

Abstract
In most eukaryotes, small RNA-mediated gene silencing pathways form complex interacting networks. In the ciliate Paramecium tetraurelia, at least two RNA interference (RNAi) mechanisms coexist, involving distinct but overlapping sets of protein factors and producing different types of short interfering RNAs (siRNAs). One is specifically triggered by high-copy transgenes, and the other by feeding cells with double-stranded RNA (dsRNA)-producing bacteria. In this study, we designed a forward genetic screen for mutants deficient in dsRNA-induced silencing, and a powerful method to identify the relevant mutations by whole-genome sequencing. We present a set of 47 mutant alleles for five genes, revealing two previously unknown RNAi factors: a novel Paramecium-specific protein (Pds1) and a Cid1-like nucleotidyl transferase. Analyses of allelic diversity distinguish non-essential and essential genes and suggest that the screen is saturated for non-essential, single-copy genes. We show that non-essential genes are specifically involved in dsRNA-induced RNAi while essential ones are also involved in transgene-induced RNAi. One of the latter, the RNA-dependent RNA polymerase RDR2, is further shown to be required for all known types of siRNAs, as well as for sexual reproduction. These results open the way for the dissection of the genetic complexity, interconnection, mechanisms and natural functions of RNAi pathways in P. tetraurelia.
INTRODUCTION
Small RNA pathways have greatly diversified in function during the evolution of eukaryotes (1,2) but generally rely on the production of 21–28 nt short RNAs (sRNAs) from single- or double-stranded RNA precursors. Once loaded onto Argonaute-containing effector complexes, sRNAs can target complementary transcripts and silence homologous sequences by diverse mechanisms, such as posttranscriptional or transcriptional inhibition or even DNA elimination (1,3). In RNA interference (RNAi), short interfering RNAs (siRNAs) are produced by endonucleases of the Dicer family from long double-stranded RNA (dsRNA) (4,5). The initial dsRNA triggers may be products of RNA-dependent RNA polymerases (RdRPs), enzymes that also act at downstream steps in some organisms to amplify the silencing response through the synthesis of secondary siRNAs (6,7).
Different small RNA pathways often coexist in a single organism, involving distinct types of sRNAs. In plants, animals and fungi, miRNAs and endogenous siRNAs regulate the expression of somatic and germline-specific genes, and specific endogenous siRNAs control transposons in somatic cells (2,3). Various organisms can also respond to environmental dsRNA, for instance some nematode and insect species that process dsRNA ingested within food bacteria or entering cells by endocytosis (8). Exogenously induced RNAi pathways were shown in several species to mediate antiviral defense (9–12). Despite the diversity of trigger molecules and sRNA responses, the different pathways involved can be interconnected by sharing some protein factors (3).
In ciliates, a phylum characterized by nuclear dimorphism, a meiosis-specific class of small RNAs (scnRNAs) targets the elimination of specific sequences during development of the somatic macronucleus (MAC) from the germline micronucleus (MIC) (13–17). As a result, the MAC genome is cleared of transposable elements and derived single-copy internal eliminated sequences (IESs) (18; for review see 19). In addition, siRNA pathways are active throughout the life cycle, as shown in Tetrahymena thermophila (20,21) and Paramecium tetraurelia (15,22,23). In P. tetraurelia, experimental induction of gene silencing revealed that different trigger molecules recruit different RNAi factors and produce different classes of sRNAs. It was first discovered that transformation of the MAC with high-copy transgenes lacking the 3′ UTR results in aberrant transcripts on both strands (22,24) and silencing of homologous endogenous genes (24,25). Transgene-induced silencing requires the Dicer protein Dcr1 (15), the Piwi proteins Ptiwi13 and Ptiwi14 (23), and the RdRP Rdr3 (22). Although Rdr3 appears to be catalytically inactive, it was implicated in the regulation of surface antigen gene expression and in the accumulation of endogenous siRNAs of unknown function (22). Transgene-induced siRNAs are 5′ monophosphate and modified at the 3′ end, presumably by 2′-O-methylation (22).
A second pathway, which allows Paramecium to process exogenous dsRNA, was found to involve a distinct but overlapping set of RNAi factors. Feeding cells with Escherichia coli bacteria engineered to produce dsRNA, equivalent to the ‘feeding’ technique in Caenorhabditis elegans (26,27), induces silencing of endogenous genes homologous to the dsRNA (28). In this pathway, Dcr1 (15) and the two RdRPs Rdr1 and Rdr2 (22) are required for accumulation of two types of siRNAs: primary siRNAs, predominantly 23 nt in length and produced from the original dsRNA trigger, and secondary siRNAs, presumably produced from the target mRNA (15). In contrast to the primer-independent, 5′ triphosphate secondary siRNAs produced by direct RdRP activity in C. elegans (29–31), P. tetraurelia dsRNA-induced siRNAs are 5′ monophosphate. Three Piwi proteins (Ptiwi13, Ptiwi12 and Ptiwi15) appear to be involved in this pathway (23).
The MIC genome of P. tetraurelia has undergone at least three whole genome duplications (WGDs), resulting in ∼40,000 genes in the MAC genome, which is responsible for all gene expression. 68% of genes still retain their duplicates from the most recent WGD (WGD1), and the conservation of protein sequences suggests that most duplicate pairs are functionally redundant (32). WGDs may explain in part the large number of genes encoding core RNAi factors: eight Dicer- and Dicer-like genes (including one WGD1 duplicate pair), 17 Piwi genes (two unpublished) (six WGD1 pairs) and four RdRP genes (no WGD1 duplicate). Only a few of these genes have been assigned any function, making P. tetraurelia a very interesting model to study the genetic and mechanistic complexity of small RNA-mediated pathways. Due to the lack of an established method to transform the MIC genome, however, reverse genetics approaches have been restricted to RNAi thus far, providing only limited possibilities for the functional analysis of genes involved in RNAi. Targeting a gene involved in its own silencing (recursive RNAi (33)) is a self-defeating process which cannot be completed, and is not conclusive when no effect is observed.
In this study, we describe the outcome of a forward genetic screen for mutants deficient in dsRNA-induced silencing, based on lethal dsRNA ‘feeding’, which was expected to yield loss-of-function alleles for genes that do not have functionally redundant paralogs. A combined strategy, including the development of a powerful method to identify mutations by whole-genome resequencing, allowed us to characterize 71 RNAi-deficient mutants and yielded a total of 47 alleles for three known and two previously unknown genes. Analyses of allelic diversity revealed a non-essential dsRNA processing machinery but provided evidence for an essential RNAi function during the life cycle, and suggested that the screen is saturated for non-essential, single-copy genes. The dsRNA- and transgene-induced RNAi pathways are further shown to share essential protein factors.
MATERIALS AND METHODS
Paramecium strains, cultivation and genetic analyses
Mutagenesis and other experiments were carried out with wild-type strain 51 of P. tetraurelia, mating type E. RNAi mutants were back-crossed either with strain 51 or with strains carrying the nd7–1 mutation (34) as a genetic marker (after three rounds of back-cross into strain 51). Unless otherwise specified, cells were grown at 27°C in wheat grass powder (Pines International Co., Lawrence, KS, USA) infusion medium bacterized with Klebsiella pneumoniae the day before use and supplemented with 0.8 µg/ml β-sitosterol. Genetic analyses of mutants were carried out according to standard procedures (35). F1 phenotypes were recorded for each of the two karyonides resulting from the two exconjugants of at least two conjugating pairs, and at least 30 F2 clones resulting from autogamy of F1 heterozygotes were studied.
Random mutagenesis and screening for RNAi-deficient mutants
UV mutagenesis (254 nm) was carried out on several independent batches of 200,000 pre-autogamous cells, according to Cohen (36), with a modified dose of 650 J/m2. Cells were allowed to undergo two divisions before autogamy was triggered by starvation to make MIC mutations homozygous in the MICs and MACs of progeny. Three batches in which autogamy reached >90% of the cells, and showing a post-autogamous survival rate of 75–80%, were selected for screening. To isolate mutants deficient in dsRNA-induced RNAi, post-autogamous cells were first fed with three volumes of Klebsiella medium (∼1–2 vegetative divisions) and starved again to eliminate fragments of the old MAC (monitored by DAPI staining), which would otherwise complement mutations in the new MAC. Cells were then washed and transferred to three volumes of N-ethylmaleimide-sensitive factor (NSF) ‘feeding medium’ (E. coli producing NSF dsRNA, see below) and grown until mass lethality (∼36 h, four divisions). Surviving cells were isolated and grown in standard K. pneumoniae-bacterized medium. Each clone was replicated for storage and further experiments. RNAi deficiency was verified by feeding each clone with dsRNAs from the unrelated genes ND7 and ND169 (34,37) both required for trichocyst discharge, a quantitative phenotype that allows to distinguish full and partial RNAi deficiencies (see below).
Induction of RNAi and phenotypic analyses
Production of dsRNA in E.coli strain HT115DE3, feeding to Paramecium cells, transgene-induced silencing of ND169 and monitoring of trichocyst discharge phenotypes were carried out as described previously (22,38). NSF-depleted cells were checked for viability after 36 h, the time at which typically all cells were dead or dying in a successful silencing. To reveal partial loss-of-function phenotypes, ND169 dsRNA producing E. coli were typically mixed in 1:5 ratio with ICL7a dsRNA producing bacteria.
Plasmid constructs and re-sequencing of known genes
To induce silencing of NSF and of ND169 by dsRNA feeding or by transgene (pTI-) plasmid constructs produced previously (22,28) were used. For silencing of PDS1 and CID1-like genes, fragments of the coding region were cloned into the plasmid L4440 and dsRNA was synthesized in E. coli HT115 DE3 (Supplementary Figure S1). Complementation of mutants was achieved by microinjection of the entire open reading frame and flanking sequences, including the endogenous promotor, cloned into the pUC18 plasmid vector. For details of all plasmid constructs see Supplementary Table S1. For re-sequencing of known genes, coding sequences including flanking regions were amplified by polymerase chain reaction using a proof-reading polymerase (Phusion® High Fidelity, Thermo Fisher Scientific, Waltham, MA, USA). Products were purified by phenole extraction and polyethylene glycol 8000 precipitation (7,5% final) and sequenced with marginal and internal primers (oligonucleotide sequences on request).
Purification of MAC DNA, whole-genome sequencing and identification of SNPs
RNAi mutants carrying an unknown mutation were backcrossed to wild type and one karyonide of each exconjugant F1 clone was brought to F2 by autogamy. Thirty F2 clones were isolated and tested for RNAi deficiency by feeding of ND169 or NSF dsRNA. Mutant clones showing silencing deficiency were raised to large cultures. Immediately prior to DNA extraction, vegetative mutant cultures of both mating types were fused and MAC DNA was extracted from this pool (∼3 Mio cells) according to the protocol used for DNA purification from developing MACs (18). Briefly, cells were concentrated, washed, supplemented with three volumes of lysis buffer (0.25 M sucrose, 10 mM MgCl2, 10 mM Tris pH 6.8, 0.2% Nonidet P-40) and lysed at 4°C with a Potter–Elvehjem homogenizer, allowing cell- but not MAC lysis. MACs were collected and purified from bacteria and cell debris by ultracentrifugation through a 2.1 M sucrose cushion. Recovered MACs were lysed and DNA was extracted as described (39). 5 μg of RNaseA-treated genomic DNA were used for library construction. DNA was sequenced by single-end (75 bp lead length) or paired-end (50 or 100 bp read length) strategies using Illumina GAII and HiSeq next-generation sequencers (for overview of sequence output for each mutant F2 pool, see Supplementary Table S2). The reads were mapped to the reference P. tetraurelia strain 51 genome (18,32) using the Burrows-Wheeler Alignment tool (BWA) (40) with default parameters and the mapping indexed with SAMtools (41). Custom Perl scripts and the SAMtools mpileup function were used to identify positions where at least 80% of the calls differed from the reference genome, at positions with at least 10-X coverage and less than 300-X coverage. The list of single nucleotide polymorphisms (SNPs) was then curated to identify candidate mutations for experimental validation.
Small RNA analysis by northern blot
Total RNA extraction and small RNA northern blots were carried out as described (22). As previously, two adjacent 50 nt sense oligonucleotide probes matching to the dsRNA region were used to detect ND169 siRNAs, as most of the produced siRNAs are antisense to the target transcript. Similarly, endogenous cluster22 siRNAs were detected with two adjacent 50 nt oligonucleotide probes, complementary to the top strand (relative to transcription of the 5′ marginal open reading frame).
RESULTS
Lethal dsRNA feeding allows isolation of Mendelian mutants fully or partially deficient in RNAi
To isolate mutants of the dsRNA-induced silencing pathway, we first treated wild-type cells with UV light to induce random mutations in the MIC genome. Cell populations were then allowed to undergo autogamy, a self-fertilization sexual process in which only one of the parental MIC alleles is retained, and made homozygous in the MICs and MACs of progeny (Figure 1A). Cultures were then screened by feeding them an E. coli strain producing dsRNA homologous to NSF, an essential gene involved in exocytosis and membrane traffic (42,43). NSF dsRNA feeding rapidly kills the wild type (28), so that only mutant cells deficient in dsRNA-induced RNAi would be able to survive. One hundred and fifty surviving cells were isolated, and the RNAi-deficient phenotypes were confirmed by dsRNA feeding targeting different genes (NSF; ND7 and ND169, two single-copy, unrelated genes involved in the exocytosis of secretory granules called trichocysts (34,37); for details, see below, Figure 7B). After sorting out non-viable, false positive or strongly hypomorphic clones, a set of 79 RNAi-deficient cell lines was established (detailed outcome of the screen in Supplementary Table S3).
Figure 1.
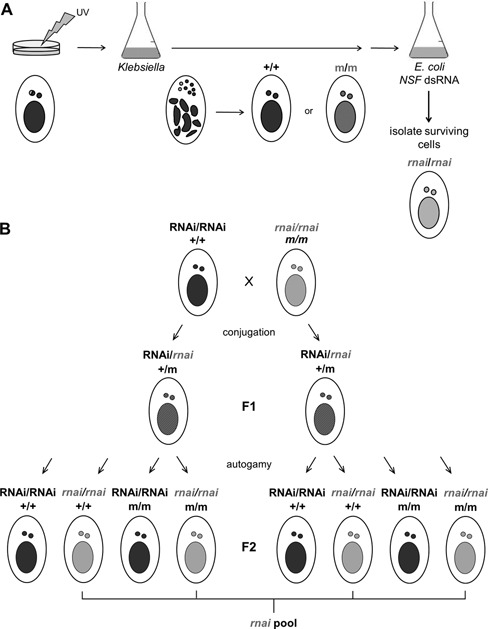
Mutagenesis screen and identification of unknown RNAi factors. (A) UV-irradiated cell cultures were starved to induce autogamy, a self-fertilization process that leads to complete homozygocity of the MIC genome. Re-feeding cells with K. pneumoniae medium allowed development of the new MACs from the resulting homozygous zygote within two cell divisions, leading to expression of the mutated genes. Transfer of cells into NSF dsRNA feeding medium allowed isolation of potential rnai mutants, as silencing of the essential NSF gene leads to lethality only in wild type cells (28,42,43). (B) To identify mutations in unknown RNAi factors, silencing deficient mutants were crossed with the wild type, resulting in heterozygous F1 progeny. After autogamy, F2 clones become homozygous for the rnai mutant (gray) or wild type (black) allele. Most other (UV-induced) mutations (m) are not linked with the target mutation (rnai) and segregate independently. F2 clones carrying the rnai allele were pooled for MAC DNA extraction and whole genome-sequencing. In this data set only the rnai mutation is covered by 100% of SNP-containing reads, whereas all other mutations are ideally covered by 50% SNP-containing and 50% wild type reads.
Figure 7.
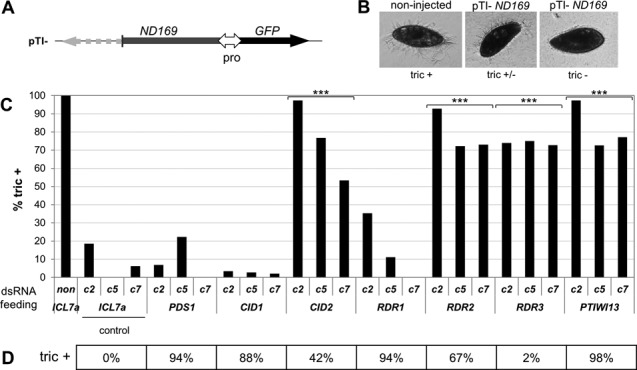
dsRNA-induced RNAi factors sharing functions with the transgene-induced silencing pathway. (A)ND169 transgene construct pTI- carrying a 3′ truncated version of the ND169 coding sequence driven by a bidirectional constitutive promotor. Green fluorescent protein (GFP) was monitored as control for injection and expression (not shown). (B) Nd169 is involved in the exocytosis of secretory granules (trichocysts) docked at the plasma membrane. Wild-type cells expell trichocysts upon treatment with picric acid (tric+). By microinjection of pTI- into the MAC, strongly silenced clones showing complete trichocyst discharge defects were obtained (tric−) (not shown), as well as three clones with moderate silencing effect (tric+/−), c2, c5, and c7. Note that clone c2 showed up to 18% of cells with tric+ phenotypes in control cultures (ICL7a feeding). (C) Knock down of RNAi factor genes by dsRNA feeding in clones with moderate transgene-induced silencing phenotypes. De-repression of ND169 silencing was measured as the percentage of cells showing a complete wild type phenotype (tric+). A significant difference to the ICL7a control was detected upon silencing of CID2, RDR2, RDR3 and PTIWI13 (P-values < 10e-5 (***), one-way ANOVA, significance level 0.005). Phenotypes recorded 120 h after the first feeding are shown. Note that pTI- induced silencing efficiencies varied slightly in different cultures of the same injected clone. (D) The effect of RNAi factor depletion on dsRNA-induced silencing was verified in parallel as a positive control by double knock down, mixing RNAi factor and ND169 dsRNA-producing bacteria in equal amounts. The percentage of tric+ cells, i.e. the degree of inhibition of ND169 silencing, is shown (mean of two replicates for CID1 and PDS1). ICL7a control dsRNA feeding does neither inhibit dsRNA- nor transgene-induced silencing; PTIWI13 is involved in both pathways, whereas RDR3 is specific for transgene- and RDR1 for dsRNA-induced silencing, as previously described (22,23).
Seventy-one lines were further analyzed. Of these, 64 were fully deficient in RNAi, showing normal growth rates in NSF feeding medium and wild-type levels of trichocyst discharge upon ND7 and ND169 feeding. Seven lines were only partially compromised in RNAi, suggesting hypomorphic mutations. We reasoned that feeding cells with a smaller amount of dsRNA would be more sensitive to partial defects in the RNAi machinery. Consistently, diluting the silencing trigger (e.g. ND169 dsRNA bacteria in a 1:5 ratio with bacteria producing dsRNA from ICL7a, which encodes a non-essential centrin (25)) revealed stronger phenotypes in hypomorphic mutants, comparable to the full RNAi deficiency, although this still led to complete silencing in wild-type cells.
Back-crossing selected lines to the wild type confirmed that RNAi phenotypes were due to Mendelian and recessive loss-of-function mutations: F1 heterozygotes showed a wild-type phenotype, and F2 homozygotes obtained by autogamy of the F1s showed the expected 1:1 segregation of wild-type and mutant phenotypes upon dsRNA feeding (Supplementary Table S4A). Although these segregation analyses would not be sufficient to dissociate closely linked mutations, the total number of UV-induced mutations in the genome was later estimated to be ∼30 per irradiated cell on average after autogamy (Supplementary Table S5A; for mutations, see Supplementary Table S5B), indicating that in most cell lines the RNAi deficiency is due to a single mutation.
The mutant screen reveals known and new genes involved in dsRNA-induced RNAi
To identify the mutations in RNAi-deficient lines, we developed a whole-genome sequencing procedure to distinguish the mutation causing the phenotype from other, irrelevant UV-induced mutations, after a single round of back-cross to the wild type (Figure 1B). Briefly, F1 heterozygotes were taken through autogamy, and sets of ∼30 homozygous F2 clones were tested for their capacity to respond to dsRNA feeding. Deep sequencing of MAC DNA from pools of ≥15 independent RNAi-deficient F2s will identify the RNAi mutation as being present in 100% of reads, while other, unlinked mutations show up only in 50% of reads on average. This strategy was used in combination with complementation tests and resequencing of genes previously implicated in the pathway to identify candidate mutations in all 71 lines. 45 of them contained mutations in RDR1, RDR2 or DCR1 (Table 1), confirming the role of these genes in dsRNA-induced RNAi.
Table 1.
Mutants obtained in a screen for dsRNA-induced RNAi deficiency
| mutants | alleles | function | homologs | |
|---|---|---|---|---|
| RDR1 | 41 | 26 | RdRP | Tt, At, Ce, Sp |
| RDR2 | 2 | 2 | RdRP | Tt, At, Ce, Sp |
| DCR1 | 2 | 2 | RNase III Dicer | Tt, At, Ce, Sp, Dm, Mm, Hs |
| CID1* | 16 | 7 | nucleotidyl transferase | Tt, At, Ce, Sp, Dm, Mm, Hs |
| PDS1* | 10 | 10 | unknown | Paramecium |
The first alleles to be discovered for the two new genes (*) CID1 and PDS1 were identified by whole-genome sequencing of pooled F2 clones (see Figure 1).
The remaining 26 lines were found to carry mutations in either of two new genes, for which one allele was initially identified by the whole-genome sequencing procedure. The missense mutation 1.8 (Figure 2; Supplementary Table S6) is located in the predicted gene PTETG9100013001 (ParameciumDB), encoding a protein of the nucleotidyl transferase family (also referred to as non-canonical polyA/U RNA polymerases (44)) (Figure 3A). It is related to the first described non-canonical polyA polymerase, Cid1 of S. pombe, as well as to S. pombe's Cid12 (Supplementary Figure S2), the first protein among others of that family shown to be involved in small RNA mediated silencing (45). The gene was therefore named CID1. Cid1 is closely related to Rdn2 of T. thermophila (Supplementary Figure S2; Figure 6A), which physically interacts with the RdRP Rdr1 in an ‘RdRC’ complex (46,47). CID1 is expressed at constant levels throughout the life cycle (Supplementary Figure S3).
Figure 2.
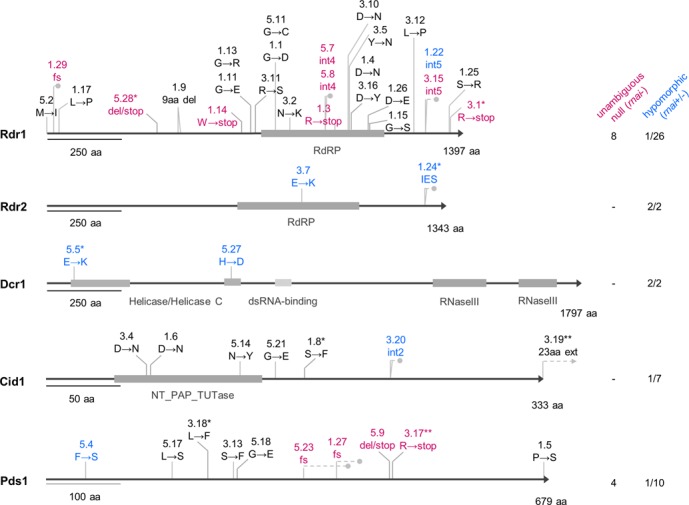
Alleles identified from a screen for mutants deficient in dsRNA-inducible RNAi. Unambiguous null alleles (purple, rnai- phenotype as defined in the text) were obtained only for RDR1 and PDS1. The location of premature stop codons resulting from frameshifting mutations is shown by gray dots. The 5′ TA boundary of the only IES (76 bp) in the RDR2 gene is mutated to AA in the rdr2–1.24 allele, leading to complete retention of the IES in the MAC (Supplementary Figure S8B). Hypomorphic alleles are shown in blue (rnai+/- phenotype). Gene accession numbers are available in supplementary data (Supplementary Table S1). Co-segregation of the phenotype with the mutation in F2 was verified for alleles labelled with an asterisk (*). Alleles complemented by injection of linear DNA encoding the wild-type gene with its natural flanking regions are labeled with two asterisks (**).
Figure 3.

Two newly identified proteins involved in dsRNA-induced RNAi. Protein alignments were performed using the MUSCLE v4 software (78). (A) Cid1 is a nucleotidyl transferase. Aspartic-acid residues of the predicted catalytic triad (*) required for nucleotidyl transferase activity of Arabidopsis thaliana Heso-1 (52) and Tetrahymena thermophila Rdn1 and Rdn2 (47) are mutated in cid1–1.6 and cid1–3.4. Pt, Paramecium tetraurelia; Tt, T. thermophila; Sp, Schizosaccharomyces pombe; Ce, Caenorhabditis elegans; At, A. thaliana; Hs, Homo sapiens. (B) Pds1 is conserved in Paramecium species. Examples of missense alleles obtained for Pt-Pds1 in the RNAi mutant screen are indicated with black arrowheads (for substituting amino acids, see Supplementary Table S6). Pt, P. tetraurelia; Ppr, P. primaurelia; Pb, P. biaurelia; Ppe, P. pentaurelia; Ps, P. sexaurelia; Pm, P. multimicronucleatum, Pc, P. caudatum.
Figure 6.
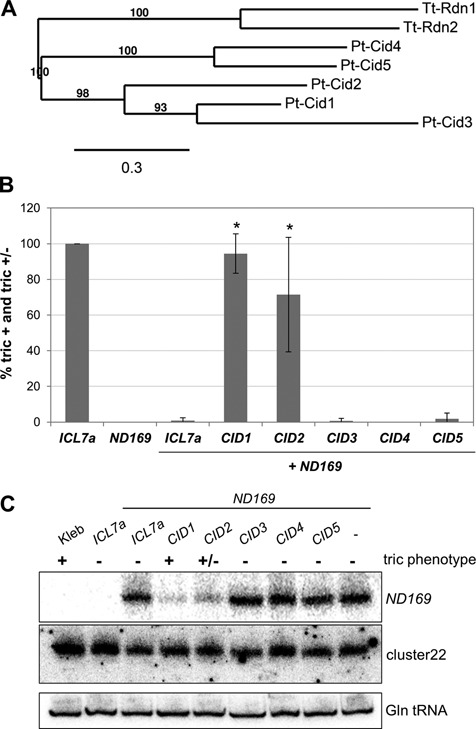
Cid2 is also involved in dsRNA-induced silencing. (A) Phylogenetic relationship of the Cid1-like proteins clustering with Pt-Cid1 (all Cid1-like genes identified in the P. tetraurelia MAC genome and generation of phylogenetic trees, see Supplementary Figure S2 and Table S7). (B) Double knock down experiment of Pt-Cid1-like genes and ND169 by dsRNA feeding. ND169 was used as a reporter for silencing; the gene was considered as silenced when cells showed complete deficiency of trichocyst discharge (tric-) and inhibition of silencing was measured as proportion of cells in the culture showing partial (tric+/−) or complete (tric+) reversion of silencing. Silencing was significantly inhibited upon knock down of CID1 and CID2 (P-values < 0.03 (*), Mann–Whitney U test, significance level 0.05; n = 3; standard deviation is shown). Phenotypes were recorded 72 h after the first feeding. Bacteria were fed in equal amounts. Triple knock down experiments (CID3+CID4+ND169; CID3+CID5+ND169; CID4+CID5+ND169) did not show inhibition of ND169 reporter silencing (data not shown). Cross-silencing between CID1 and CID2 genes is unlikely, as the maximum length of perfect homology is 12 nt, and 18 nt with one central mismatch. (C) Northern blot analysis of associated ND169 siRNAs. The ND169 probe corresponds to a 100 bp region of the dsRNA and is sense oriented. The lower panel shows hybridization to glutamine tRNA as a loading control.
Mutation 3.18 lies in a 2-kb single-copy gene on MAC scaffold 6 (PTETG600032001) (Figure 2; Supplementary Table S6), expressed throughout the life cycle (Supplementary Figure S3). Blast and conserved domain searches did not reveal any significant hit, even in other available ciliate genomes (Tetrahymena thermophila, T. borealis, T. elliotti, T. malaccensis (Oligohymenophora) and Oxytricha trifallax (Hypotricha). In contrast, the gene is present in all sequenced species of the Paramecium genus, allowing identification of conserved amino acids (Figure 3A; Supplementary Figure S4). Deduced from its specificity for dsRNA-induced RNAi (see below), we named this gene PDS1 (Paramecium dsRNA-induced RNAi-specific protein 1).
These candidate mutations were shown to co-segregate with the RNAi-deficient phenotype in each of the F2 clones included in the genome-sequencing pools, and microinjection of the cloned wild-type genes into the MAC of mutants (cid1–3.19 and pds1–3.17) rescued the RNAi deficiency (data not shown). In addition, depletion of these proteins by recursive RNAi experiments (see below, Figure 7D) resulted in an RNAi-deficient phenotype. We conclude that CID1 and PDS1 are required for dsRNA-induced silencing.
The RNAi response to dsRNA from food bacteria is not essential for viability
Our screen for RNAi-deficient mutants was unlikely to reveal genes with functionally redundant WGD1 ohnologs, unless disruption of one copy led to a dosage effect. For single-copy genes, we could expect null alleles of non-essential genes and hypomorphic alleles of essential genes. Unambiguous null alleles were here defined as those containing nonsense mutations or frameshifts resulting in premature stop codons, due to indels or mutations in intron splice sites (48), except in cases where the best part of the protein is conserved and where there is experimental evidence for a partial RNAi deficiency. An allele was categorized as hypomorphic if the RNAi deficient phenotype was only partial and/or dsRNA feeding-induced siRNAs were reduced, but still detectable on northern blots. Indeed, the diversity of alleles obtained for each of the genes hit appeared to reflect their importance for cellular viability (Figure 2): 41 rdr1 mutants were found, representing 26 different alleles. Among these were eight putative null alleles (e.g. rdr1–5.28 or rdr1–5.7). Missense alleles were mostly non-conservative substitutions in conserved residues, two of which in the putative catalytic core region (rdr1–1.4 D1021N and rdr1–3.16 D1021Y) (49–51) (Supplementary Table S6). Similarly, the 10 alleles obtained for PDS1 included null alleles, suggesting that this gene, like RDR1, is not required for viability. Although no unambiguous null allele was found among the seven cid1 alleles, some of the mutations changed highly conserved amino acids (cid1–3.4 D68N and cid1–1.6 D70N) shown to be required for uridylyl transferase activity (47,52). In all putative null mutants tested, a complete loss of dsRNA-induced siRNAs was observed on northern blots (Figure 4A and Supplementary Figure S6). RNAi-deficient rdr1, pds1 and cid1 null mutants are fully viable throughout the life cycle (vegetative growth and sexual events), and no other phenotypic anomaly was observed. Furthermore, the F2 (Supplementary Table S4B) and F3 generations of an rdr1–3.1/cid1–1.8 double homozygote showed normal vegetative growth in standard conditions and at high temperature (34°C). We conclude that the capacity to synthesize siRNAs from dsRNA ingested with food is not essential in laboratory conditions.
Figure 4.
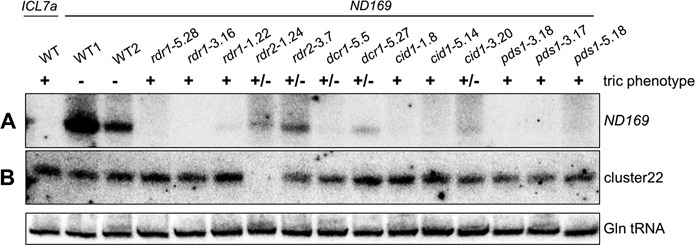
Abolished siRNA accumulation in RNAi mutants. (A) Northern blot analysis of ND169 siRNAs revealed failure or strong reduction of accumulation of exogenously induced siRNAs in all mutants, consistent with previous findings from depletion of Dcr1, Rdr1 and Rdr2 by RNAi (15,22,23). This suggests a role of these RNAi factors upstream or within siRNA biosynthesis or stabilization. It is of note that silencing efficiencies can vary from one dsRNA feeding experiment to another, possibly due to contamination by traces of the standard food bacterium Klebsiella (showing partial Ampicillin resistance) which overgrows dsRNA-producing E. coli. This may explain differences in the total level of dsRNA-induced siRNAs in different wild-type cultures. The ND169 probe corresponds to a 100 bp region of the dsRNA and is sense oriented, as antisense siRNAs represent the predominant fraction of siRNAs in the dsRNA region (22). (B) Detection of endogenous siRNAs from an intergenic region of scaffold 22 (cluster22) revealed that rdr2 mutants are unable to accumulate these siRNAs. The probe is complementary to the predominant fraction of siRNAs (top strand) of this region. The lower panel shows hybridization to glutamine tRNA as a loading control.
Nevertheless, the dsRNA-induced RNAi pathway appears to be highly conserved in the Paramecium genus, since tblastn searches identified at least one copy of the RDR1, CID1 and PDS1 genes in all sequenced species of the P. aurelia complex, as well as in P. multimicronucleatum and P. caudatum (Supplementary Figure S5). Essential factors playing a role in this pathway in P. tetraurelia (see below) are also highly conserved (not shown). This suggests that the pathway may fulfill an important function required for long-term survival of these species in their natural environment.
The screen is saturated for non-essential single-copy genes
The large numbers of alleles obtained for three non-essential, single-copy genes suggest that the screen is saturated for this category of genes. For RDR1, PDS1 and CID1, the number of alleles is roughly proportional to the length of the gene; the relationship indicates that, with the number of mutant lines analyzed here, one mutation was obtained for every 172 bp of coding sequence on average (Figure 5). Thus, there may not be any other non-essential, single-copy gene involved in the dsRNA-induced RNAi pathway.
Figure 5.
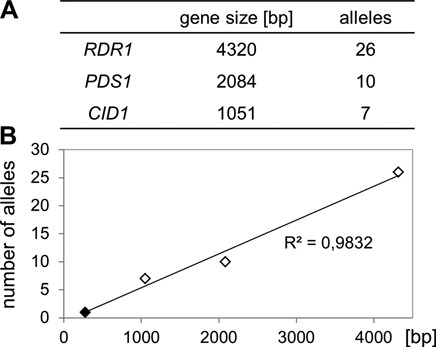
Saturation of the screen for non-essential single-copy genes involved in dsRNA-induced RNAi. Gene sizes of non-essential RNAi factors (A) plotted against the number of identified mutant alleles obtained (B). Extrapolation by linear regression determined the mutation frequency to 1 in 172 bp of coding sequence.
Essential genes may be involved in dsRNA-induced RNAi
In contrast to RDR1, CID1 and PDS1, only two alleles of RDR2 were identified. rdr2–3.7 is an amino acid substitution (E859K) within a less conserved region of the RdRP domain, and rdr2–1.24 retains an intragenic IES (MIC-specific) located downstream of the conserved RdRP domain region, due to a T-to-A mutation in one of the TA boundaries that are required for excision during macronuclear development (53) (Supplementary Figure S8B). Both alleles are hypomorphic, as indicated by partial RNAi deficiency and by partial loss of siRNAs upon dsRNA feeding (Figure 4A and Supplementary Figure S6). The greater reduction in siRNA levels in rdr2–1.24 was paralleled by a weaker silencing of the ND169 target using dilute dsRNA trigger (not shown). Similarly, for DCR1 only two hypomorphic missense alleles were identified. Interestingly, the mutations are located within the N-terminal helicase (dcr1–5.5 E87K) or helicase C domains (dcr1–5.27 H622D). Dcr1–5.5 shows a stronger RNAi deficiency than dcr1–5.27, judging from both phenotypes and siRNA levels (Figure 4A and Supplementary Figure S6). These findings suggest that the helicase and helicase C domains play a role in the biosynthesis of dsRNA-derived siRNAs, unlike the helicase domain of C. elegans’ Dcr-1, which is only required for a subset of endo-siRNAs (54). The fact that RDR2 and DCR1 are only represented in the mutant collection by a small number of hypomorphic alleles suggests that they are essential genes for which null alleles would lead to lethality. Thus, RNAi may fulfill essential functions in P. tetraurelia.
Another, putatively essential nucleotidyl transferase is involved in dsRNA-induced silencing
Whereas no other PDS1-related gene could be identified in the P. tetraurelia genome, 22 Cid1-like non-canonical poly(A)-polymerases were found (Supplementary Table S7). According to phylogenetic analyses (Supplementary Figure S2), five of them, including Cid1, fall in a common branch with Rdn1 and Rdn2 of T. thermophila (Figure 6A). None of them is an ohnolog to another. To analyze their function in dsRNA-induced RNAi, we used a recursive RNAi approach, i.e. we silenced each of them by dsRNA feeding, simultaneously with ND169 as a reporter (22). Apart from CID1, targeting CID2 also prevented silencing of ND169 by dsRNA feeding (Figure 6B). This was correlated with a reduced accumulation of ND169 siRNAs (Figure 6C). It is of note that silencing of a gene involved in its own silencing has only limited efficiency, explaining the partial impairment of ND169 silencing and siRNA accumulation. The results indicate that, like CID1, CID2 is involved in dsRNA-induced RNAi. Although it is a single-copy gene, our screen did not yield a mutant allele, raising the hypothesis that CID2 is essential for viability.
Putatively essential RNAi factors are shared with the transgene-induced silencing pathway
Although transgene-induced siRNAs are biochemically different from dsRNA-induced siRNAs, Dcr1 and Ptiwi13 are shared between these two pathways. Like DCR1, PTIWI13 is possibly an essential gene, as mutants were not obtained in our screen, probably due to inefficient recovery of hypomorphic mutants. Rdr3, a factor specifically involved in transgene-induced silencing, also seems to be essential, as its silencing results in a reduced vegetative growth rate (22). To test whether the newly identified genes are also involved in transgene-induced silencing, we induced silencing of ND169 by injecting a transgene construct (pTI-) truncated at the 3’end of the coding sequence (Figure 7A), as previously described (22). Candidate genes were then silenced in injected clones by dsRNA, and the ND169 silencing phenotype was monitored (recursive RNAi, as described previously (22)). To increase the sensitivity of the silencing response to depletion of potentially involved factors, we used injected clones that showed only moderate ND169 silencing efficiency (tric+/−) (Figure 7B). Cells showed a strong decrease in silencing efficiency upon CID2 silencing and, as expected, upon RDR3 and PTIWI13 silencing (Figure 7C). Surprisingly, using this experimental setup we were also able to detect a significant reduction of transgene-induced silencing efficiency when RDR2 was co-silenced, which had not been observed previously on strongly silenced clones using the same RDR2 silencing construct (22). In contrast, targeting CID1, PDS1 or RDR1 did not result in reduced silencing of the ND169 transgene, although dsRNA-induced RNAi was inhibited in control experiments (Figure 7D). This suggests that these genes are not involved in the transgene-induced RNAi pathway. The involvement of the putative essential gene DCR1 in silencing induced by the ND169 transgene construct (as well as ND169 dsRNA) could also be confirmed (not shown), as previously shown for a promotor-less transgene (15). We conclude that RDR1, CID1 and PDS1 are specific for the dsRNA-induced pathway, while genes hypothesized to be essential according to the above criteria (RDR2, CID2, DCR1, PTIWI13) are involved in both dsRNA- and transgene-induced RNAi pathways.
Rdr2 mutants are compromised in endogenous siRNA accumulation and show reduced viability after sexual reproduction
Factors of the transgene-induced RNAi pathway may have essential functions in any phase of the life cycle, including vegetative growth, sexual events and development. We checked whether mutants are impaired in the accumulation of a cluster of endogenous siRNAs of unknown function (cluster22) which are produced throughout the life cycle from an intergenic locus (15). Northern blot analysis revealed that their accumulation was completely suppressed in the rdr2–1.24 mutant (Figure 4B). Signal quantification (Supplementary Figure S6) showed only a slight, if any, reduction for the rdr2–3.7 allele, consistent with its strongly hypomorphic nature. Tested mutations in RDR1, CID1 and PDS1 had little or no effect. Surprisingly, dcr1 mutants showed only slight (if any) reduction in cluster22 siRNA levels as well, as also observed after targeting DCR1 by dsRNA-induced RNAi (not shown). Similarly, targeting CID2 did not result in any detectable reduction (Figure 6C), though this may be due to inefficient depletion by recursive RNAi. Despite the molecular phenotype evidenced here, the rdr2 mutants did not differ from the wild type in viability or growth rate during vegetative growth.
In contrast, rdr2 mutants showed a range of defects during sexual reproduction. Survival of homozygotes for the stronger allele rdr2–1.24 was found to be affected by a long starvation period (≥3 days), but only when this immediately followed autogamy, and not when the same starvation was experienced by vegetative clones that had already undergone 7–8 divisions (Supplementary Figure S7). New MAC development nevertheless appeared to be cytologically normal, as judged from DAPI staining, and the reason for this post-autogamous defect remains unknown.
Conjugation was also impaired in both rdr2 mutants. After a cross to the wild type, the F1 clones derived from one or both parents were unviable for 35% (18/51) of conjugating pairs and the frequency of proper formation of heterozygous new MACs seemed reduced (Supplementary Figure S8A, Table S4A). The most striking effect was that autogamy of the viable, heterozygous F1s most often (9/10; P = 0.01) gave rise to F2 progeny in which close to 100% of clones were unviable or showed regeneration of the F1 MAC (Supplementary Figure S8, Table S4A). This was observed for F1s deriving from both parents and suggests a dominant-negative maternal effect of the heterozygous F1 MAC with incomplete penetrance, because the rare cases where the F2 progeny survived gave the expected 1:1 segregation of rdr2 and wild-type F2 homozygotes, and both types of clones survived equally well. The reason why post-autogamous lethality is much stronger for heterozygotes than for rdr2 homozygotes, and the nature of the cellular process affected, remain unclear. Testing excision of some IESs in post-autogamous, newly developed MACs (rdr2–1.24) did not reveal any DNA rearrangement defect for maternally controlled (scnRNA-dependent) or non-maternally controlled IESs (data not shown). Nevertheless, our observations indicate that RDR2 has essential functions during sexual reproduction. A similar phenotype was not observed in any other RNAi mutant.
DISCUSSION
A whole-genome sequencing method to identify mutations potentiates a powerful screen
In this study, we have used a powerful and straightforward mutagenesis screen to identify genes required for the RNAi response to dsRNA feeding in P. tetraurelia: dsRNA homologous to the essential gene NSF efficiently kills the wild type, and can be fed to large populations of cells. Mutational studies have long benefited from the easy genetics and short generation time of this organism (55–59), but the identification of mutations relied on functional complementation and sib-selection sorting of an indexed library (60,61), a time-consuming method ill-suited for large mutant collections. We have designed an efficient procedure to identify phenotype-causing mutations after a single round of back-cross to the wild type. Other, unlinked mutations segregate independently among F2 homozygotes, so that whole-genome sequencing of pools of phenotypically mutant and wild-type F2s will show the RNAi mutation in 100% and 0% of reads, respectively, while most irrelevant mutations should be present in 50% of reads from both pools. In practice, sequencing of the wild-type pool was found to be unnecessary; 15-X coverage of pools of ∼20 mutant clones, with 75-bp single reads, was often sufficient to narrow down the list of candidate mutations to a single one, although 100-bp paired-end sequencing proved more efficient. Distinguishing closely linked mutations would require more F2s and a higher coverage, but the standard mutagenesis conditions used (∼20% lethality in homozygous progeny of irradiated cells) were shown to result in an average of ∼30 mutations in the 72-Mb MAC genome of viable progeny, making cases of tight linkage infrequent. Similar approaches have been used in C. elegans and A. thaliana (62,63), but here the possibility to generate large numbers of entirely homozygous recombinant F2s by autogamy makes the method faster and simpler, dispensing with the need for extensive back-crossing or pre-mapping.
Mutations causing full or partial RNAi deficiency identify known and new genes
Mutations were identified in 71 RNAi-deficient cell lines, yielding a total of 47 different alleles for only five genes (Figure 8). Three of them (RDR1, RDR2 and DCR1) were previously implicated in dsRNA-induced RNAi on the basis of recursive RNAi experiments. The two new genes included the nucleotidyl transferase CID1 and the Paramecium-specific gene PDS1. Among the 47 alleles, 14 were isolated more than once; in three cases the same alleles were recovered from different mutagenized cultures and thus originated from different mutation events. This suggests that our analysis provides a rather exhaustive view of the genes and alleles that can be identified by the experimental scheme used.
Figure 8.
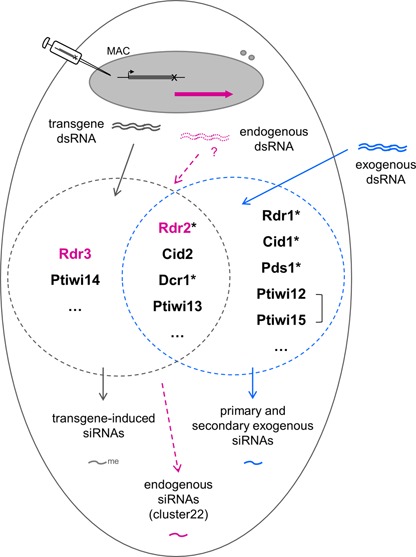
Sharing of RNAi core factors by non-essential exogenously and essential endogenously triggered RNAi pathways in P. tetraurelia. In this study, mutants were obtained for genes labelled with an asterisk, and genes shared between the transgene-induced (dark gray circle) and the dsRNA-induced pathway (blue circle) were found to be essential. Brackets connecting two genes symbolize WGD1 duplicate genes. CID2 was identified by recursive RNAi in this study. Other genes were identified previously (15,22,23). The RDR2 gene is required for the accumulation of endogenous siRNAs (purple) of the cluster22 locus, as previously found for RDR3 (22). Note that production of dsRNA from this locus is speculative, and that other endogenous siRNAs may require different factors. The figure is only meant to depict genetic requirements, not the subcellular localization or mechanistic roles of the corresponding proteins.
The latter allowed recovery of some hypomorphic alleles (seven out of 47) causing only partial RNAi deficiencies, as confirmed by functional tests using dilute dsRNA trigger. The rather low frequency of such alleles may be due to the ∼7 vegetative divisions allowed for fully resistant mutants in NSF dsRNA feeding medium, before cell isolation: although hypomorphic mutants were able to survive that step, their growth rate was probably greatly reduced. The experimental scheme would thus have to be modified to yield a higher frequency of hypomorphic alleles, the only ones that can be obtained for essential genes.
The dsRNA- and transgene-induced RNAi pathways share essential genes
Analysis of the diversity of alleles obtained for each of these five genes showed that they fall in two groups. For RDR1, CID1 and PDS1, the many alleles recovered included unambiguous null ones such as nonsense mutations and/or non-conservative changes of conserved residues, which all abolished accumulation of siRNAs upon dsRNA feeding. Although these mutants were completely deficient in the silencing response to dsRNA, they did not show any other phenotype at any stage of the life cycle, indicating that the capacity to synthesize siRNAs from dsRNA ingested as food is not required for short-term viability in laboratory conditions. In contrast, only two alleles were obtained for each of DCR1 and RDR2 and these were hypomorphic, only partially impairing the silencing response and siRNA accumulation. This strongly suggests that these are essential genes in which null mutations would be lethal at some stage of the life cycle. In particular, these results establish a clear distinction between RDR1 and RDR2, two RdRPs which could not be distinguished in previous recursive RNAi tests applied during the vegetative phase (22).
Strikingly, these two groups of genes also differ in the specificity of their involvement in RNAi pathways (Figure 8). We confirmed that DCR1 is also required for transgene-induced silencing and showed that the same is true of RDR2, although this had not been seen previously. Here, recursive RNAi tests (by dsRNA feeding) were made more sensitive by using a transformed clone showing only moderate silencing of the ND169 reporter (as proposed in a model for recursive RNAi dynamics (33)). In the same conditions, recursive silencing of RDR1, CID1 and PDS1 did not affect transgene-induced silencing. Thus, the only essential genes uncovered by our screen are those also involved in transgene-induced RNAi. Two more genes likely belong to this group. By testing close paralogs of CID1, we found that CID2 is involved in both pathways, as previously shown for PTIWI13. The fact that no mutation was recovered in these genes is consistent with the idea that they are also essential.
A third, presumably non-catalytic RdRP gene previously implicated in transgene-induced silencing, RDR3, is further required for normal vegetative growth rate and accumulation of cluster22 endogenous siRNAs. The latter differ from transgene-induced siRNAs in that they lack a modification of the terminal ribose, and interestingly, were also absent or reduced in the hypomorphic rdr2 mutants. RDR2 thus appears to be required for all known types of siRNAs in Paramecium (Figure 8). However, the wild-type growth rate of the mutants suggests that its putative essential function may not be needed during the vegetative phase.
In contrast to rdr2, no effect was observed in dcr1 mutants or CID2 knock down cells on cluster22 siRNAs. Although we cannot completely exclude a false-negative result due to the hypomorphic nature of the dcr1 alleles and the inefficient knock down of CID2, this may indicate that these endogenous siRNAs are not dependent on all components of the transgene-induced pathway and are thus produced by a different pathway (Figure 8). This is supported by the observation that they do not carry a 3′ modification.
RDR2 is involved in sexual reproduction
The crosses of rdr2 mutants to the wild type revealed defects in sexual reproduction. F1s showed a high lethality rate and occasional failure of the formation of heterozygous MACs. However, it is unclear whether this was significant in F1s deriving from the wild-type parent (which would point to defects in pre-zygotic stages such as karyogamy). More striking was the frequent unviability of most or all F2 clones produced by autogamy of some—though not all—viable heterozygotes, regardless of F2 genotype or F1 parental origin. The stochastic nature of the effect raises the possibility that it is due to defects in maintenance of the germline in mutant homozygotes, such as defects in chromosome segregation during MIC mitosis, which would have no impact on vegetative growth of mutants, but would lead to aneuploidy in the F1 and therefore lethality in the F2. Such defects have been observed for instance in RNAi mutants in S. pombe, including the RdRP Rdp1 (64,65) and the nucleotidyl transferase Cid12 (66), which are both essential for pericentromeric heterochromatin formation (45,67). Stochastic loss of MIC chromosomes during vegetative growth would make each mutant clone potentially unique in its ability to foster viable F2 progeny.
The genetic complexity of the dsRNA-induced RNAi pathway
Previous studies have probed the genetic complexity of RNAi pathways (68–72). In C. elegans, a genome-wide RNAi screen for genes involved in silencing induced by RNA hairpin expression identified 90 genes, 54 of which are essential for viability (69). In our study, the screen seems to be saturated only for the sub-category of non-essential, single-copy genes, all three of which are specifically involved in dsRNA-induced RNAi. Additional non-essential genes are likely to have been missed because they retain their WGD1 duplicates, which very often have redundant functions. Indeed, 68% of all P. tetraurelia genes still have their WGD1 duplicate (32). Such may be the case of PTIWI12/PTIWI15, previously shown to be involved in dsRNA-induced silencing but for which co-silencing of both duplicates was required to detect an effect (23). Other genes may have been missed because they are essential, since our scheme yielded only a low frequency of hypomorphic alleles. Essential genes may be shared with the transgene-induced pathway; in addition, processing of dsRNA from food bacteria could be coupled to other cellular processes such as uptake of nutrients, as shown in D. melanogaster S2 cells and C. elegans (73,74). Thus, the total number of genes contributing to dsRNA-induced RNAi might be much higher than the nine known so far.
Environmental RNAi in Paramecium
Environmental RNAi, i.e. the processing of exogenous dsRNA into functional siRNAs, has been observed in a number of organisms (8), but has not yet been reported to be essential for cellular viability. It is also dispensable for short-term viability of P. tetraurelia in laboratory conditions. However, the genes involved are highly conserved in the Paramecium genus, suggesting they may have important functions for the long-term survival of these species in their natural environment. Exogenous RNAi may serve as an antiviral defense, a major function of the pathway in D. melanogaster, C. elegans and other species (9–12), even though no Paramecium virus has been identified so far. If most of the genes in the pathway are conserved for the processing of viral RNA, only a few more may be required for the uptake of dsRNA from phagosomes, and these are not necessarily conserved. Such a situation is seen with some Caenorhabditis species that lack the dsRNA uptake mechanism but respond to exogenous dsRNA when the C. elegans dsRNA transporter SID-2 is expressed (75). The identification of novel genes and the availability of mutants will help investigations into the mechanism and natural functions of dsRNA uptake.
Biosynthesis of dsRNA triggers and siRNAs
Although it is still unclear how RdRPs are specifically recruited to their target template during biogenesis or maintenance of silencing triggers in vivo, recent studies in S. pombe, C. elegans and T. thermophila point to the formation of RdRP complexes (RdRC) as an important step (45,47,76,77). In the ciliate T. thermophila, a single RdRP (Rdr1) interacts in a mutually exclusive manner with either of two nucleotidyl transferases, forming an essential RdRC with Rdn1, or a non-essential one with Rdn2. The latter contains an associated factor, either Rdf1 or Rdf2 (47,77). The role of these specific RdRCs in vivo is still unclear (21,47). In P. tetraurelia, it is tempting to speculate that Rdr1 and Cid1 form a non-essential RdRC which specifically acts in dsRNA-induced silencing, while Rdr2 and Cid2 form an essential RdRC acting in both pathways (Rdf1 and Rdf2 are not conserved in the P. tetraurelia genome). The reason why two RdRCs should be required for dsRNA-induced silencing, however, will remain puzzling until their precise functions are identified. They may have distinct roles in the production of primary and secondary siRNAs (15,22). The present work opens the way for an analysis of siRNAs and their precursors in each mutant, which is likely to provide insight into the processing mechanisms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including: Supplementary Figures S1–S8 and Supplementary Tables S1–S7 including Supplementary References (79–81).
Acknowledgments
We thank Jean Cohen for providing plasmid constructs and advice in UV-induced mutagenesis, Michael Lynch, Thomas G. Doak and Casey McGrath for sharing unpublished sequence data of Paramecium species, Linda Sperling, Martin Simon and Vincent Récamier for comments on the manuscript, and all lab members for critical discussions and continuous support. The sequencing of RNAi mutant MAC genomes benefited from the facilities and expertise of the high throughput sequencing platform of IMAGIF (Centre de Recherche de Gif—www.imagif.cnrs.fr).
Footnotes
Present address: Centre for Human and Molecular Biology, Molecular Cell Dynamics, Saarland University, Campus A2 4, 66123 Saarbrücken, Germany.
FUNDING
Agence Nationale de la Recherche [‘Investissements d'Avenir’ program ANR-10-LABX-54 MEMO LIFE/ANR-11-IDEX-0001–02 Paris Sciences et Lettres* Research University, ANR-08-BLAN-0233 ‘ParaDice’, ANR-12-BSV6–0017 ‘INFERNO’ to E.M.]; Fondation pour la Recherche Médicale [‘Equipe FRM’ to E.M.] and Fondation Pierre Gilles de Gennes [post-doctoral fellowship to S.M.]. National Science Foundation [NSF EF-0328516-A006]. This study was carried out in the context of the CNRS-supported European Research Group ‘Paramecium Genome Dynamics and Evolution’ and the European COST Action BM1102. Funding for open access charge: Agence Nationale de la Recherche.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cerutti H., Casas-Mollano J.A. On the origin and functions of RNA-mediated silencing: from protists to man. Curr. Genet. 2006;50:81–99. doi: 10.1007/s00294-006-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghildiyal M., Zamore P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ketting R.F. The many faces of RNAi. Dev. Cell. 2011;20:148–161. doi: 10.1016/j.devcel.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Meister G., Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 5.Tomari Y., Zamore P.D. Perspective: machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- 6.Baulcombe D.C. Molecular biology. Amplified silencing. Science. 2007;315:199–200. doi: 10.1126/science.1138030. [DOI] [PubMed] [Google Scholar]
- 7.Voinnet O. Use, tolerance and avoidance of amplified RNA silencing by plants. Trends Plant Sci. 2008;13:317–328. doi: 10.1016/j.tplants.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Whangbo J.S., Hunter C.P. Environmental RNA interference. Trends Genet. 2008;24:297–305. doi: 10.1016/j.tig.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Félix M.A., Ashe A., Piffaretti J., Wu G., Nuez I., Belicard T., Jiang Y., Zhao G., Franz C.J., Goldstein L.D., et al. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 2011;9:e1000586. doi: 10.1371/journal.pbio.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu R., Maduro M., Li F., Li H.W., Broitman-Maduro G., Li W.X., Ding S.W. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature. 2005;436:1040–1043. doi: 10.1038/nature03870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkins C., Dishongh R., Moore S.C., Whitt M.A., Chow M., Machaca K. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature. 2005;436:1044–1047. doi: 10.1038/nature03957. [DOI] [PubMed] [Google Scholar]
- 12.Wang X.-H., Aliyari R., Li W.-X., Li H.-W., Kim K., Carthew R., Atkinson P., Ding S.-W. RNA interference directs innate immunity against viruses in adult drosophila. Science. 2006;312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mochizuki K., Fine N.A., Fujisawa T., Gorovsky M.A. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in tetrahymena. Cell. 2002;110:689–699. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- 14.Lepère G., Bétermier M., Meyer E., Duharcourt S. Maternal noncoding transcripts antagonize the targeting of DNA elimination by scanRNAs in Paramecium tetraurelia. Genes Dev. 2008;22:1501–1512. doi: 10.1101/gad.473008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lepère G., Nowacki M., Serrano V., Gout J.F., Guglielmi G., Duharcourt S., Meyer E. Silencing-associated and meiosis-specific small RNA pathways in Paramecium tetraurelia. Nucleic Acids Res. 2009;37:903–915. doi: 10.1093/nar/gkn1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mochizuki K., Gorovsky M.A. Conjugation-specific small RNAs in Tetrahymena have predicted properties of scan (scn) RNAs involved in genome rearrangement. Genes Dev. 2004;18:2068–2073. doi: 10.1101/gad.1219904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garnier O., Serrano V., Duharcourt S., Meyer E. RNA-mediated programming of developmental genome rearrangements in Paramecium tetraurelia. Mol. Cell. Biol. 2004;24:7370–7379. doi: 10.1128/MCB.24.17.7370-7379.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnaiz O., Mathy N., Baudry C., Malinsky S., Aury J.M., Wilkes C.D., Garnier O., Labadie K., Lauderdale B.E., Le Mouël A., et al. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet. 2012;8:e1002984. doi: 10.1371/journal.pgen.1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chalker D.L., Meyer E., Mochizuki K. Epigenetics of ciliates. Cold Spring Harbor Perspectives in Biology. 2013;5:a017764. doi: 10.1101/cshperspect.a017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S.R., Collins K. Two classes of endogenous small RNAs in Tetrahymena thermophila. Genes Dev. 2006;20:28–33. doi: 10.1101/gad.1377006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couvillion M.T., Lee S.R., Hogstad B., Malone C.D., Tonkin L.A., Sachidanandam R., Hannon G.J., Collins K. Sequence, biogenesis, and function of diverse small RNA classes bound to the Piwi family proteins of Tetrahymena thermophila. Genes Dev. 2009;23:2016–2032. doi: 10.1101/gad.1821209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marker S., Le Mouël A., Meyer E., Simon M. Distinct RNA-dependent RNA polymerases are required for RNAi triggered by double-stranded RNA versus truncated transgenes in Paramecium tetraurelia. Nucleic Acids Res. 2010;38:4092–4107. doi: 10.1093/nar/gkq131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouhouche K., Gout J.F., Kapusta A., Bétermier M., Meyer E. Functional specialization of Piwi proteins in Paramecium tetraurelia from post-transcriptional gene silencing to genome remodelling. Nucleic Acids Res. 2011;39:4249–4264. doi: 10.1093/nar/gkq1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galvani A., Sperling L. Transgene-mediated post-transcriptional gene silencing is inhibited by 3′ non-coding sequences in Paramecium. Nucleic Acids Res. 2001;29:4387–4394. doi: 10.1093/nar/29.21.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruiz F., Vayssie L., Klotz C., Sperling L., Madeddu L. Homology-dependent gene silencing in Paramecium. Mol. Biol. Cell. 1998;9:931–943. doi: 10.1091/mbc.9.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timmons L., Court D.L., Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- 27.Timmons L., Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854–854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 28.Galvani A., Sperling L. RNA interference by feeding in Paramecium. Trends Genet. 2002;18:11–12. doi: 10.1016/s0168-9525(01)02548-3. [DOI] [PubMed] [Google Scholar]
- 29.Sijen T., Steiner F.A., Thijssen K.L., Plasterk R.H. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science. 2007;315:244–247. doi: 10.1126/science.1136699. [DOI] [PubMed] [Google Scholar]
- 30.Aoki K., Moriguchi H., Yoshioka T., Okawa K., Tabara H. In vitro analyses of the production and activity of secondary small interfering RNAs in C. elegans. EMBO J. 2007;26:5007–5019. doi: 10.1038/sj.emboj.7601910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pak J., Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315:241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- 32.Aury J.M., Jaillon O., Duret L., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- 33.Marshall W.F. Modeling recursive RNA interference. PLoS Comput. Biol. 2008;4 doi: 10.1371/journal.pcbi.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skouri F., Cohen J. Genetic approach to regulated exocytosis using functional complementation in Paramecium: identification of the ND7 gene required for membrane fusion. Mol. Biol. Cell. 1997;8:1063–1071. doi: 10.1091/mbc.8.6.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonneborn T.M. Methods in Paramecium research. Methods Cell Physiol. 1970;4:241–339. [Google Scholar]
- 36.Cohen J. Cytotoxic versus mutagenic effect of ethyl methanesulfonate on Paramecium tetraurelia. Mutat. Res. 1980;70:251–254. doi: 10.1016/0027-5107(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 37.Froissard M., Keller A.M., Dedieu J.C., Cohen J. Novel secretory vesicle proteins essential for membrane fusion display extracellular-matrix domains. Traffic. 2004;5:493–502. doi: 10.1111/j.1600-0854.2004.00194.x. [DOI] [PubMed] [Google Scholar]
- 38.Simon M.C., Marker S., Schmidt H.J. Posttranscriptional control is a strong factor enabling exclusive expression of surface antigens in Paramecium tetraurelia. Gene Expr. 2006;13:167–178. doi: 10.3727/000000006783991809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gratias A., Bétermier M. Processing of double-strand breaks is involved in the precise excision of Paramecium internal eliminated sequences. Mol. Cell. Biol. 2003;23:7152–7162. doi: 10.1128/MCB.23.20.7152-7162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Subgroup G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Froissard M., Kissmehl R., Dedieu J.C., Gulik-Krzywicki T., Plattner H., Cohen J. N-ethylmaleimide-sensitive factor is required to organize functional exocytotic microdomains in paramecium. Genetics. 2002;161:643–650. doi: 10.1093/genetics/161.2.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kissmehl R., Froissard M., Plattner H., Momayezi M., Cohen J. NSF regulates membrane traffic along multiple pathways in Paramecium. J. Cell Sci. 2002;115:3935–3946. doi: 10.1242/jcs.00079. [DOI] [PubMed] [Google Scholar]
- 44.Stevenson A.L., Norbury C.J. The Cid1 family of non-canonical poly(A) polymerases. Yeast. 2006;23:991–1000. doi: 10.1002/yea.1408. [DOI] [PubMed] [Google Scholar]
- 45.Motamedi M.R., Verdel A., Colmenares S.U., Gerber S.A., Gygi S.P., Moazed D. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 46.Talsky K.B., Collins K. Initiation by a eukaryotic RNA-dependent RNA polymerase requires looping of the template end and is influenced by the template-tailing activity of an associated uridyltransferase. J. Biol. Chem. 2010;285:27614–27623. doi: 10.1074/jbc.M110.142273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S.R., Talsky K.B., Collins K. A single RNA-dependent RNA polymerase assembles with mutually exclusive nucleotidyl transferase subunits to direct different pathways of small RNA biogenesis. RNA. 2009;15:1363–1374. doi: 10.1261/rna.1630309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaillon O., Bouhouche K., Gout J.F., Aury J.M., Noel B., Saudemont B., Nowacki M., Serrano V., Porcel B.M., Segurens B., et al. Translational control of intron splicing in eukaryotes. Nature. 451:359–362. doi: 10.1038/nature06495. [DOI] [PubMed] [Google Scholar]
- 49.Makeyev E.V., Bamford D.H. Cellular RNA-dependent RNA polymerase involved in posttranscriptional gene silencing has two distinct activity modes. Mol. Cell. 2002;10:1417–1427. doi: 10.1016/s1097-2765(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 50.Sugiyama T., Cam H., Verdel A., Moazed D., Grewal S.I. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc. Natl. Acad. Sci. U. S. A. 2005;102:152–157. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Curaba J., Chen X. Biochemical activities of Arabidopsis RNA-dependent RNA polymerase 6. J. Biol. Chem. 2008;283:3059–3066. doi: 10.1074/jbc.M708983200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y., Yu Y., Zhai J., Ramachandran V., Dinh T.T., Meyers B.C., Mo B., Chen X. The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr. Biol. 2012;22:689–694. doi: 10.1016/j.cub.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayer K.M., Forney J.D. A mutation in the flanking 5’-TA-3’ dinucleotide prevents excision of an internal eliminated sequence from the Paramecium tetraurelia genome. Genetics. 1999;151:597–604. doi: 10.1093/genetics/151.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Welker N.C., Pavelec D.M., Nix D.A., Duchaine T.F., Kennedy S., Bass B.L. Dicer's helicase domain is required for accumulation of some, but not all, C. elegans endogenous siRNAs. RNA. 2010;16:893–903. doi: 10.1261/rna.2122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Houten J., Preston R.R. Eukaryotic unicells: How useful in studying chemoreception? Ann. N. Y. Acad. Sci. 1987;510:16–22. doi: 10.1111/j.1749-6632.1987.tb43460.x. [DOI] [PubMed] [Google Scholar]
- 56.Preer J.R., Preer L.B., Rudman B., Barnett A. Molecular biology of the genes for immobilization antigens in Paramecium1. J. Protozool. 1987;34:418–423. doi: 10.1111/j.1550-7408.1987.tb03205.x. [DOI] [PubMed] [Google Scholar]
- 57.Sonneborn T.M. Paramecium aurelia. In: King R., editor. Handbook of Genetics. New York: Plenum press; 1974. pp. 469–594. [Google Scholar]
- 58.Vayssie L., Skouri F., Sperling L., Cohen J. Molecular genetics of regulated secretion in paramecium. Biochimie. 2000;82:269–288. doi: 10.1016/s0300-9084(00)00201-7. [DOI] [PubMed] [Google Scholar]
- 59.Saimi Y., Kung C. Behavioral genetics of Paramecium. Annu. Rev. Genet. 1987;21:47–65. doi: 10.1146/annurev.ge.21.120187.000403. [DOI] [PubMed] [Google Scholar]
- 60.Haynes W.J., Vaillant B., Preston R.R., Saimi Y., Kung C. The cloning by complementation of the pawn-A gene in Paramecium. Genetics. 1998;149:947–957. doi: 10.1093/genetics/149.2.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keller A.M., Cohen J. An indexed genomic library for Paramecium complementation cloning. J. Eukaryot. Microbiol. 2000;47:1–6. doi: 10.1111/j.1550-7408.2000.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 62.Greenberg M.V.C., Ausin I., Chan S.W.L., Cokus S.J., Cuperus J.T., Feng S., Law J.A., Chu C., Pellegrini M., Carrington J.C., et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics. 2011;6:344–354. doi: 10.4161/epi.6.3.14242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zuryn S., Le Gras S., Jamet K., Jarriault S. A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics. 2010;186:427–430. doi: 10.1534/genetics.110.119230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hall I.M., Noma K., Grewal S.I. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc. Natl. Acad. Sci. U. S. A. 2003;100:193–198. doi: 10.1073/pnas.232688099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Volpe T., Schramke V., Hamilton G.L., White S.A., Teng G., Martienssen R.A., Allshire R.C. RNA interference is required for normal centromere function in fission yeast. Chromosome Res. 2003;11:137–146. doi: 10.1023/a:1022815931524. [DOI] [PubMed] [Google Scholar]
- 66.Win T.Z., Stevenson A.L., Wang S.W. Fission yeast Cid12 has dual functions in chromosome segregation and checkpoint control. Mol. Cell. Biol. 2006;26:4435–4447. doi: 10.1128/MCB.02205-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Volpe T.A., Kidner C., Hall I.M., Teng G., Grewal S.I., Martienssen R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 68.Vastenhouw N.L., Fischer S.E.J., Robert V.J.P., Thijssen K.L., Fraser A.G., Kamath R.S., Ahringer J., Plasterk R.H.A. A genome-wide screen identifies 27 genes involved in transposon silencing in C. elegans. Curr. Biol. 2003;13:1311–1316. doi: 10.1016/s0960-9822(03)00539-6. [DOI] [PubMed] [Google Scholar]
- 69.Kim J.K., Gabel H.W., Kamath R.S., Tewari M., Pasquinelli A., Rual J.F., Kennedy S., Dybbs M., Bertin N., Kaplan J.M., et al. Functional genomic analysis of RNA interference in C. elegans. Science. 2005;308:1164–1167. doi: 10.1126/science.1109267. [DOI] [PubMed] [Google Scholar]
- 70.Elmayan T., Balzergue S., Beon F., Bourdon V., Daubremet J., Guenet Y., Mourrain P., Palauqui J.C., Vernhettes S., Vialle T., et al. Arabidopsis mutants impaired in cosuppression. Plant Cell. 1998;10:1747–1758. doi: 10.1105/tpc.10.10.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cogoni C., Macino G. Isolation of quelling-defective (qde) mutants impaired in posttranscriptional transgene-induced gene silencing in Neurospora crassa. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10233–10238. doi: 10.1073/pnas.94.19.10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matzke M., Aufsatz W., Kanno T., Daxinger L., Papp I., Mette M.F., Matzke A.J. Genetic analysis of RNA-mediated transcriptional gene silencing. Biochim. Biophys. Acta. 2004;1677:129–141. doi: 10.1016/j.bbaexp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 73.Saleh M.C., van Rij R.P., Hekele A., Gillis A., Foley E., O'Farrell P.H., Andino R. The endocytic pathway mediates cell entry of dsRNA to induce RNAi silencing. Nat. Cell Biol. 2006;8:793–802. doi: 10.1038/ncbl439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ulvila J., Parikka M., Kleino A., Sormunen R., Ezekowitz R.A., Kocks C., Ramet M. Double-stranded RNA is internalized by scavenger receptor-mediated endocytosis in Drosophila S2 cells. J. Biol. Chem. 2006;281:14370–14375. doi: 10.1074/jbc.M513868200. [DOI] [PubMed] [Google Scholar]
- 75.Nuez I., Félix M.A. Evolution of susceptibility to ingested double-stranded RNAs in Caenorhabditis nematodes. PLoS One. 2012;7:e29811. doi: 10.1371/journal.pone.0029811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duchaine T.F., Wohlschlegel J.A., Kennedy S., Bei Y., Conte D., Jr, Pang K., Brownell D.R., Harding S., Mitani S., Ruvkun G., et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124:343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 77.Lee S.R., Collins K. Physical and functional coupling of RNA-dependent RNA polymerase and Dicer in the biogenesis of endogenous siRNAs. Nat. Struct. Mol. Biol. 2007;14:604–610. doi: 10.1038/nsmb1262. [DOI] [PubMed] [Google Scholar]
- 78.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.