

Abstract
Melanoma represents a significant malignancy in humans and dogs. Different from genetically engineered models, sporadic canine melanocytic neoplasms share several characteristics with human disease that could make dogs a more relevant preclinical model. Canine melanomas rarely arise in sun-exposed sites. Most occur in the oral cavity, with a subset having intra-epithelial malignant melanocytes mimicking the in situ component of human mucosal melanoma. The spectrum of canine melanocytic neoplasia includes benign lesions with some analogy to nevi, as well as invasive primary melanoma, and widespread metastasis. Growing evidence of distinct subtypes in humans, differing in somatic and predisposing germ-line genetic alterations, cell of origin, epidemiology, relationship to ultraviolet radiation and progression from benign to malignant tumors, may also exist in dogs. Canine and human mucosal melanomas appear to harbor BRAF, NRAS, and c-kit mutations uncommonly, compared with human cutaneous melanomas, although both species share AKT and MAPK signaling activation. We conclude that there is significant overlap in the clinical and histopathological features of canine and human mucosal melanomas. This represents opportunity to explore canine oral cavity melanoma as a preclinical model.
Keywords: melanoma, animal model, comparative study, clinical trial design, image analysis, digital telepathology, signal transduction
Introduction
Melanoma represents a significant health problem with over 76 690 newly diagnosed cases and over 9480 deaths annually in the United States alone (Howlader et al., 2013). While there has been significant progress in delineating the underlying genetic alterations and developing small molecule inhibitors to block key signaling pathways as well as in harnessing the immune system to kill melanoma cells, metastatic melanoma remains mostly an untreatable, ultimately fatal disease. A series of oncogenic alterations in signaling components primarily of the MAP-kinase pathway have been identified. They affect genes such as BRAF, NRAS, KIT, HRAS, GNAQ, and GNA11 and, at least early in progression, are found in a mutually exclusive pattern. The individual mutations are associated with distinct clinical, histopathological, and epidemiological features, suggesting that melanocytic neoplasia is comprised of biologically distinct subtypes (Broekaert et al., 2010). The subtypes differ in pathogenetic factors such as ultraviolet radiation, mutational processes shaping the cancer genomes, and their cell of origin (reviewed in Tsao et al., 2012; Whiteman et al., 2011; D. C. Whiteman and B.C. Bastian, manuscript in preparation).
These differences indicate that therapeutic or preventative strategies have to be tailored to individual subtypes. Animal models play a key role in evaluating therapeutic strategies to treat or prevent cancer. Multiple genetically engineered mouse models of melanoma have been developed as preclinical models (reviewed in Damsky and Bosenberg, 2010; Walker et al., 2011). While most of these models are valuable to investigate certain aspects of the disease, they typically lack the genetic complexity that accompanies naturally evolving cancers in which single cells may acquire mutations and undergo waves of clonal expansion to finally reach a fully evolved malignant state. In particular, animal models that demonstrate the entire spectrum of a cancer reaching from benign neoplasms, primary tumors, and metastases are rare. Furthermore, most of the contemporary melanoma models are driven by constitutive activation of BRAF and NRAS.
Melanocytic neoplasms occur sporadically in many animals and are particularly frequent in certain breeds of horses, pigs, and dogs (Goldschmidt and Hendrick, 2002). In dogs, melanomas are most commonly observed in Scottish terrier, poodle, golden retriever, dachshund, cocker spaniel, miniature poodle, Chow Chow, Gordon setter, and Anatolian Sheepdog breeds, although the true incidence in individual breeds of dogs is poorly established (Bergman et al., 2013). Fatal melanomas in dogs typically originate from the oral cavity and acral (foot pads and nail apparatus) sites. They occasionally occur in the hair-bearing skin, but with much less frequency (Goldschmidt and Hendrick, 2002). While the pathogenesis of canine melanomas is not known, the anatomic distribution suggests that ultraviolet radiation is not a causative factor. Similar to human melanocytic neoplasms, the unequivocal differentiation of benign and malignant lesions is not always possible.
The noteworthy presence of a frequent lentiginous intra-epithelial component in canine melanomas, a feature documented to precede invasive melanomas in humans that subsequently spread to regional lymph nodes and eventually to visceral sites, implicates a similar progression cascade in the dog. In fact, canine oral cavity melanomas mimic the evolution and clinical progression of the human disease originating from several mucosal sites, having similar propensity for invasion and dissemination (Bergman et al., 2013; Piliang, 2011; Prasad et al., 2004). Additionally, as their human counterparts, canine melanomas are resistant to chemotherapy and radiation therapy (Bergman and Wolchok, 2008).
Genetic alterations in canine malignant melanomas from mucosal or acral sites have not been fully delineated. Activating mutations in BRAF exon 15 are not found (Fowles et al., 2013; Shelly et al., 2005), similar to human mucosal melanoma (Maldonado et al., 2003). Activating mutations of NRAS and c-kit appear to be absent in canine mucosal melanoma (Chu et al., 2012; Fowles et al., 2013; Murakami et al., 2011), in contrast to human mucosal melanoma where these genes are mutated in 15% of tumors (Curtin et al., 2006).
Apparent commonalities in the clinical and histopathological features of mucosal melanoma in dogs and humans raised the possibility that investigational studies in dogs could lead to insight into the human condition. Therefore, we assembled a panel of oncologists, pathologists, and researchers with expertise in melanocytic neoplasia in dogs and humans to examine similarities between the disease spectra and how such could be leveraged to accelerate treatment in both species. Several previous studies have explored clinical trials in canine cancer as a preclinical model to inform the design of clinical trials in humans (Gordon et al., 2009; Paoloni and Vail, 2013; Rusk et al., 2006). Similarly, naturally occurring non-neoplastic diseases in dogs have also yielded information relevant to human diseases (Grall et al., 2012; Shearin and Ostrander, 2010).
Naturally occurring canine melanoma model for human disease
Basis for consensus
Physician and veterinary pathologists (Table S1) compared histopathological features of 28 human and 139 canine melanoma specimens (Table 1). Melanomas, contributed by 11 medical and veterinary institutions representing national treatment centers, were obtained with appropriate consent and according to institutional review. Anonymized patient information was reviewed as available. Canine melanoma specimens were predominantly from mucosal sites, but included melanomas from other sites (Table 1). Canine specimens were compared with both human mucosal and cutaneous melanomas, including features illustrated within web-based and other atlases [e.g. www.skinpathology.org/].
Table 1.
Specimens obtained for comparative melanoma tumor board review
Nineteen canine amelanotic oral sarcomas were excluded from further study based upon the absence of melanocyte differentiation marker expression by IHC (see Table S2). Twenty-seven of 111 oral melanomas were considered low malignant potential (see narrative). An additional 44 low malignant potential melanocytic neoplasms were studied by a board subpanel.
Includes 11 benign cutaneous melanocytomas.
Acral/ungual/scrotal cutaneous sites.
These represent additional melanoma patient specimens, including acral sites, available for comparison.
Anorectal, vulvovaginal, gall bladder, sinonasal, and esophagus. Board participants agreed dogs have rare nasal and anorectal melanoma.
No well-recognized classification scheme exists for mucosal melanomas from either species; therefore, evaluation included review of melanoma features previously documented (Goldschmidt and Hendrick, 2002; Patel et al., 2002; Pfister et al., 2012; Prasad et al., 2004; Smedley et al., 2011b). The salient histopathological features of mucosal melanoma were tabulated for both species (Table S2). Near universal concordance of these features was observed between canine and human melanoma (Table S2). Analogous architectural features important for diagnosing and staging melanoma were noted in both species (Figure 1). As recognized for cutaneous melanomas, both human and canine mucosal melanomas included the range of epithelioid, spindloid, mixed epithelioid/spindloid, or small round blue cell melanocyte morphologies. Some dog specimens included a lentiginous-like growth pattern within stratified squamous mucosal epithelium and a significant radial growth phase involving mucosal epithelium flanking the vertical growth phase (Figure 2). By the time of clinical recognition, mucosal melanomas are typically advanced with considerable local invasion, ulceration, focal necrosis, and even metastasis, particularly in the dog. In both species, there was considerable pleomorphism with significant variation in cell and nuclear size, shape, and presence of nucleoli (Figure S1 and Table S2). Given the substantial difference in incidence between various anatomic sites, the board chose not to compare frequencies of all features.
Figure 1.
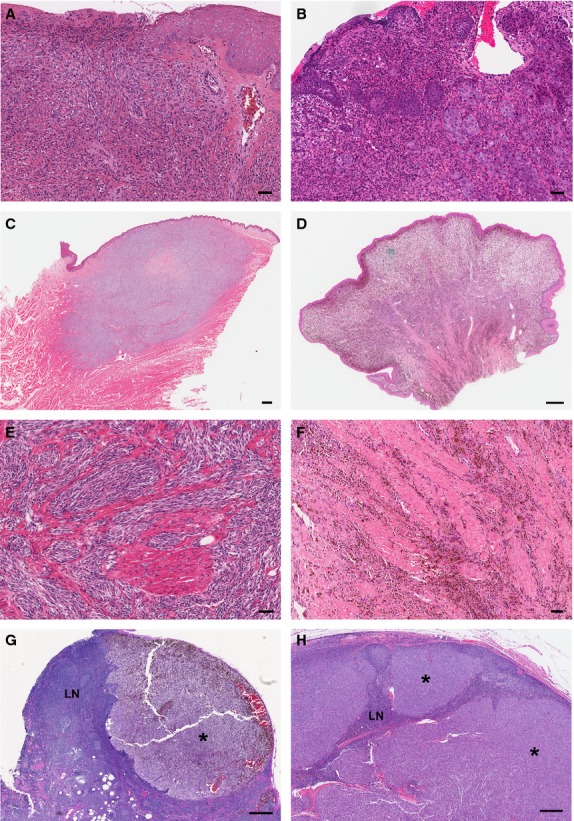
Similarities between histopathological features of mucosal melanomas in dogs and humans. Photomicrographs of representative human (left side column A, C, E, G) and dog (right side column B, D, F, H) melanomas are shown. (A, B) Ulceration in amelanotic melanomas. (C, D, E, F) Extensive vertical growth phase with malignant melanocytes infiltrating the proprial/submucosal muscle and/or collagen bundles. (G, H) Extensive invasion of lymph node (LN) parenchyma by metastatic melanoma (*). Hematoxylin and eosin stain. (A, B, E, F, Bar = 50 μm; C, D, G, H, Bar = 500 μm).
Figure 2.
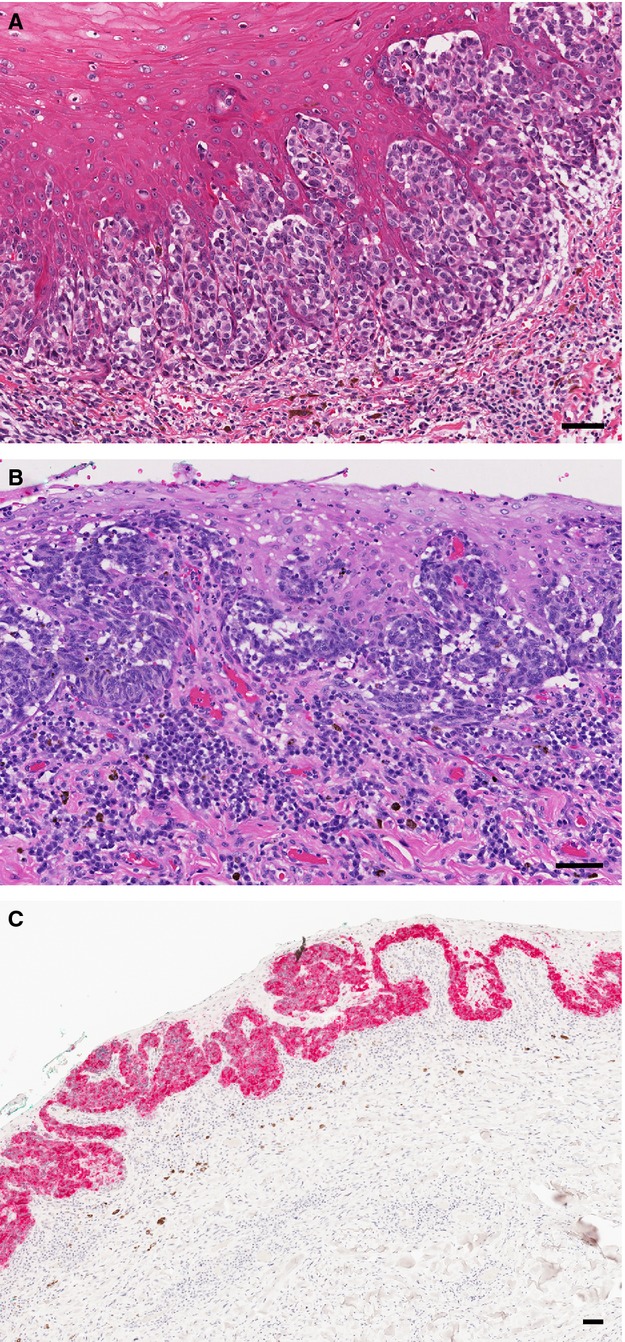
Lentiginous-like in situ involvement in mucosal melanomas by malignant melanocytes in the mucosal epithelium. Clusters of malignant melanocytes occur in the epithelial stratum basale and ascend into superficial strata. Photomicrographs of hematoxylin–eosin-stained (A) human mucosal melanoma and (B) canine mucosal melanoma. (C) Radial extension of malignant melanocytes is evident in the intact mucosal epithelium lateral to the vertical tumor component in some canine melanomas; (same canine patient as in B). Antimelan-A immunohistochemistry, red chromogen label, hematoxylin counter stain. Bar = 50 μm.
Overlapping histopathological features were also noted for dog and human acral and cutaneous melanomas. Less than 20% of all cutaneous melanocytic neoplasms in dogs are malignant, most of the rest being benign canine melanocytomas (Goldschmidt and Hendrick, 2002). Sufficient numbers of canine cutaneous and acral malignant melanomas were not available for this study for histopathological comparison. They are comparatively rare and therefore more difficult to enroll in clinical trials stratified by subtype.
Both dog and human malignant melanomas exhibited a wide range of melanin pigmentation, from intense pigmentation obscuring cellular detail to no microscopically detectable melanin (Figure 1 and Figure S1). Intense pigmentation was more frequently seen in canine melanoma. Immunohistochemical (IHC) evaluation of melanocytic differentiation (melan-A, PNL2, and TRP2) was performed on 30 amelanotic and on 78 pigmented canine (Table S3) and human melanomas. Directly concordant comparisons of phenotypic differentiation by immunohistochemistry (IHC) are limited by the fact that some antibodies commonly used to phenotype human melanoma specimens are not sufficiently reactive for dog, or lack specificity for differential diagnosis (eg, S100, HMB45) (Smedley et al., 2011a). Nineteen of 30 canine amelanotic tumors lacked labeling with any of the melanocyte differentiation antibodies (Table S3). As the diagnosis of melanoma could not be definitively confirmed in this subset of tumors, they were excluded from further evaluation in this study by the board. These likely represent multiple tumor types as dogs experience a variety of oral cavity mesenchymal tumors requiring differential diagnosis, but may also include a subset of amelanotic melanomas failing to immunoreact by IHC.
Human and dog oral/mucosal melanomas have a poor prognosis, even with limited extent of primary tumor burden (Goldschmidt and Hendrick, 2002; Prasad et al., 2004). However, the board discussed the existence of a category with infrequent cancer-associated mortality, having previously described characteristics in dogs (Esplin, 2008), which share features with blue nevi in humans (Buchner and Hansen, 1987). In contrast to most canine melanomas (Smedley et al., 2011b), these melanocytic neoplasms have limited size (generally < 2 cm in diameter), are often intensely pigmented, lack significant cellular atypia, have infrequent mitotic figures (absent in 56/71 neoplasms), and are rarely ulcerated (< 15% having only minor ulceration). Such melanocytic neoplasms with low malignant potential may represent up to 10% of mucosal melanocytic tumors in dogs and must be recognized for appropriate clinical management. Additionally, these site-relevant melanocytic neoplasms with low malignant potential are useful, once excised and diagnosed, for contrast in molecular assays of primary canine melanomas (see below). The relationship between these oral melanocytic neoplasms with low malignant potential and benign cutaneous melanocytic lesions in the human, as well as various subtypes of human melanocytic nevi, requires additional evaluation.
Limited access to specimens from surgical excisions of dog tumors was a major limiting factor for our histopathological comparison of the complexity of dog and human melanomas. Many samples were obtained from tissue-banked specimens, representing only portions of the tumor rather than the entire specimen. In some instances, these tumors lacked a contiguous mucosal component due to ulceration and/or having been subdivided to provide for preservation of multiple specimen types within the biorepository. Either circumstance compromised evaluation of the in situ or radial growth components. Only a limited number of specimens, mostly obtained from board members' surgical pathology practices, provided an opportunity for a more comprehensive histopathological analysis. These limitations precluded us from assessing the relative frequency of the presence of a notable in situ or radial growth in more of the canine cases. This is relevant for evaluating differentiation and potential behavior, particularly of amelanotic tumors. While access to biobanked canine accessions provided the board with a readily available national tissue source for study, complexity of tumor sampling of biobanked specimens used for histopathology comparisons accentuated the difficulty in considering lateral as well as vertical extent of patient disease. Consideration of such circumstances is important when developing biorepositories and designing comparative pathology studies.
Activation of key oncogenic signaling pathways was assessed by IHC using tissue microarrays (TMA) of primary canine melanomas. Expression levels of p-AKTSer473, KIT, and p-ERK1/2 varied significantly, and no correlation with disease-specific survival was found (Figure 3, and Table S4). Phosphorylation of AKT was evident in all 44 (100%) melanomas on the TMA, although 15 of these (34%) had only rare cells or weak-positive cell labeling (immunolabeling score < 50, see methods). Phospho-ERK1/2 expression occurred in 34 of 44 (77%) canine malignant melanomas. Twenty-three of 44 (52%) melanomas exhibited both phosphorylation of ERK1/2 and moderate (or greater) AKT phosphorylation (≥ 50 AKT immunolabeling score), a distinction shared with some human mucosal melanomas (Omholt et al., 2011). Weak or absent PTEN expression (defined as PTEN immunolabeling score < 42, see methods) was observed in 21 of 44 (48%) of these (Table S4), a characteristic also seen in human melanoma (Bogenrieder and Herlyn, 2010; Davies et al., 2009; Zhou et al., 2000). Presence of diminished PTEN expression did not correlate with survival in dogs (P = 0.47). KIT was expressed in 37 of 44 (85%) canine melanomas, but only a few cells were labeled in 27 of the 37.
Figure 3.
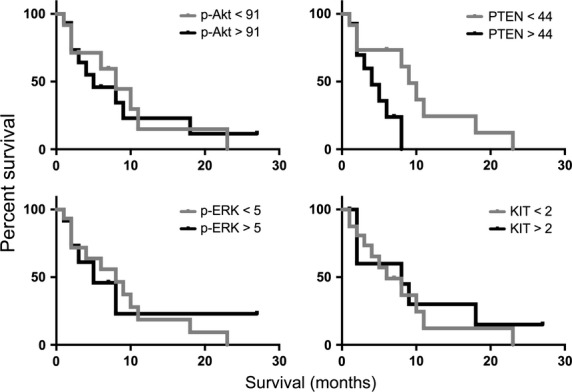
Analysis of quantitative expression intensity for p-AKT, PTEN, p-ERK1/2, and KIT, and disease-specific survival within a subset of 27 canine melanoma patients with clinical follow-up. Kaplan–Meier survival curves were generated using patient groups defined as above or below the median expression for each marker (determined by color deconvolution image analysis as immunolabeling scores of brightfield chromogenic IHC from TMA tissue cores; see also Table S4). Expression of these proteins in this cohort was not significantly correlated with survival, as assessed using Mantel–Cox test (p-AKT, P = 0.90; PTEN, P = 0.14; p-ERK, P = 0.86, and KIT, P = 0.68). Primary melanoma tissue specimens were surgically collected from dogs at the time of initial diagnosis prior to further treatment.
The entity classified as canine oral melanocytic neoplasia with low malignant potential was noted to show a virtual absence of KIT expression, absent to low p-ERK1/2 and p-AKT expression (Table S4), and had similarly low relative expression of pathway mediators downstream of PI3K/AKT (eg, mTOR, pS6, eIF4E), in contrast to malignant melanomas (H.T. Michael and R. M. Simpson, manuscript in preparation). Overall, outcome information was available for only a limited number of canine patients, which received a variety of interventions. Although our evaluations comparing survival in dogs must be considered preliminary, it is noteworthy that human patient outcomes are likewise essentially unlinked to activation status of these signaling pathways (Dai et al., 2005; Oba et al., 2011; Slipicevic et al., 2005). Extended studies, also taking into account genetic alterations, are necessary for more thorough comparison with human melanoma.
Board consensus perspective
Substantial common characteristics exist in canine and human mucosal melanomas, indicating that the dog could serve as a model for human mucosal melanoma. Mucosal melanomas in dogs and humans share clinical and histopathological commonalities in their clinical course, and the need for improved therapeutic modalities to impact the consistently poor therapeutic response of metastatic or locally unresectable disease. These parallels provide rationale for the investment needed to explore dogs with melanoma as a clinical model for human melanoma. Additionally, although published mutation analyses of canine melanomas cover limited gene regions on small numbers of patients and are not conclusive to date (Table S5), the results suggest that overlapping genetic causes exist.
Absence of evidence of BRAF mutation in dogs (Fowles et al., 2013; Shelly et al., 2005) is analogous to predominantly wild-type status in human mucosal melanomas (Buery et al., 2011; Maldonado et al., 2003). While approximately 15% of human cases harbor NRAS mutations, they appear to be rare in canine mucosal melanoma, with a single cell line exhibiting heterozygosity for Q61R in exon 2 (Fowles et al., 2013; Mayr et al., 2003; Murua Escobar et al., 2004). Reports indicate that expression of KIT (CD117) in canine melanoma varies considerably, with approximately half or more of cases exhibiting KIT expression; however, activating c-kit mutations in dogs have yet to be clearly documented (Chu et al., 2012; Gomes et al., 2012; Murakami et al., 2011; Newman et al., 2012). The lack of correlation between KIT expression and the presence of activating c-kit mutations has also been documented in human melanoma (Beadling et al., 2008; Curtin et al., 2006). There is conflicting information regarding association between KIT expression and survival in dogs (Gomes et al., 2012; Newman et al., 2012). KIT expression (in the absence of activating c-kit mutations) is not predictive of clinical response to KIT inhibitors in humans (Ugurel et al., 2005; Wyman et al., 2006). Furthermore, the presence of activation of signaling pathways in many canine melanomas is similar to that seen in human melanoma (Bogenrieder and Herlyn, 2010; Dai et al., 2005; Fowles et al., 2013; Garrido and Bastian, 2010; Kent et al., 2009; Mikhail et al., 2005; Shelly et al., 2005; Slipicevic et al., 2005). Detection of AKT and MAPK signaling pathway activation in primary melanomas from otherwise untreated dogs in this study corroborates findings from five canine melanoma cell lines (Fowles et al., 2013). Corresponding activation of AKT and MAPK in human and canine melanomas supports the existence of similar constitutive signaling, although the activation may be the consequence of alternative, as yet undiscovered, genetic alterations in the dog. Thus, canine melanoma may also prove useful in the discovery of novel driver mutations in mucosal melanoma. In the interim, exploration of existing therapies targeting components of the MAPK and PI3K/AKT pathways could be evaluated in dogs, particularly in modeling therapeutics for human melanomas lacking typical BRAF and NRAS mutations.
Currently, deficiencies in canine genome sequence annotation hamper progress in comparing human and canine melanoma genetics. More thoroughly annotated versions of dog genome are forthcoming. Similar to human melanoma (Curtin et al., 2005; Whiteman et al., 2011), the board anticipates the existence of distinct melanoma subtypes in the dog, with differing molecular aberrations linked to histopathological phenotypes and outcomes. Defining these entities, through correlations of not only mutations, but also evaluations of chromosomal, epigenetic, and expression changes between dog and human melanoma, will be important to future goals for modeling human melanoma and improving canine cancer care.
Melanoma clinical trial initiative in dogs
The interdisciplinary synergy exemplified by this board's proceedings can serve as a paradigm for fostering a substantive canine clinical trial initiative for human melanoma. Evaluating and credentialing canine melanoma as a surrogate clinical (preclinical) model should be developed with a veterinary and human clinical and basic science team approach. Canine clinical oncology trials are possible using existing infrastructure. For example, 20 academic veterinary medical centers are part of a consortium organized within the National Cancer Institute's Comparative Oncology Program. This program designs and executes clinical trials in dogs with selected cancers to inform human drug development. [https://ccrod.cancer.gov/confluence/display/CCRCOPWeb/Comparative+Oncology+Trials+Consortium].
As melanoma case management relies on pathology guidance (Ehrhart and Withrow, 2013), developing the dog model and formulating clinical trials must include aims to improve understanding of disease pathogenesis and define correlative outcomes as a part of evaluating therapy. In the past, veterinary therapy has often been pursued in the absence of sufficient evidence of efficacy (Butler et al., 2013). Comparison between studies has been hampered by inadequate quality and consistency of reporting and due to the small numbers of patients included. Much remains to be learned about melanoma in animals that can be enhanced by minimizing patients lost to follow up in a clinical research setting. Advances in both human and veterinary patient care have been chronically hindered by lack of sufficient autopsy evaluation in clinical trials to assess disease status and treatment consequences at end of life. Comprehension of what is possible, economic issues, and elected euthanasia based upon prognosis and/or quality of life contribute to these difficulties in veterinary medicine. Future approaches must address these issues. Consequently, the board discussed strategic planning for clinical scientific development of the dog model useful in both modeling human disease comparisons and canine patient care, as well as leading to new knowledge on underlying melanoma pathobiology (Table 2). It is essential that pathologists partner with clinicians in trial design and management of patient specimens for diagnosis and classification based upon an amalgam of contextual clinical, cellular, and molecular genetic features.
Table 2.
Consideration for canine melanoma surrogate clinical trial development strategya
| Elements of strategy | Fundamental action/procedure | Constructive consideration |
|---|---|---|
| Clinical documentation | ||
| Patient data | Presentation/history, duration, previous workup, management | Breed and other background information useful to generate data on incidence |
| Gross lesion documentation | Extent of disease. Description of specific anatomic location (not just indication of oral cavity); dimensions in mm, two axes; ulceration, evidence of dissemination. | Photograph lesion with a ruler if possible |
| Biopsy | Inclusion for diagnostic intent/therapeutic intent (excisional, incisional); preservation for correlative molecular analysis. | Consideration of lateral extent as well as vertical depth of invasion; attention paid to quality of sampling, preservation, QA, and utilization |
| Pathology review | Development of features of malignancy for initial assessment for trial enrollments: differentiation, proliferation, growth pattern, invasion, and dissemination, etc. Continue refining prognostic summation; Inclusion of IHC panel if needed for diagnosis | Incorporate Table 3. Smedley et al. (2011b) Capture classical features outlined – Adapt how used initially versus what becomes useful from adjunct molecular data and outcomes |
| Clinical staging/prognosis and monitoring | ||
| Imaging for dissemination | Ultrasound of lymph nodes to detect metastasis (includes submandibular) | +/− consideration of removal for staging; alternative consideration ultrasound-guided fine needle aspirate cytology for staging |
| CT (MRI) imaging evaluation | Lung particularly; lymph node; abdomen | Consideration of monitoring for brain involvement; inclusion of cranial imaging |
| Biopsy | Monitoring response to therapy, as appropriate | Lymph nodes or other palpable disease is recommended |
| Endpoint assessment | Necropsy examination, with collection of tissue for research, and documentation of extent of disease/host response. | |
| Quality-of-life measures | Assessments of fatigue, cardiac function, mucositis, altered mentation, serial assessments of metabolic and hematological toxicity, threshold of toxicity versus response | Harmonized approach for multicenter trials; similar to Paoloni and Vail (2013) |
| Client education | Informed consent; necropsy education; should include education on how the initiative intended to benefit both dogs and humans relies upon evidence obtained from patient specimens | Necropsy education; emphasis on historical shortcomings impediment to progress. Education design beyond pro forma consent for necropsy |
| Follow-up | Directly with owner/clients and indirectly with primary care clinician | |
| Genomics | Global discovery genomics, proteomics and informatic methods: develop and apply. Database and clinical monitoring integration | |
Strategic approach for trial design represents an initial outline to be developed further with medical and veterinary oncologists, pathologists, and basic and clinical melanoma research investigators for use in developing multidisciplinary trials for piloting therapeutics for human melanoma. Research outcomes are anticipated to produce parallel benefits for canine melanoma patients.
Prospectus
As an ideal clinical model for the human disease would share a common cell of origin, pathogenesis, disease progression, clinical, and histopathological features and responses to therapy, we realize such an ideal model is yet to be identified for any human disease. Notwithstanding, and in contrast to genetically engineered mouse models, dogs represent a unique opportunity to investigate certain subtypes of spontaneous melanoma formation, progression, metastasis, and disease intervention in a large mammal that sporadically develops melanoma in an immunocompetent setting, and in an environment that is largely shared with humans. Advantages gained by incorporating a surrogate clinical trial perspective include ability to obtain serial biopsies of tumor tissues during therapy for pharmacodynamic and pharmacokinetic analysis as well as the capacity to identify and validate biomarkers and medical imaging applications. It also encompasses evaluation of safety profiles in a species historically used in pharmaceutical development. Established classical standards of therapy are rare in veterinary oncology; therefore, it is considered acceptable to offer clients investigational new cancer treatments for naïve disease in their dogs, rather than to expect failure of previously tried cancer drugs first, with ethical guidelines being adhered to (Paoloni and Vail, 2013).
Canine clinical trials could inform novel therapeutic strategies and influence phase I and II studies for human melanoma including targeted therapies, administered singly or in combination, as well as immune-based melanoma therapeutics, the preclinical evaluation of which is of particular interest. For example, the existence of recombinant canine CTLA4-Ig, shown previously to induce long-lived tolerance (Graves et al., 2009), and also to suppress Th1 cytokines, lymphocyte proliferation, and thyroglobulin antibody production in experimental autoimmune thyroiditis (Choi et al., 2008), could be compared in a variety of canine mucosal melanoma treatment settings. Similarly, development of other canine antibody-based therapeutics, such as anti-PD-1, would help to pilot optimization of relevant human therapeutic approaches, providing opportunity to mechanistically evaluate immune regulation, tolerance, and all too frequent eventual loss of effect or activity in tumor immunology. Therapeutic effectiveness and optimized schedules can be evaluated much more rapidly in spontaneous melanoma in an immune competent setting. The board believes that attaining the true value of the canine model will necessitate significant focus on appropriate patient stratification by further delineating the defining genetic, histopathological, molecular, and clinical features of the different types of melanocytic neoplasms in dogs to accurately identify the subtypes that best match their counterparts in humans.
Analysis approach and methods
The National Cancer Institute Comparative Melanoma Tumor Board is a collaboration of diagnostic/investigative experts representing scientific and clinical experience with canine and human melanocytic lesions (Table S1). Melanomas were acquired from the Canine Comparative Oncology and Genomics Consortium (CCOGC, Inc., Rockville, MD, USA) biospecimen repository [http://www.ccogc.net/] and from the diagnostic services of board members (Table 1). The majority of canine melanomas were surgically resected from the oral mucosa preceding other treatments, as most canine malignant melanomas originate there. A series of canine well-differentiated oral melanocytic neoplasms (Esplin, 2008) was evaluated comparatively.
Histopathology slides were routinely prepared, stained with hematoxylin and eosin, and optically scanned as digital image files with either Aperio (Leica, Vista, CA, USA) or Nanozoomer whole-slide imagers (Hamamatsu, Bridgewater, NJ, USA) at 20× resolution (approximately 0.43 μm/pixel) (Barisoni et al., 2013; Webster et al., 2011). Images were reviewed using Digital Image Hub (DIH, Slidepath-Leica, Dublin, Ireland). Tumor board members reviewed slides individually via web browser, and DIH was used for discussing images concurrently. Tumor board members participated in multiple telepathology webinar conferences and in two meetings at the National Cancer Institute, Bethesda, Maryland, during which the panel also met with veterinary and human oncologists, and melanoma basic and clinical scientists [http://nih-cbstp.nci.nih.gov/resources_pathology/NCIMelanomaTumorBoard.asp#TumorBoard].
A tissue microarray (TMA) incorporating 44 melanomas and 8 mucosal melanocytic neoplasms with low malignant potential was constructed as described (Takikita et al., 2009). Additional TMA tissues included human melanomas and normal dog and human tissues (control). These were analyzed by immunohistochemistry (IHC) using three melanocyte differentiation markers to confirm histogenesis (Smedley et al., 2011a), and using antibodies to KIT, PTEN, and phosphorylated forms of AKT, and ERK1/2 (Table S6). IHC was adapted from previous methods (Custer et al., 2006). Reagents and conditions are provided in Table S6. Use of known positive and known negative tissues was evaluated to ensure appropriate reactions for all antibodies used in IHC. Immunolabeling signal was developed using avidin-/biotin-conjugated alkaline phosphatase, Vector Red chromogen substrate (Vector Laboratories, Burlingame, CA, USA), with hematoxylin counterstain. For canine tumors in which melanocytic markers were negative or equivocal on TMA, IHC was repeated on whole-tissue sections to rule out sampling/heterogeneity issues. Human mucosal melanomas were diagnostically confirmed within the submitting institutions.
Digital image files of IHC-labeled melanoma TMA slides were used for relative quantitation of cell signal pathway protein expression from 44 malignant and 8 canine mucosal melanocytic neoplasms with low malignant potential. Image analysis was performed using color deconvolution software (Aperio, Color deconvolution, version 9, Vista, CA, USA). An individual analysis area region of interest was created for each patient melanoma tissue core using manual image segmentation. Areas of non-specific/off-target chromogenic labeling were excluded. Optical density measurements for the red, green, and blue components of the red chromogen were obtained on representative tissues using the software to optimize detection of chromogen (Vector Red) and its differentiation from melanin pigment. Three fixed-threshold tiers corresponding to mild, moderate, and strong immunolabeling intensity were independently established for each marker using the selected optical density color vectors. Individual color deconvolution channels as well as intensity-range pseudo-color markup images were assessed post-processing to confirm appropriate detection of chromogen, differentiation of the chromogen from melanin, and accurate classification of labeling intensity. Relative immunolabeling scores ([1 ×% mildly labeled pixel threshold tier] + [2 ×% moderately labeled pixel threshold tier] + [3 ×% strongly labeled pixel threshold tier]) were calculated for each melanoma core using TMA slides. Maximum weighted immunolabeling score possible = 300, when all (100%) pixels were strongly immunolabeled. Values were rounded to whole numbers. Assessments furthermore incorporated subjective immunolabeling score thresholds for p-AKT and PTEN intended to indicate tissue cores with weak expression and/or labeling of rare malignant cells (< 50 for p-AKT and < 42 for PTEN, respectively). Quantified immunolabeling intensity extent was useful for comparing relative expression/activation among canine melanomas assayed with a given marker, but was considered less appropriate for comparisons between different markers. Canine patient survival information was obtained from the CCOGC, Inc., but was only available for 27 of 44 melanoma specimens [http://www.ccogc.net/]. In addition to the limited number of patients with follow-up, we noted comparisons with patient outcomes in this study involved probe of comparative signal transduction protein expression applied to size-limited 1-mm-diameter TMA tumor tissue cores. Thus, additional studies would be prudent. Graphs and statistical analyses were performed using Prism software (version 6.00; Graph Pad, San Diego, CA, USA) and Excel (version 12.3.3; Microsoft Corporation, Redmond, WA, USA).
Acknowledgments
This research was supported by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland. Additional funding support was provided by The Animal Cancer Foundation, Norwalk, Connecticut [www.acfoundation.org/], and The Canine Comparative Oncology and Genomics Consortium, Inc., Rockville, Maryland [http://www.ccogc.net/]. NCI Comparative Melanoma Tumor Board meeting support was provided by the NCI Center for Cancer Research and The Animal Cancer Foundation. Dr. B. C. Bastian is supported by awards R01- CA131524, and P01 CA025874. Drs. J. M. Gary and H. T. Michael are currently molecular pathology fellows in the NIH Comparative Biomedical Scientist Training Program supported by the National Cancer Institute, in partnership with Michigan State University, East Lansing (Gary) and University of Maryland, College Park (Michael). Dr. M. R. Anver receives support through the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The authors thank Christina Mazcko, NCI, for assistance with study specimens and patient data, and John Hickerson, Kelly Government Solutions, for logistical support of tumor board study meetings. The views of the authors and tumor board members are their own, and no commercial endorsement is attributable to them or their affiliations.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Pleomorphic cytomorphologies of human (A, C, E) and canine (B, D, F) mucosal melanomas.
National Cancer Institute Comparative Melanoma Tumor Board Members.
Mucosal melanoma histopathological features in study patients.
Immunohistochemical evidence of melanocyte differentiation in canine malignant melanomas.
Immunohistochemical labeling scores for signal transduction pathway molecule expression in canine melanomas evaluated on tissue microarray.
Summary of previous molecular/genetic findings in canine melanoma.
Canine melanoma immunohistochemistry reagents and conditions.
References
- Barisoni L, Nast CC, Jennette JC. Digital pathology evaluation in the multicenter Nephrotic Syndrome Study Network (NEPTUNE) Clin. J. Am. Soc. Nephrol. 2013;8:1449–1459. doi: 10.2215/CJN.08370812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadling C, Jacobson-Dunlop E, Hodi FS. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008;14:6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- Bergman P. Wolchok J. Of Mice and Men (and Dogs): development of a xenogeneic DNA vaccine for canine oral malignant melanoma. Cancer Ther. 2008;6:817–826. [Google Scholar]
- Bergman PJ, Kent MS. Farese JP. Melanoma. In: Page RL, editor; Withrow SJ, Vail DM, editors. Withrow and MacEwen's Small Animal Clinical Oncology. St. Louis, MO: Elsevier/Saunders; 2013. pp. 321–334. [Google Scholar]
- Bogenrieder T. Herlyn M. The molecular pathology of cutaneous melanoma. Cancer Biomark. 2010;9:267–286. doi: 10.3233/CBM-2011-0164. [DOI] [PubMed] [Google Scholar]
- Broekaert SM, Roy R, Okamoto I. Genetic and morphologic features for melanoma classification. Pigment Cell Melanoma Res. 2010;23:763–770. doi: 10.1111/j.1755-148X.2010.00778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner A. Hansen LS. Pigmented nevi of the oral mucosa: a clinicopathologic study of 36 new cases and review of 155 cases from the literature. Part I: a clinicopathologic study of 36 new cases. Oral Surg. Oral Med. Oral Pathol. 1987;63:566–572. doi: 10.1016/0030-4220(87)90229-5. [DOI] [PubMed] [Google Scholar]
- Buery RR, Siar CH, Katase N, Gunduz M, Lefeuvre M, Fujii M, Inoue M, Setsu K. Nagatsuka H. RAS and BRAF mutation frequency in primary oral mucosal melanoma. Oncol. Rep. 2011;26:783–787. doi: 10.3892/or.2011.1385. [DOI] [PubMed] [Google Scholar]
- Butler LM, Bonnett BN. Page RL. Epidemiology and the evidence-based medicine approach. In: Page RL, editor; Withrow SJ, Vail DM, editors. Withrow and MacEwen's Small Animal Clinical Oncology. St. Louis, MO: Elsevier/Saunders; 2013. pp. 68–82. [Google Scholar]
- Choi EW, Shin IL, Lee CW. Youn HY. The effect of gene therapy using CTLA4Ig/silica-nanoparticles on canine experimental autoimmune thyroiditis. J. Gene Med. 2008;10:795–804. doi: 10.1002/jgm.1203. [DOI] [PubMed] [Google Scholar]
- Chu PY, Pan SL, Liu CH, Lee J, Yeh LS. Liao AT. KIT gene exon 11 mutations in canine malignant melanoma. Vet. J. 2012;196:226–230. doi: 10.1016/j.tvjl.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Busam K, Pinkel D. Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006;23:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- Custer MC, Risinger JI, Hoover S, Simpson RM, Patterson T. Barrett JC. Characterization of an antibody that can detect the Kai1/CD82 murine metastasis suppressor. Prostate. 2006;66:567–577. doi: 10.1002/pros.20386. [DOI] [PubMed] [Google Scholar]
- Dai DL, Martinka M. Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J. Clin. Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- Damsky WE. Bosenberg M. Mouse melanoma models and cell lines. Pigment Cell Melanoma Res. 2010;23:853–859. doi: 10.1111/j.1755-148X.2010.00777.x. [DOI] [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Lin E, et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin. Cancer Res. 2009;15:7538–7546. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhart N. Withrow S. Biopsy principles. In: Page RL, editor; Withrow SJ, Vail DM, editors. Withrow and MacEwen's Small Animal Clinical Oncology. St. Louis, MO: Elsevier/Saunders; 2013. pp. 143–148. [Google Scholar]
- Esplin DG. Survival of dogs following surgical excision of histologically well-differentiated melanocytic neoplasms of the mucous membranes of the lips and oral cavity. Vet. Pathol. 2008;45:889–896. doi: 10.1354/vp.45-6-889. [DOI] [PubMed] [Google Scholar]
- Fowles JS, Denton CL. Gustafson DL. Comparative analysis of MAPK and PI3K/AKT pathway activation and inhibition in human and canine melanoma. Vet. Comp. Oncol. 2013 doi: 10.1111/vco.12044. doi: 10.1111/vco.12044. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Garrido MC. Bastian BC. IT as a therapeutic target in melanoma. J. Invest. Dermatol. 2010;130:20–27. doi: 10.1038/jid.2009.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt M. Hendrick M. Tumors of the skin and soft tissues. In: Meuten DJ, editor; Tumors in Domestic Animals. Ames, IA: Iowa State Press; 2002. pp. 78–84. [Google Scholar]
- Gomes J, Queiroga FL, Prada J. Pires I. Study of c-kit immunoexpression in canine cutaneous melanocytic tumors. Melanoma Res. 2012;22:195–201. doi: 10.1097/CMR.0b013e32835273f9. [DOI] [PubMed] [Google Scholar]
- Gordon I, Paoloni M, Mazcko C. Khanna C. The Comparative Oncology Trials Consortium: using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS Med. 2009;6:e1000161. doi: 10.1371/journal.pmed.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grall A, Guaguere E, Planchais S, et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat. Genet. 2012;44:140–147. doi: 10.1038/ng.1056. [DOI] [PubMed] [Google Scholar]
- Graves SS, Stone D, Loretz C, Peterson L, McCune JS, Mielcarek M. Storb R. Establishment of long-term tolerance to SRBC in dogs with recombinant canine CTLA4-Ig. Transplantation. 2009;15:317–322. doi: 10.1097/TP.0b013e3181ae3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review 1975–2010. Bethesda, MD: National Cancer Institute; 2013. http://seer.cancer.gov/csr/1975_2010/, based on November 2012 SEER data submission, posted to the SEER web site, 2013. [Google Scholar]
- Kent MS, Collins CJ. Ye F. Activation of the AKT and mammalian target of rapamycin pathways and the inhibitory effects of rapamycin on those pathways in canine malignant melanoma cell lines. Am. J. Vet. Res. 2009;70:263–269. doi: 10.2460/ajvr.70.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D. Bastian BC. Determinants of BRAF mutations in primary melanomas. J. Natl Cancer Inst. 2003;95:1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- Mayr B, Schaffner G, Reifinger M, Zwetkoff S. Prodinger B. N-ras mutations in canine malignant melanomas. Vet. J. 2003;165:169–171. doi: 10.1016/s1090-0233(02)00245-9. [DOI] [PubMed] [Google Scholar]
- Mikhail M, Velazquez E, Shapiro R, et al. PTEN expression in melanoma: relationship with patient survival, Bcl-2 expression, and proliferation. Clin. Cancer Res. 2005;11:5153–5157. doi: 10.1158/1078-0432.CCR-05-0397. [DOI] [PubMed] [Google Scholar]
- Murakami A, Mori T, Sakai H, Murakami M, Yanai T, Hoshino Y. Maruo K. Analysis of KIT expression and KIT exon 11 mutations in canine oral malignant melanomas. Vet. Comp. Oncol. 2011;9:219–224. doi: 10.1111/j.1476-5829.2010.00253.x. [DOI] [PubMed] [Google Scholar]
- Murua Escobar H, Gunther K, Richter A, Soller JT, Winkler S, Nolte I. Bullerdiek J. Absence of ras-gene hot-spot mutations in canine fibrosarcomas and melanomas. Anticancer Res. 2004;24:3027–3028. [PubMed] [Google Scholar]
- Newman SJ, Jankovsky JM, Rohrbach BW. Leblanc AK. C-kit expression in canine mucosal melanomas. Vet. Pathol. 2012;49:760–765. doi: 10.1177/0300985811414032. [DOI] [PubMed] [Google Scholar]
- Oba J, Nakahara T, Abe T, Hagihara A, Moroi Y. Furue M. Expression of c-Kit, p-ERK and cyclin D1 in malignant melanoma: an immunohistochemical study and analysis of prognostic value. J. Dermatol. Sci. 2011;62:116–123. doi: 10.1016/j.jdermsci.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Omholt K, Grafstrom E, Kanter-Lewensohn L, Hansson J. Ragnarsson-Olding BK. KIT pathway alterations in mucosal melanomas of the vulva and other sites. Clin. Cancer Res. 2011;17:3933–3942. doi: 10.1158/1078-0432.CCR-10-2917. [DOI] [PubMed] [Google Scholar]
- Paoloni M. Vail D. Clinical trials and developmental therapeutics. In: Page RL, editor; Withrow SJ, Vail DM, editors. Withrow and MacEwen's Small Animal Clinical Oncology. St. Louis, MO: Elsevier/Saunders; 2013. pp. 293–304. [Google Scholar]
- Patel SG, Prasad ML, Escrig M, Singh B, Shaha AR, Kraus DH, Boyle JO, Huvos AG, Busam K. Shah JP. Primary mucosal malignant melanoma of the head and neck. Head Neck. 2002;24:247–257. doi: 10.1002/hed.10019. [DOI] [PubMed] [Google Scholar]
- Pfister DG, Ang KK, Brizel DM, et al. Mucosal melanoma of the head and neck. J. Natl. Compr. Canc. Netw. 2012;10:320–338. doi: 10.6004/jnccn.2012.0033. [DOI] [PubMed] [Google Scholar]
- Piliang MP. Acral lentiginous melanoma. Clin. Lab. Med. 2011;31:281–288. doi: 10.1016/j.cll.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Prasad ML, Patel SG, Huvos AG, Shah JP. Busam KJ. Primary mucosal melanoma of the head and neck: a proposal for microstaging localized, Stage I (lymph node-negative) tumors. Cancer. 2004;100:1657–1664. doi: 10.1002/cncr.20201. [DOI] [PubMed] [Google Scholar]
- Rusk A, Mckeegan E, Haviv F, Majest S, Henkin J. Khanna C. Preclinical evaluation of antiangiogenic thrombospondin-1 peptide mimetics, ABT-526 and ABT-510, in companion dogs with naturally occurring cancers. Clin. Cancer Res. 2006;12:7444–7455. doi: 10.1158/1078-0432.CCR-06-0109. [DOI] [PubMed] [Google Scholar]
- Shearin AL. Ostrander EA. Leading the way: canine models of genomics and disease. Dis. Model Mech. 2010;3:27–34. doi: 10.1242/dmm.004358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly S, Chien MB, Yip B, Kent MS, Theon AP, Mccallan JL. London CA. Exon 15 BRAF mutations are uncommon in canine oral malignant melanomas. Mamm. Genome. 2005;16:211–217. doi: 10.1007/s00335-004-2441-x. [DOI] [PubMed] [Google Scholar]
- Slipicevic A, Holm R, Nguyen MT, Bohler PJ, Davidson B. Florenes VA. Expression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcome. Am. J. Clin. Pathol. 2005;124:528–536. doi: 10.1309/YT58WWMTA6YR1PRV. [DOI] [PubMed] [Google Scholar]
- Smedley RC, Lamoureux J, Sledge DG. Kiupel M. Immunohistochemical diagnosis of canine oral amelanotic melanocytic neoplasms. Vet. Pathol. 2011a;48:32–40. doi: 10.1177/0300985810387447. [DOI] [PubMed] [Google Scholar]
- Smedley RC, Spangler WL, Esplin DG, Kitchell BE, Bergman PJ, Ho HY, Bergin IL. Kiupel M. Prognostic markers for canine melanocytic neoplasms: a comparative review of the literature and goals for future investigation. Vet. Pathol. 2011b;48:54–72. doi: 10.1177/0300985810390717. [DOI] [PubMed] [Google Scholar]
- Takikita M, Altekruse S, Lynch CF, et al. Associations between selected biomarkers and prognosis in a population-based pancreatic cancer tissue microarray. Cancer Res. 2009;69:2950–2955. doi: 10.1158/0008-5472.CAN-08-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Chin L, Garraway LA. Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26:1131–1155. doi: 10.1101/gad.191999.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br. J. Cancer. 2005;92:1398–1405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GJ, Soyer HP, Terzian T. Box NF. Modeling melanoma in mice. Pigment Cell Melanoma Res. 2011;24:1158–1176. doi: 10.1111/j.1755-148X.2011.00923.x. [DOI] [PubMed] [Google Scholar]
- Webster JD, Simpson ER, Michalowski AM, Hoover SB. Simpson RM. Quantifying histological features of cancer biospecimens for biobanking quality assurance using automated morphometric pattern recognition image analysis algorithms. J. Biomol. Tech. 2011;22:108–118. [PMC free article] [PubMed] [Google Scholar]
- Whiteman DC, Pavan WJ. Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res. 2011;24:879–897. doi: 10.1111/j.1755-148X.2011.00880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman K, Atkins MB, Prieto V, Eton O, McDermott DF, Hubbard F, Byrnes C, Sanders K. Sosman JA. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006;106:2005–2011. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ. Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am. J. Pathol. 2000;157:1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pleomorphic cytomorphologies of human (A, C, E) and canine (B, D, F) mucosal melanomas.
National Cancer Institute Comparative Melanoma Tumor Board Members.
Mucosal melanoma histopathological features in study patients.
Immunohistochemical evidence of melanocyte differentiation in canine malignant melanomas.
Immunohistochemical labeling scores for signal transduction pathway molecule expression in canine melanomas evaluated on tissue microarray.
Summary of previous molecular/genetic findings in canine melanoma.
Canine melanoma immunohistochemistry reagents and conditions.