with peroxidase-conjugated secondary antibodies. Protein bands were visualized using enhanced chemiluminescence (Pierce) and ChemiDoc MP Imaging System (BIorad). Protein quantification was determined by densitometric analysis using Image Lab (Bio-Rad) software.
2.6 Staining for fibrosis
After conducting the optical mapping experiments, the hearts were fixed in 10% neutral buffered formalin and transmural samples of the anterior LV free wall were taken. Samples were paraffin-embedded and sectioned (4 μm thickness), and H&E staining was conducted by core facilities at the University of Michigan. Picrosirius red staining was performed according to previously published methods [31]. Quantitative evaluations of fibrosis were performed on images taken at 20x magnification using a 12bit color CCD camera (Qicam, Qimaging) installed on a Zeiss Axio imager A1 microscope. Areas occupied by fibrotic tissue were automatically measured with Bioquant’s Life Science package software by employing a color thresholding technique. Images were obtained from immediately beneath the LV epicardium across the full length of each sample (7–17 images per sample). Perivascular and epicardial fibrosis were excluded from the analysis by setting regions of interest.
2.7 Computer simulations
We performed simulations for cardiac propagation using a very thin, rectangular sheet (dimensions of 0.3 mm× 10 mm, or 3 × 100 cells), that incorporated the Luo-Rudy ventricular ionic model [32], and solved for the reaction diffusion equation
to understand and interpret the experimental results. The numerical techniques were similar to those employed in previous studies [33]. Here V denotes membrane voltage, Cm is membrane capacitance, Iion is the sum of all ionic currents in the ventricular cell, and D is the diffusion coefficient.
2.8 Statistical analysis
Values are reported as mean ± standard error of the mean (SEM). Control and diabetic data were compared using unpaired Student’s t-test. Differences were considered significant at p<0.05.
3. Results
Optical mapping was used to record electrical activity in Langendorff-perfused hearts. At a pacing cycle length of 250 ms (4 Hz) under normal extracellular K+ concentration ([K+]o =4 mM), APD70 in controls was 139.5±2.958 ms (N=8), compared to 137.3±1.618 ms (N=7) in diabetic hearts (p=NS) (Fig. 1A). In contrast, the CV at [K+]o=4 mM was significantly slower (by ≈13%) in diabetic hearts, compared to controls (CV = 0.53±0.02 m/s in control vs. 0.46±0.02 m/s in diabetes, p<0.05). Some earlier studies suggested that IKr was reduced in diabetic hearts [13,14], whereas others observed no change [12,15]. Since the conductance of IKr is proportional to the square root of [K+]o [34], we hypothesized that stressing the heart with changes in [K+]o would exacerbate any differences in APD between control and diabetic hearts. Therefore, we initially subjected the hearts to hypokalemia ([K+]o = 2mM). The expected increase in APD and reduction in CV in hypokalemia compared to normokalemia was observed in both control and diabetic hearts. At a pacing cycle length of 250 ms under hypokalemic conditions, APD70 in control hearts was 152.0±2.376 ms (N=7), and not different compared to diabetic hearts, at 150.8±2.363 ms (N=6; p=NS) (Fig. 1B). The CV in hypokalemic conditions was again significantly slower (by ≈17%) in diabetic hearts, compared to controls (CV = 0.464±0.016 m/s in control vs. 0.385±0.006 m/s in diabetes, p<0.05). The hearts were then subjected to hyperkalemia ([K+]o = 12mM). The expected decrease in APD and reduction in CV in hyperkalemia compared to normokalemia was observed in both control and diabetic hearts. In hyperkalemia ([K+]o=12mM), we were able to obtain 1:1 pacing at 4Hz in 5 control hearts, and in 3 diabetic hearts. At a pacing cycle length of 250 ms under hyperkalemic conditions, the APD70 in control hearts was 117.2±4.497 ms, compared to 126.2±5.037 ms in diabetic hearts (p=NS) (Fig. 1C). The difference in CV was further enhanced in these hyperkalemic conditions, with ≈33% slower CV in diabetic versus control hearts; CV = 0.33±0.02 m/s in control (n=5) vs. 0.22±0.02 m/s in diabetes (n=3), p<0.05 (Figure 1C).
Figure 1. Quantifying basic electrophysiology parameters via optical mapping in isolated hearts.

Comparison of APD70 (left panels) and conduction velocity (CV) (right panels), at BCL=250 ms, in control versus diabetic hearts in (A.) normal [K+]o levels (4mM), (B.) low [K+]o levels (2mM), hypokalemia, and (C.) high [K+]o levels (12mM), hyperkalemia.
Figure 2 depicts representative activation maps on the anterior epicardial ventricular surface of a control and a diabetic rabbit heart during hyperkalemia at a pacing cycle length of 250 ms; the slower impulse propagation is evident in the diabetic heart, compared to controls (* represents the pacing site).
Figure 2. Representative activation maps.
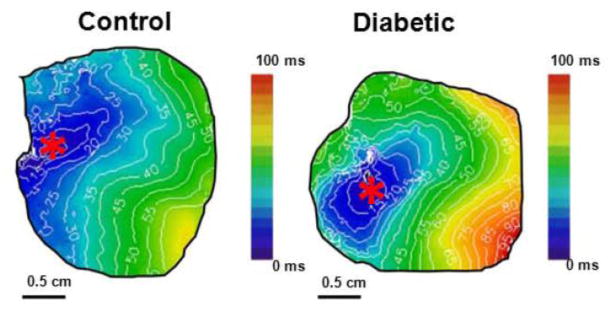
Comparison of representative activation maps of cardiac propagation in control and diabetic hearts in hyperkalemia ([K+]o=12mM) at BCL=250 ms. “*” = pacing site.
Since our optical mapping experiments showed a slower CV in diabetic hearts, we conducted patch clamp experiments in isolated ventricular myocytes from control (N=3) and diabetic hearts (N=3), focusing on the main determinant of the cardiac excitability, i.e. the Na+ current, INa, and the data are shown in Figure 3. The cell capacitance (Cm) values were 126.10±7.62 pF and 144.35±6.66 pF in control versus diabetic cells (p=0.04). Thus Cm was increased by ~14% in diabetic cells. Fig. 3A shows representative voltage-clamp traces of INa in a control versus diabetic ventricular myocyte. The peak current density at different membrane voltages is quantified in a current-voltage (I–V) plot in Fig 3B. At −40 mV, the peak current INa density was −22.59±1.83 pA/pF in control cells (n=12), compared to −15.42±1.17 pA/pF in diabetic cells (n=17) (p<0.02). Thus there was ~32% reduction in INa peak current density in the diabetic group at this voltage. In contrast, there were no differences in the steady-state activation and inactivation properties Fig. 3C (the V1/2 values were not different), or the recovery from inactivation kinetics (Fig. 3D). To confirm that the observed reduction in INa was not due to age differences between control and diabetic hearts, we conducted further additional experiments in young (~16 weeks old, 2.5 Kg weight) vs. more older (~43 weeks old, 3.4 Kg weight) rabbits. There were no age-dependent changes in the biophysical properties and the density of INa (supplemental figure S1).
Figure 3. Patch clamp experiments for INa in control versus diabetic ventricular cells.

(A.) Representative recordings of INa. (B.) I–V relationship. (C.) Steady-state activation and inactivation curves. (D.) Recovery from inactivation. [n = 12–17 (N=3)].
To gain insights into the possible molecular basis of slower propagation in diabetic hearts, we assessed the mRNA levels of important ionic determinants of CV, including that of INa (Nav1.5; the α subunit of the sodium channel responsible for INa), and connexin-43 (Cx43). Analysis of gene expression by qPCR revealed no significant differences in expression of Nav1.5 and Cx43 between control and diabetic groups (Fig. 4A). To account for variability in the ratio of myocytes to other cell types in the tissue samples, ion channel mRNA levels were normalized to cardiac myosin heavy chain (MHC) expression levels. The MHC primers used recognize a region of sequence identity between the two cardiac MHC isoforms. There was no difference in MHC expression level between diabetic and control groups (supplemental figure S4). If GAPDH was instead used for normalization of expression levels, variability (standard deviation) increased but the results were not fundamentally different (supplemental figure S4). Data presented in Fig. 4B and 4C show the expression levels of Nav1.5 and Cx43 proteins in samples of epicardial left ventricle from control and diabetic rabbits. In diabetic rabbits the relative expression levels of Nav1.5 show a reduction compared to those from control rabbits, although the aggregate difference is not statistically significant (p=0.114, N=4). The expression of Cx43 appears unchanged in the diabetic rabbits, when compared to controls.
Figure 4. mRNA and protein expression of Nav1.5 and Cx43.

(A.) mRNA Expression levels of Nav1.5, and Cx43 in control vs. diabetic rabbit LV epicardial tissue. N=5 per group. (B.) Immunoblot showing immunodetection of Nav1.5 (Top), Cx43 expression (Middle), and GAPDH (loading control; Bottom) from epicardial left ventricle samples of control (C) and diabetic (D) rabbits. (C.) Densitometric analysis of Cx43 and Nav1.5 expression normalized to GAPDH levels in control and diabetic rabbits. Values represent data (mean ± SEM) from 4 animals in each condition.
In addition to changes in ionic currents, a major determinant of CV is the degree of interstitial fibrosis. For this reason, we stained for fibrosis with picrosirius red in ventricular epicardial tissue from control and diabetic rabbit hearts, and representative images are shown in Fig. 5A. Quantification of fibrosis levels from control (N=7) and diabetic rabbits hearts (N=7) revealed no statistically significant differences (Fig. 5B).
Figure 5. Quantification of interstitial fibrosis in LV rabbit epicardial tissue.
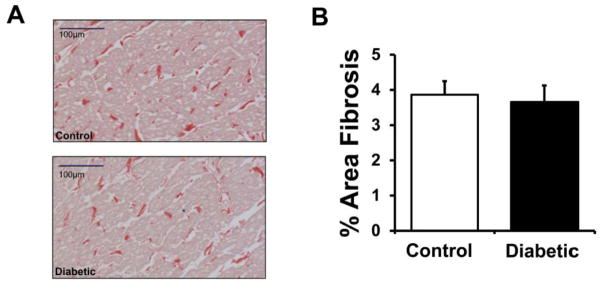
(A.) Representative examples of stained sections (red = collagen). (B.) Quantification of % area fibrosis in control (N=7) and diabetic (N=7) rabbit samples.
To interpret the experimental results, we conducted computer simulations using the Luo-Rudy ionic model [32], in a thin rectangular sheet, whose dimensions, the location of the stimulus, and the measurements points (for calculating CV) are shown in Fig. 6A. When [K+]o was decreased (hypokalemia, 2 mM) or increased (hyperkalemia, 12 mM), compared to controls, the simulated CV was changed as expected, in both control conditions and diabetes, in accordance with experimental results as shown in Fig. 6B (the diabetes condition was simulated by a decrease in the conductance of INa). The effects of changes in individual variables on the simulated CV are shown (Fig. 6C). In accordance with patch clamp experiments, when the maximum conductance of INa was reduced by 30%, this modification was sufficient to explain a 25% reduction in CV during hyperkalemia, and a 10–12% reduction in normal conditions and hypokalemia. Increasing Cm by 14% (as was seen in the patch-clamp experiments) in the reaction diffusion equation also decreased the CV, but to a much smaller extent than that seen due to changes in INa. A reduction in the diffusion coefficient D alone by 20%, to partially mimic gap junctional uncoupling, reduced CV by ~ 10% across all K+ concentrations (Fig. 6C). Further, it has been suggested that the Na+-K+ pump function is impaired in diabetes [20]. To assess its potential impact, we reduced the Na+/K+ pump current in the model by 30%. This had little impact on the CV in normo- and hypokalemia, but reduced the CV by ~5% at 12mM K+ (Fig. 6C). Thus, the numerical results support the hypothesis that a reduction in INa is largely responsible for the reduced CV in the diabetic hearts.
Figure 6. Computer simulations.

(A.) Schematic of 2D model. (B.) Conduction velocity (CV) in simulation vs. experiments in normokalemia, hypokalemia, hyperkalemia in both control and diabetic hearts; diabetes was simulated by reducing the density of INa by 30%. (C.) Reduction in CV when Na+ current magnitude is reduced by 30%, gap junction coupling is reduced by 20%, Na+-K+ pump current is reduced by 30%, and Cm is increased by 14% in the reaction diffusion equation.
4. Discussion
We have used a combination of optical mapping, patch clamp, immunohistochemistry, molecular biology and mathematical modeling techniques to provide mechanistic insights into the electrophysiological alterations produced by alloxan-induced diabetes in the isolated rabbit heart. The main new findings from our study are the following: 1.) there is no change in the APD, but the CV is slower in the ventricle of the diabetic rabbit hearts, in normokalemia, hypo- and hyperkalemia. 2.) Cm is increased, and the density of INa is reduced in diabetic ventricle when compared to controls, but the other biophysical properties of INa remain unchanged. 3.) We found no changes in fibrosis, or the mRNA/protein levels of Cx43 in diabetic versus control hearts, 4.) The mRNA levels of Nav1.5 were not changed, and though the protein levels tended to be less in diabetic compared to control hearts, the difference was not statistically significantly. 5.) Computer simulations showed that the reduced INa density could largely mimic the experimentally observed reduction in CV in the diabetic rabbit heart across variations in [K+]o.
Our results regarding no changes in APD in the diabetic ventricle compared to controls are more similar to the reports of Lengyel et al in rabbits [12] and Xie in guinea pigs [16]. Further, the APD remains unchanged between diabetic and control hearts under conditions of hypo- and hyperkalemia. This result indirectly suggests that the density of IKr is not likely changed, since the conductance of IKr is proportional to the square root of [K+]o [34], and hypo/hyperkalemia would have further amplified the APD differences with respect to control, if the IKr density was indeed reduced in diabetic hearts. Our finding of a reduced CV is similar to that reported in studies conducted in diabetic guinea pigs [16] and rats [17,18]. It was also shown that the Cx43 was lateralized in diabetic rat hearts [17]. Further, using computer simulations, these investigators predicted that changes in INa properties could possibly explain their experimental findings [36]. Indeed, our experiments and simulations confirm an important role for INa in the impaired propagation in diabetes. Further when Cm is increased, or the parameter “D” is reduced in the reaction diffusion equation, or the density of Na-K pump is reduced, the CV is reduced, but to a much smaller extent. Although an increased Cm may represent larger cellular dimensions and by itself may contribute to an increased CV [37], this can be offset by gap junction lateralization, and the net result may result in a reduced CV [38].
In an elegant simulation study published a number of years ago [25], the relationship between a decrease in the maximum conductance of INa and a reduction in the CV was plotted (please see Fig. 4 in reference 25). The reduction in CV was approximately linear initially, but was more severe and highly non-linear in the later stages, as the maximum conductance of INa was gradually reduced. When [K+]o is increased in the setting of diabetes, the combination of a reduced density, in addition to the decreased availability (on account of the depolarized membrane potential in high [K+]o) likely pushes the magnitude of INa much earlier into the non-linear range, leading to a much larger reduction in the CV in diabetic, compared to control healthy hearts. Our findings of reduced INa are also supported indirectly by findings in long-duration (3–4 months) STZ-induced diabetic rat models, where it was found that the maximum upstroke velocity of the action potential (Vmax) was reduced more prominently, as the duration of diabetes was increased [39]; Vmax is known to be mainly influenced by the biophysical properties/density of INa [40]. A recent study also showed similar conduction slowing in the diabetic rat atria [41]. Taken together, impaired impulse propagation resulting from sustained hyperglycemia in the diabetic heart seems to be an emerging phenotype. We note that our observations of no change in APD and reduced INa density are inconsistent with a previous study in diabetic rabbits [13]. Although the reasons for this discrepancy are not clear to us, it could be related to the duration of diabetes (~10 weeks in the study by Zhang compared to ~21 weeks in our study), despite similar levels of hyperglycemia (mean value of 399 mg/dL in our study, versus 406 mg/dL in their study).
The precise molecular determinants of a reduced density in INa remain unclear. Further studies investigating the possible role of post-translational modifications in INa from diabetic hearts are needed. The protein levels of Nav1.5 were decreased in 75% of the samples examined (3/4), but unchanged in the remaining 25% (1/4). This may reflect variability in the disease severity of the diabetic cohort. Additionally, oxidative stress, which is known to be upregulated in diabetic hearts [42], and which is now known to modify the properties of INa [43], could also play an important role. Finally, a reduced INa will likely increase the susceptibility to arrhythmias, although this hypothesis needs to be tested.
5. Limitations
A number of clear limitations must be kept in mind while interpreting the experimental results. For our control hearts, we did not inject saline (in lieu of alloxan in diabetic hearts), and observe them for a similar duration, to limit experimental costs. However, the age difference arising as a result does not confound our results with regards to INa, as shown in our studies (Fig. S1). We have used blebbistatin to arrest contraction, while conducting our optical imaging studies. The speed of electrical propagation in the myocardium has four major determinants: INa, gap junctional conductance, the inward rectifier current (IK1), and fibrosis. Although we measured INa and fibrosis, we have not directly measured IK1 (although mRNA levels of Kir2.1 were not different between control and diabetic ventricles; see online supplement Fig. S3). Investigating changes in IK1 is also important, since this could influence the resting membrane potential (RMP), thereby reducing the CV by making less INa available. Thus another limitation is that we have not measured the RMP in control versus diabetic rabbit ventricles. We have also not measured gap junction conductance or investigated whether the phosphorylation of Cx43 was altered. Optical mapping in isolated hearts can give us an indication of CV, but because of the 3D nature of the tissue, the exact direction of the electrical impulse cannot be determined. Thus, we are measuring apparent CV over the epicardial surface. We attempted to limit variability and also minimize influence of the specialized conduction system (purkinje fibers) on conduction by pacing from the right ventricular (RV) epicardial surface. Further, our mapping does not allow for dissecting the transverse/longitudinal components of the CV. The alloxan-induced diabetic rabbit model has very high uncontrolled blood glucose levels. Thus, results from this study should be extrapolated with caution to other species, including humans. Further, the alloxan model mimics type-1 diabetes more faithfully, and whether similar findings can be seen in type-2 diabetes, which is more common, needs to be investigated. We have not investigated whether controlling glucose via insulin injections would prevent the reduced CV/INa in diabetes in our experimental model. In our computer simulations, we have not taken into account changes in cell size, and lateralization in gap junction proteins, which may contribute in complex, non-linear ways to reduce CV, as was shown in earlier modeling work [38]. Thus more detailed experimental data regarding changes in cellular dimensions, gap junction conductance, and lateralization occurring in diabetes, in conjunction with sophisticated simulations that incorporate these changes, is needed to fully elucidate the ionic determinants of a reduced CV in diabetes. Despite these various limitations, our study provides novel and useful insights into the mechanisms by which hyperglycemia can affect cardiac electrophysiology.
Supplementary Material
Highlights.
Optical mapping in diabetic rabbit hearts showed no change in APD
However, cardiac impulse propagation was slower in diabetes
The density of Na+ current (INa) was reduced in diabetes
No changes in connexin 43 or fibrosis were found in diabetes
Simulations show that reduced INa can account for slower propagation in diabetes
Acknowledgments
This work was supported by a Pilot & Feasibility Grant awarded to Dr. Sandeep V. Pandit, via the Michigan Diabetes Research and Training Center at the University of Michigan, which is funded by NIH5P60 DK20572 from the National Institute of Diabetes and Digestive and Kidney Diseases. This work was performed during Dr. Catherine Stables’ tenure as the Michael Bilitch Fellowship in Cardiac Pacing and Electrophysiology Fellow of the Heart Rhythm Society. We would like to thank Drs. José Jalife and Todd Herron for useful discussions and support. Also supported by NHLBI Grants P01HL039707, P01HL087226, Gilead Inc., and the Leducq Foundation (to Dr. José Jalife), and by Gilead Inc. (to Dr. Sandeep V. Pandit).
Footnotes
Disclosure
Dr. Sandeep V. Pandit has received funding from the Industry (Gilead).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Centers for disease control and prevention. National diabetes fact sheet: National estimates and general information on diabetes and prediabetes in the united states, 2011. Atlanta, GA: U.S. Department of health and human services, centers for disease control and prevention; 2011. [Google Scholar]
- 2.Association AD. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2011;34:S62–S69. doi: 10.2337/dc11-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dillmann WH. Diabetes and thyroid-hormone-induced changes in cardiac function and their molecular basis. Annu Rev Med. 1989;40:373–394. doi: 10.1146/annurev.me.40.020189.002105. [DOI] [PubMed] [Google Scholar]
- 4.El-Atat FA, McFarlane SI, Sowers JR, Bigger JT. Sudden cardiac death in patients with diabetes. Curr Diab Rep. 2004;4:187–193. doi: 10.1007/s11892-004-0022-8. [DOI] [PubMed] [Google Scholar]
- 5.Bergner DW, Goldberger JJ. Diabetes mellitus and sudden cardiac death: what are the data? Cardiol J. 2010;17(2):117–29. Review. [PubMed] [Google Scholar]
- 6.Rossing P, Breum L, Major-Pedersen A, Sato A, Winding H, Pietersen A, Kastrup J, Parving HH. Prolonged qtc interval predicts mortality in patients with type 1 diabetes mellitus. Diabet Med. 2001;18:199–205. doi: 10.1046/j.1464-5491.2001.00446.x. [DOI] [PubMed] [Google Scholar]
- 7.Christensen PK, Gall MA, Major-Pedersen A, Sato A, Rossing P, Breum L, et al. Qtc interval length and qt dispersion as predictors of mortality in patients with non-insulin-dependent diabetes. Scand J Clin Lab Invest. 2000;60:323–332. doi: 10.1080/003655100750046486. [DOI] [PubMed] [Google Scholar]
- 8.Shimoni Y, Firek L, Severson D, Giles W. Short-term diabetes alters k+ currents in rat ventricular myocytes. Circ Res. 1994;74:620–628. doi: 10.1161/01.res.74.4.620. [DOI] [PubMed] [Google Scholar]
- 9.Shimoni Y, Severson D, Giles W. Thyroid status and diabetes modulate regional differences in potassium currents in rat ventricle. J Physiol. 1995;488 (Pt 3):673–688. doi: 10.1113/jphysiol.1995.sp020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):1205–53. doi: 10.1152/physrev.00002.2005. Review. [DOI] [PubMed] [Google Scholar]
- 11.Lengyel C, Iost N, Virag L, Varro A, Lathrop DA, Papp JG. Pharmacological block of the slow component of the outward delayed rectifier current (i(ks)) fails to lengthen rabbit ventricular muscle qt(c) and action potential duration. Br J Pharmacol. 2001;132:101–110. doi: 10.1038/sj.bjp.0703777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lengyel C, Virag L, Kovacs PP, Kristof A, Pacher P, Kocsis E, et al. Role of slow delayed rectifier k+-current in qt prolongation in the alloxan-induced diabetic rabbit heart. Acta Physiol (Oxf) 2008;192:359–368. doi: 10.1111/j.1748-1716.2007.01753.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Xiao J, Lin H, Luo X, Wang H, Bai Y, et al. Ionic mechanisms underlying abnormal qt prolongation and the associated arrhythmias in diabetic rabbits: A role of rapid delayed rectifier K+ current. Cell Physiol Biochem. 2007;19:225–238. doi: 10.1159/000100642. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, et al. Restoring depressed herg k+ channel function as a mechanism for insulin treatment of abnormal qt prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol. 2006;291:H1446–1455. doi: 10.1152/ajpheart.01356.2005. [DOI] [PubMed] [Google Scholar]
- 15.Lengyel C, Virag L, Biro T, Jost N, Magyar J, Biliczki P, et al. Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res. 2007;73:512–520. doi: 10.1016/j.cardiores.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Xie C, Biary N, Tocchetti CG, Aon MA, Paolocci N, Kauffman J, et al. Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic guinea pigs. Am J Physiol Heart Circ Physiol. 2013;304:H916–26. doi: 10.1152/ajpheart.00026.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nygren A, Olson ML, Chen KY, Emmett T, Kargacin G, Shimoni Y. Propagation of the cardiac impulse in the diabetic rat heart: Reduced conduction reserve. J Physiol. 2007;580:543–560. doi: 10.1113/jphysiol.2006.123729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahnema P, Shimoni Y, Nygren A. Reduced conduction reserve in the diabetic rat heart: role of iPLA2 activation in the response to ischemia. Am J Physiol Heart Circ Physiol. 2011;300(1):H326–34. doi: 10.1152/ajpheart.00743.2010. [DOI] [PubMed] [Google Scholar]
- 19.Punske BB, Rossi S, Ershler P, Rasmussen I, Abel ED. Optical mapping of propagation changes induced by elevated extracellular potassium ion concentration in genetically altered mouse hearts. J Electrocardiol. 2004;37 (Suppl):128–134. doi: 10.1016/j.jelectrocard.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 20.Hansen PS, Clarke RJ, Buhagiar KA, Hamilton E, Garcia A, White C, et al. Alloxan-induced diabetes reduces sarcolemmal na+-k+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol. 2007;292:C1070–1077. doi: 10.1152/ajpcell.00288.2006. [DOI] [PubMed] [Google Scholar]
- 21.Pandit SV, Kaur K, Zlochiver S, Noujaim SF, Furspan P, Mironov S, et al. Left-to-Right Ventricular Differences in IKATP Underlies Epicardial Repolarization Gradient During Global Ischemia. Heart Rhythm. 2011;8(11):1732–9. doi: 10.1016/j.hrthm.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandit SV, *, Zlochiver S, *, Filgueiras-Rama D, Mironov S, Yamazaki M, Ennis SR, et al. Targeting Atrio-Ventricular Differences in Ion Channel Properties for Terminating Acute Atrial Fibrillation in Pigs. Cardiovasc Res. 2011;89(4):843–51. doi: 10.1093/cvr/cvq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, et al. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 24.Kagiyama Y, Hill JL, Gettes LS. Interaction of acidosis and increasedxtracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ Res. 1982;51(5):614–23. doi: 10.1161/01.res.51.5.614. [DOI] [PubMed] [Google Scholar]
- 25.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81(5):727–41. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 26.Tolkacheva EG, Anumonwo JM, Jalife J. Action potential duration restitution portraits of mammalian ventricular myocytes: role of calcium current. Biophys J. 2006;91(7):2735–45. doi: 10.1529/biophysj.106.083865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 109(31):E2134–43. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA, Taffet SM, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105(6):523–6. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tellez JO, Dobrzynski H, Greener ID, Graham GM, Laing E, Honjo H, et al. Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ Res. 2006;99:1384–1393. doi: 10.1161/01.RES.0000251717.98379.69. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka K, Zlochiver S, Vikstrom KL, Yamazaki M, Moreno J, Klos M, et al. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ Res. 2007;101(8):839–47. doi: 10.1161/CIRCRESAHA.107.153858. [DOI] [PubMed] [Google Scholar]
- 32.Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78(5):2392–404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deo M, Sato P, Musa H, Pandit SV, Delmar M, Berenfeld O. Relative contribution of changes in sodium current vs intercellular coupling on reentry initiation in two dimensional preparations of Plakophilin-2-deficient cardiac cells. Heart Rhythm. 2011;8(11):1740–8. doi: 10.1016/j.hrthm.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng J, Laurita KR, Rosenbaum DS, Rudy Y. Two components of the delayed rectifier K+ current in ventricular myocytes of the guinea pig type. Theoretical formulation and their role in repolarization. Circ Res. 1995;77(1):140–52. doi: 10.1161/01.res.77.1.140. [DOI] [PubMed] [Google Scholar]
- 35.Pandit SV, Giles WR, Demir SS. A mathematical model of the electrophysiological alterations in rat ventricular myocytes in type-I diabetes. Biophys J. 2003;84(2 Pt 1):832–41. doi: 10.1016/S0006-3495(03)74902-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghaly HA, Boyle PM, Vigmond EJ, Shimoni Y, Nygren A. Simulations of reduced conduction reserve in the diabetic rat heart: response to uncoupling and reduced excitability. Ann Biomed Eng. 2010;38(4):1415–25. doi: 10.1007/s10439-009-9855-2. [DOI] [PubMed] [Google Scholar]
- 37.Wiegerinck RF, Verkerk AO, Belterman CN, van Veen TA, Baartscheer A, Opthof T, et al. Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation. 2006;113(6):806–13. doi: 10.1161/CIRCULATIONAHA.105.565804. [DOI] [PubMed] [Google Scholar]
- 38.Spach MS, Heidlage JF, Barr RC, Dolber PC. Cell size and communication: role in structural and electrical development and remodeling of the heart. Heart Rhythm. 2004 Oct;1(4):500–15. doi: 10.1016/j.hrthm.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Pacher P, Ungvári Z, Nánási PP, Kecskeméti V. Electrophysiological changes in rat ventricular and atrial myocardium at different stages of experimental diabetes. Acta Physiol Scand. 1999;166(1):7–13. doi: 10.1046/j.1365-201x.1999.00538.x. [DOI] [PubMed] [Google Scholar]
- 40.Cohen CJ, Bean BP, Tsien RW. Maximal upstroke velocity as an index of available sodium conductance. Comparison of maximal upstroke velocity and voltage clamp measurements of sodium current in rabbit Purkinje fibers. Circ Res. 1984;54(6):636–51. doi: 10.1161/01.res.54.6.636. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe M, Yokoshiki H, Mitsuyama H, Mizukami K, Ono T, Tsutsui H. Conduction and refractory disorders in the diabetic atrium. Am J Physiol Heart Circ Physiol. 2012;303(1):H86–95. doi: 10.1152/ajpheart.00010.2012. [DOI] [PubMed] [Google Scholar]
- 42.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–70. doi: 10.1161/CIRCRESAHA.110.223545. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107(8):967–74. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.