


Abstract
To identify non-coding RNA (ncRNA) signals within genomic regions, a classification tool was developed based on a hybrid random forest (RF) with a logistic regression model to efficiently discriminate short ncRNA sequences as well as long complex ncRNA sequences. This RF-based classifier was trained on a well-balanced dataset with a discriminative set of features and achieved an accuracy, sensitivity and specificity of 92.11%, 90.7% and 93.5%, respectively. The selected feature set includes a new proposed feature, SCORE. This feature is generated based on a logistic regression function that combines five significant features—structure, sequence, modularity, structural robustness and coding potential—to enable improved characterization of long ncRNA (lncRNA) elements. The use of SCORE improved the performance of the RF-based classifier in the identification of Rfam lncRNA families. A genome-wide ncRNA classification framework was applied to a wide variety of organisms, with an emphasis on those of economic, social, public health, environmental and agricultural significance, such as various bacteria genomes, the Arthrospira (Spirulina) genome, and rice and human genomic regions. Our framework was able to identify known ncRNAs with sensitivities of greater than 90% and 77.7% for prokaryotic and eukaryotic sequences, respectively. Our classifier is available at http://ncrna-pred.com/HLRF.htm.
INTRODUCTION
Non-coding RNAs (ncRNAs) are involved in a variety of important biological functions in the cell, including the control of chromosome dynamics, RNA splicing, RNA editing, translational inhibition and mRNA destruction (1). ncRNAs have recently been acknowledged to be diverse and significantly more important than previously thought (2–3). In human transcriptome analysis, >70% of the human genome is likely transcribed into ncRNAs, whereas protein-coding transcripts account for only ∼2–3% of the genome (4–6). NcRNAs can be roughly classified into short ncRNAs (such as microRNA (miRNAs), short-interfering RNAs (siRNAs), piwi-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), and short hairpin RNAs (shRNAs)) or long ncRNAs (lncRNAs), depending on the transcript size (3,7–9). Short ncRNAs are shorter than 200 nucleotides (nt), while lncRNAs are longer than 200 nt (10). LncRNAs can range in size from 200 to 100 000 nt (3,11). In contrast to well-studied short ncRNAs such as miRNAs and snoRNAs, lncRNAs are relatively uncharacterized, but accumulating evidence indicates that they likely have a broad range of functions (7–9,12). lncRNAs have been proposed to constitute the major fraction of eukaryotic transcriptomes; the transcriptome is involved in the regulation of many important cellular processes as well as the epigenetic control of complex mechanisms (5,13). Dysfunctions of lncRNAs have been associated with various diseases, including cancers, cardiovascular diseases and neurodegenerative diseases (14–16). The range of ncRNAs is expanding rapidly as new ncRNAs continue to be discovered via high-throughput sequencing. However, a large number of ncRNAs likely remain to be identified (1,17). Thus, the identification and annotation of ncRNAs are important steps for the elucidation of various regulatory mechanisms in the cell.
Current experimental methods have yielded promising results but are subject to certain limitations. The expression of most ncRNAs is lower than that of mRNAs, and ncRNAs display tissue/stage-specific expression patterns (1,18,19). Moreover, high-throughput sequencing causes enormous informatics, and requires extensive computational analysis (15). Thus, computational identification methods may complement experimental methods to quickly identify ncRNAs in new genomes, particularly ncRNAs that are transcribed under specific conditions in specific cell types. Various computational methods have been proposed to predict ncRNAs, including comparative (20–23) and non-comparative methods (24–31).
Most ncRNA identification algorithms are designed to identify structural ncRNAs based on low-energy structures. However, the use of secondary structure feature alone is generally not sufficiently statistically robust enough to detect all types of ncRNA (32) because a random RNA with high GC content can also fold into a low-energy structure. Computational methods utilizing thermodynamic stability structure features have successfully identified highly structured ncRNAs such as tRNAs, snoRNAs and miRNAs but cannot identify less densely structured classes of ncRNAs, such as lncRNAs (11). LncRNAs share some features with protein-coding genes: they can be spliced, 5′-capped and/or polyadenylated (13). However, unlike protein-coding genes, lncRNAs are generally expressed at low levels and lack strong evolutionary conservation across species compared to protein-coding sequences and small RNAs (e.g. miRNAs and snoRNAs). To identify lncRNAs, ncRNA gene identification method must incorporate other signal features that can be used to characterize lncRNAs. The recognition of a wide range of ncRNAs that exhibit heterogeneity among different species and different ncRNA families remains computationally challenging. In addition, some ncRNAs have weak structure signals.
In this work, we developed a generalized classifier for ncRNAs based on an ensemble of multiple decision trees, a random forest (RF), to discriminate ncRNAs from genomic sequences. Due to the complex and heterogeneous nature of ncRNAs, the classification of ncRNAs based on structures or sequences may not be appropriate. To classify ncRNAs efficiently, we take into account various characteristics of the ncRNAs (such as modularity elements, structural robustness scores, base-pair features, sequence compositions and structural features). We hypothesized that certain combinations of significant features would improve the characterization of heterogeneous ncRNAs. Thus, we propose a new composite feature based on a logistic regression model, SCORE, to increase the sensitivity of the RF prediction. We used our framework to scan for ncRNAs in a wide range of genomes, including both bacterial and eukaryotic genomes. In particular, we focused on genomes of economic, social, public health, environmental and agricultural importance.
MATERIALS AND METHODS
Dataset
Two training datasets were used: training 1 and training 2. The training 1 dataset was composed of all ncRNA seed alignment sequences obtained from the Rfam database, version 11.0 (33). The positive data were randomly selected from 32 300 ncRNAs from various organisms. Then, ncRNA sequences shorter than 50 nt were excluded, and CD-HIT (34) was used to eliminate redundant sequences with sequence similarities above 80%. Thus, the positive data consisted of 6649 non-redundant sequences. The negative data included RefSeq (35) coding sequences and shuffled sequences of both coding and ncRNA sequences. A set of 2147 non-redundant coding sequences was extracted from the coding region sequences of human RefSeq genes, and these were used to validate the lack of annotated or unannotated ncRNA sequences. A set of 4502 shuffled sequences obtained from 2147 coding and 2355 non-coding sequences was shuffled while preserving both the mono- and di-nucleotide frequencies. The balance between the number of positive and negative training sets was maintained because an imbalanced distribution could affect the performance of the classifier. The training 2 dataset included all lncRNAs obtained from the lncRNAdb database (36), a comprehensive database of lncRNAs, as the positive training data. The negative training data were randomly selected from the negative samples of the training 1 dataset.
To verify the results, genomic data for the following species were downloaded from the NCBI GenBank genome database (ftp://ftp.ncbi.nih.gov/genomes/): Escherichia coli K12 (U00096), Acholeplasma laidlawii PG-8A (CP000896), Acidovorax sp. JS42 (CP000539), Brucella suis 1330 Chromosome (chr.) 1 (AE014291), Candidatus methanoregula boonei 6A8 (CP000780), Oryza sativa japonica chr.1 (CM000138), Arthrospira (Spirulina) platensis NIES-39 (AP011615), Penaeus monodon mitochondrial genome (AF217843), Mycobacterium tuberculosis H37Rv (AL123456), Pseudomonas aeruginosa (CP000438) and Homo sapiens genomic regions containing ncRNAs. These genomes and genomic regions were used to test the method. For each of the test genomes, with the exception of the A. platensis and P. monodon mitochondrial genomes, sets of known ncRNAs were downloaded from the Rfam database version 11.0 in generic feature format. The files were then parsed to retrieve the start and end positions of all types of ncRNAs in the test genomes. For H. sapiens, we extracted five regions containing known ncRNAs: (i) five known lncRNAs (GNAS_AS1_1–5) in the 57 417 000–57 426 000 bp region of chr. 20; (ii) nine known miRNAs in the 49 767 700–49 779 500 bp region of chr. X; (iii) six known lncRNAs (MAT2A_A-F) in the 85 770 900–85 772 300 bp region of chr. 2; (iv) five known lncRNAs (HOTAIRM1_1–5) in the 27 135 000–27 139 900 bp region of chr. 7; and (v) six known lncRNAs (HOXA11_AS1_1–6) in the 27 225 000–27 228 900 bp region of chr. 7. Furthermore, northern blot verified ncRNA candidates from the literature (37) were used to test the ability of the proposed method for the identification of novel ncRNAs.
Features
Various types of features were extracted that could be important for ncRNA prediction. In total, 369 features were considered in this work, which can be divided into five categories, as summarized in Table 1. Descriptions of the features are listed in the Supplementary Material and Method 1.
Table 1. Summary of the 369 ncRNA features.
| Feature group | No. of features | Feature symbol |
|---|---|---|
| Sequence-based features | 277 | %G+C,% A+U,%AA,%AC,%AG,%AU,%CA,%CC,%CG,%CU,%GA,%GC,%GG,%GU,%UA,%UC,%UG,%UU,%AAAA -%UUUU (256 of 4-Mer), Blast_bits score (Bits), Modular_Bits (Bits2), Coding Potential (logodds) |
| Secondary structure features | 23 | MFE, efe, MFEI1, MFEI2, MFEI3, MFEI4, dG, dQ, dD, dF, Prob, zG, zQ, zD, zF, nefe, Freq, diff, dH, dS, Tm, CM score (CM), SCORE |
| Base-pair features | 28 | dP, zP, div, tot_bp, stem, loop, A-U/L, G-U/L, G-C/L,%A-U/Stem,%G-C/Stem,%G-U/Stem, Probpair1–10, Avg_PP, NonBP_A, NonBP_C, NonBP_G, NonBP_U, Non_BPP |
| Triplet sequence-structure | 32 | A(((, A((., A(.., A(.(,A.((,A.(.,A..(, A…, C(((, C((., C(.., C(.(, C.((, C.(., C..(, C…, G(((, G((., G(.., G(.(, G.((, G.(., G..(, U…, U(((, U((., U(.., U(.(, U.((, U.(., U..(, U… |
| Structural robustness features (SC-derived features) | 9 | SC, SCxMFE/Mean_dG, SCxdP, SCxabsZG, SC/(1-dP), SC/NonBP_A, SC/NonBP_C, SC/NonBP_G, SC/NonBP_U |
| Total | 369 |
Feature selection
Due to the large number of features, which may include redundant features, a feature selection process is needed to filter out irrelevant, redundant and uninformative features and select only the most informative set for ncRNAs identification. In this work, correlation-based feature selection (CFS) (38) and a genetic algorithm (GA) search method were used to select the discriminative feature subset. The CFS+GA combination has been used for pre-miRNA data to yield a compact feature subset and an improvement in performance (31). The CFS+GA method selected a feature subset with the highest merit criterion, and the combination of those features exhibited good predictive power in ncRNA identification. A set of 20 features from the 369 ncRNA features was selected as the discriminatory set by CFS+GA, including Prob, zG, SC_score, SCxdP, Bits, CM_score, Bits2, SCORE, AAAA, AGGA, CAAC, CAGU, CUAC, CUGA, GAUA, GCAU, GUUC, UAAG, UACA and UUUU. To visualize the spread of the training data 1 for each selected feature, graphical boxplots are shown in Figure 1.
Figure 1.
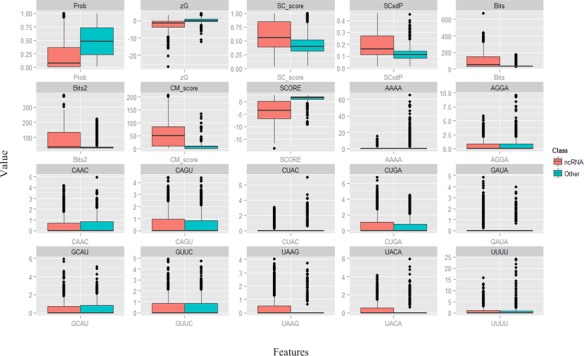
Boxplots showing the spread of the training data for each of the 20 selected features: Prob, zG, SC_score, SCxdP, Bits, CM_score, Bits2, SCORE, AAAA, AGGA, CAAC, CAGU, CUAC, CUGA, GAUA, GCAU, GUUC, UAAG, UACA and UUUU. For each plot, the left side represents the ncRNA class, and the right side represents other classes.
Logistic regression model
A logistic regression model (39) was used to define the complex relationships among important characteristic factors and to describe the ncRNA elements in the lncRNAs. The model was trained with training set 2. A combination of features was used to fit a logistic regression model represented by the following equation:
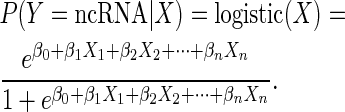 |
In this study, the logit transformation (the logarithm of the odds ratio or likelihood ratio) was used for the link function with logistic regression. The logit function, referred to as the SCORE feature, is defined as
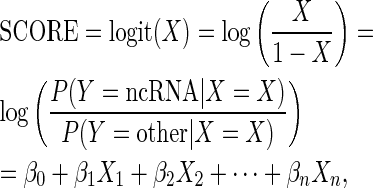 |
where βi represents the regression coefficient of the explanatory variable Xi. The logistic regression model was implemented by using the generalized linear model (GLM) function in R (40). We used GLM to describe a function of related factors to predict the possibility that a sequence was lncRNA. We defined all relevant features that might be involved in predicting lncRNAs, and ANOVA tests were used to identify the statistically significant features associated with lncRNAs. Various regression models were explored, and the optimal model was validated based on a 10-fold cross validation. A stepwise forward variable selection was used to select the significant factors based on their p-values.
The logistic model was based on five significant features: sequence similarity (Bits), structural motif similarity (CM_score), modularity sequence profile score (Bits2), coding potential (logodds) and structural robustness score (SCxabsZG). These features were included in the logistic function to describe the lncRNAs because they are statistically significant based on the Wald test (p < 0.001). The sequence similarity is a Bit score obtained from BLAST (Basic Local Alignment Search Tool) (41). Bits2 is a modular sequence similarity obtained from BLAST search against the modularity sequence profile database. To build the modularity sequence profile database, we collected the modularity pivot sequences from Rfam seed alignment by searching for portions of common RNA sequences in long non-coding alignments and curating them as custom libraries for BLAST. CM_score is an RNA secondary structure similarity score obtained by using infernal (42) to search against the covariance models (CM) (43), which are RNA secondary profiles from a custom modular CM library. In brief, our modular CM database was created in three steps: (i) manual extraction of the critical conserved substructures from selected lncRNA seed alignments and CM model construction, (ii) searches for short motifs RNA in the Rfam seed alignments using CMfinder (44) and CM model construction, and (iii) scans for and removal of redundant models. The structural robustness score was the product of a self-containment (SC) score (45) and the absolute value of the z-score (zG) (46). The coding potential based-feature (logodds) was extracted from the framefinder s/w (47,48) to identify the longest reading frame in the three forward frames of the sequences. Finally, the logistic model was used as a composite feature to discriminate between lncRNAs and other sequences in the machine learning (ML) model. All analyses were performed in the R statistical environment (40).
Machine learning algorithms
We trained an RF as the main classifier. An RF is an ensemble method that uses decision trees as its base classifiers (49,50). Each of the individual decision trees was trained on a random subset of the total features to maximize the classification criteria at each node, and the different classification hypothesis trees were then combined to form the ensemble (51,52). The predictions of the RF model represent a consensus of the predictions made by all of the individual trees. We used Weka (53) and the randomForest R package (40,51) with 10 trees (ntree = 10), and 5 randomly sampled features as candidates at each split (mtry = 5). Detailed implementation of other ML methods was used in this work, including Naïve Bayes (NB), support vector machine (SVM), K-nearest neighbors (kNN), neural network (MLP), decision tree (DT), rule induction (RIPPER) and RF. The algorithm selection procedure is described in the Supplementary Material and Method 2.
Classification framework for ncRNAs
The process of computational ncRNA gene identification is illustrated in Figure 2. The input was a sequence of any length, preferably with a size between 75 and 200 nt. For the entire genome, a sliding window module was used to split the genome into multiple overlapping sub-sequences with a size of 120 nt and a step of 40 nt. A window size of 120 nt, as used in RNAz (21) screens, is an appropriate input size because it is large enough to detect most ncRNAs, ranging from small ncRNAs to at least a substructure of a lncRNA (43). The window size was primarily tested and trained as the performance was optimized (data not shown). In the next step, a feature extraction module was used to extract the 20 most informative features from each input sequence. In the final step, the trained RF classifier was used to classify the input sequences.
Figure 2.
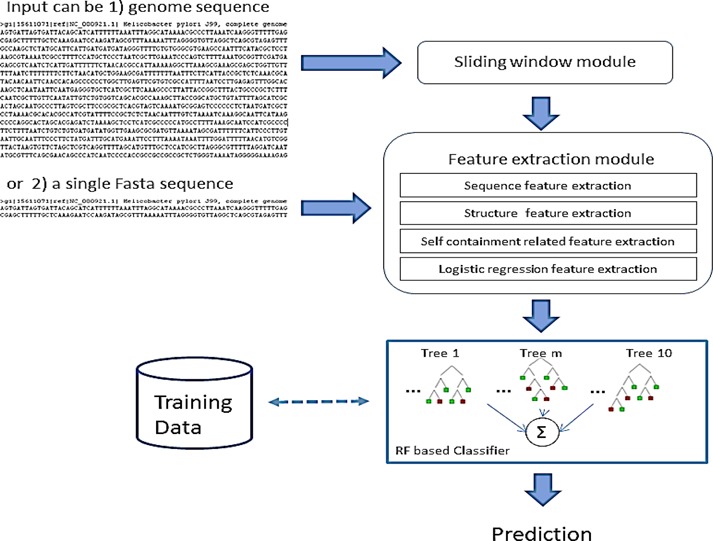
Process of computational ncRNA gene prediction.
Performance assessment
To precisely evaluate the predictive power of our classification model, several standard performance measures were used:
 |
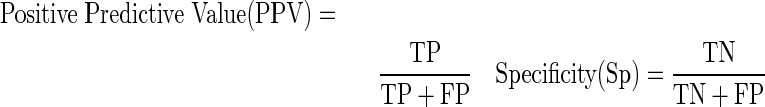 |
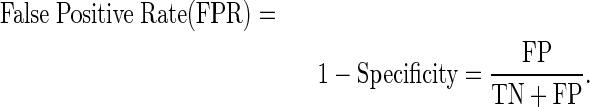 |
We evaluated the area under the ROC curve (AUC), in which the sensitivity is plotted as a function of the false positive rate (FPR) at different decision thresholds. A greater AUC value indicates a better classification result. In case of genome screening, False Discovery Rate (FDR) was calculated using the same approach as previously described in (29), which involves steps in calculating FPR in the shuffled genome. The shuffling method ensured that all unknown ncRNAs located within the genome were fragmented (30).
RESULTS AND DISCUSSION
Predictive power of the newly proposed model
The performances of ML algorithms usually depends on the task to which they are applied. Different algorithms are able to take advantage of different characteristics and relationships in a given dataset, and thus we constructed seven classifiers based on seven algorithms trained on the same training data and the same features and evaluated their performances using 10-fold cross validation (see the Algorithm selection in the Supplementary Method 2). The RF method outperformed the six other techniques for the prediction of ncRNAs. To visualize the performance of the difference algorithms, ROC curves were plotted (Figure 3). On average, RF yielded the highest AUC (of 0.972), which reflects a better balance compared to the other algorithms. The better performance of RF could be due to the robustness provided by the bootstrap aggregating (bagging) technique and the random feature selection process for building the ensemble of the DT model. Our data included a broad range of ncRNA families; thus, RF is more suitable than other methods because the heterogeneity of the ncRNA subfamilies can be captured by an ensemble of trees.
Figure 3.
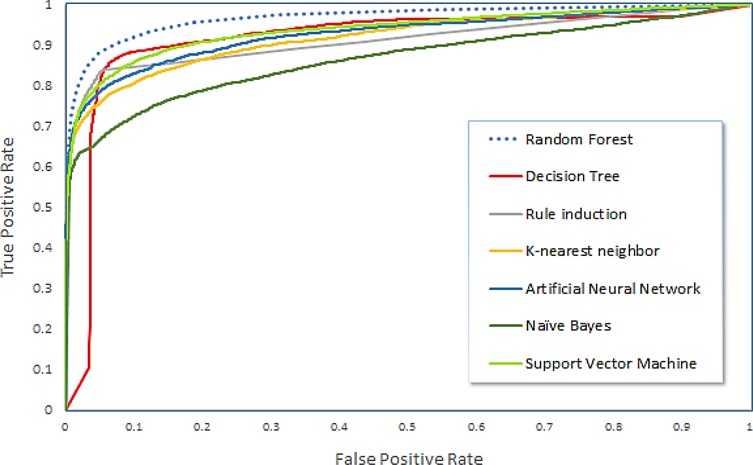
ROC curves showing the performances of the classifiers in 10-fold cross validation.
To assess the overall sensitivity of our RF method for screening previously annotated ncRNAs in various prokaryotic genomes, we began with E. coli because it is a well-studied prokaryotic model organism (with a genome size of 4 686 137 nt and 265 known ncRNA families) and has been previously used as a test genome for several de novo ncRNA prediction methods (24–26). A neural network (NN)-based method (24) outperforms all other reported methods when applied to the E. coli genome. Therefore, we compared the genome-wide performance of our method in identifying known ncRNAs in E. coli to that of NN (24). Our RF model performed significantly better than NN in terms of both sensitivity and specificity for identifying ncRNAs on both strands of the E. coli genome (Table 2).
Table 2. Performance comparison of our prediction method and the neural network (NN) method (24) for identifying known ncRNAs on both strands of the E. coli genome.
We also performed an independent test to compare the prediction performance of our method with the performances of two recently developed approaches that used SVMs: smyRNA (29) and ncRNAscout (30). The smyRNA method uses primary sequence motif features to detect ncRNAs, and the ncRNAscout uses both a primary sequence motif and secondary structure-based features for ncRNA discovery. The results of these methods for various compact microbial genomes with varying lengths and %GC contents are summarized in Table 3. Our RF-based method performed very well in identifying well-characterized, previously annotated ncRNAs with a broad range of GC contents and nucleotide lengths.
Table 3. ncRNAs detected in four genomes: Acholeplasma laidlawii PG-8A (CP000896), Acidovorax sp. JS42 (CP000539), Brucella suis 1330 Chromosome (chr.) 1 (AE014291) and Candidatus methanoregula boonei 6A8 (CP000780).
| Genome source | Nucleotide length (nt) | GC content (%) | Based on Rfam 10 | Based on Rfam 11 | ||||
|---|---|---|---|---|---|---|---|---|
| No. of known ncRNAs | Detected known ncRNAs (%) | No. of known ncRNAs | Detected by our model (%) | |||||
| ncRNA scout | smyRNA | Our model | ||||||
| A. laidlawii PG-8A | 1 496 992 | 31.92 | 42 | 92.857 | 95.238 | 95.238 | 64 | 96.875 |
| Acidovorax sp. JS42 | 4 448 856 | 66.17 | 134 | 96.269 | 70.149 | 97.015 | 181 | 95.580 |
| B. suis 1330 chromosome1 | 2 107 794 | 57.21 | 49 | 89.796 | 71.429 | 100 | 63 | 93.650 |
| C. methanoregula boonei 6A8 | 2 542 943 | 54.51 | 23 | 73.913 | 56.522 | 100 | 40 | 97.500 |
Our method was compared to ncRNAscout (30) and smyRNA (29).
The high FPR is another important aspect of computational ncRNA identification in genomes. It is impossible to calculate the FPR in real genomes because knowledge about ncRNAs is incomplete, and real genome sequences may contain some unknown novel ncRNAs. Thus, to evaluate the FDR, a shuffled genome was used to ensure that all unknown ncRNAs in the genome were broken up (29,30). An FDR of 16.9% was achieved by our method for the detection of known ncRNAs in the shuffled Acidovorax sp. JS42 genome, which indicates that the model can discriminate real ncRNAs from shuffled genomic sequences.
Newly proposed feature improving prediction accuracy of our model
To determine which features play the most important roles in prediction, we plotted the importance of each variable based on permutation and gini criteria. The RF method provides the importance of each feature measure, which is called the relative variable importance. As shown in Figure 4, the SCORE feature ranked as the top discriminative feature, demonstrating the effectiveness of this feature for ncRNA identification. However, it has been suggested that the importance of an RF variable may be misleading if it is biased toward variables with many categories and continuous predictor variables (54,55). A revised RF model (cforest) based on conditional inference trees using subsampling without replacement has been proposed to provide a more reliable measure of variable importance. Thus, a conditional feature importance strategy was also plotted by using the cforest function (56) as shown in Supplementary Figure S1. The revised RF variable importance was in agreement with the original RF variable importance.
Figure 4.
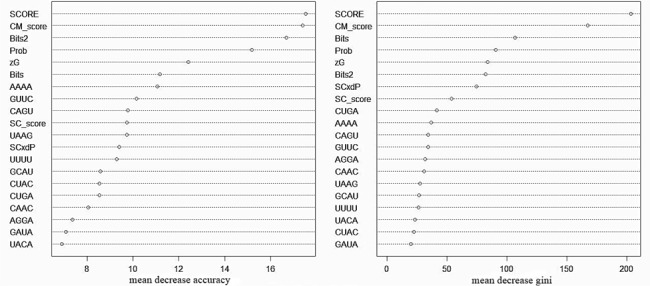
The variable importance based on the RF method (randomforest package). Left: permutation importance. The RF algorithm estimates the importance of a variable based on the increases in prediction error when the out-of-bag (OOB) error for that variable is permuted while all others are left unchanged. Right: gini importance. The RF algorithm estimates the decrease in impurity in the splitting criterion produced by each variable.
Different features are useful for identifying different classes of ncRNAs; thus the integration of multiple types of data can improve the sensitivity of computational methods for ncRNA identification (57). To address the diversity in ncRNA families, our feature selection method selected various types of features: sequence, structure, modularity, robustness and composite features. Sequence- and structure-based features are used extensively for ncRNA identification, including features such as MFE, zG (a structure or thermodynamic stability-based feature) and k-mer (a sequence-based feature). However, these commonly used features are not sufficiently general to encompass all ncRNA families. For example, algorithms that use only thermodynamic stability appear to have an advantage in detecting simple hairpin structural RNAs (58). Certain RNA families do indeed exhibit thermodynamic stability. However, this concept cannot be applied to all ncRNA families (46,59). Some features, such as nucleotide composition (k-mer), have been used with some success for ncRNA gene prediction (e.g. in smyRNA and ncRNAscout). A statistically significant signal based on CG content can be applied restrictively in AT-rich hyper-thermophiles (60–63). However, these sequence features are limited to compact bacterial organisms with base-composition biases, and their associated signals are generally insufficient for generalized ncRNA gene identification in most organisms (58).
To address the diversity in ncRNA families, our selected feature subset was composed of various complementary features that could enhance RF performance by capturing most possible aspects of heterogeneity in ncRNAs. These different features are useful for identifying different types of ncRNAs. For example, the CM score is advantageous for identifying ncRNAs with a high degree of conservation in sequence and structure. The BLAST score is advantageous for identifying ncRNAs with a high degree of sequence similarity. For example, SRP RNAs, U5 RNAs and U3 RNAs are well conserved at the sequence level (64). The SC_score or SC feature is advantageous for the detection of pre-miRNAs, which have high structural robustness and remain stable through two cleavage steps during their biogenesis (45). The pre-miRNAs generally exhibit a greater SC value, between 0.85 and 0.98. Moreover, pre-miRNAs are also anticipated to have higher SCxdP values compared to other ncRNA sequences (31). The Prob and zG features are useful for identifying ncRNAs with high thermodynamic stability. However, some classes of ncRNAs appear to be more difficult to detect than others (58), such as those with complex structural and highly variable lncRNAs. lncRNAs lack strong conservation (only 5% of lncRNA bases are evolutionarily constrained (8,17)) because they consist of multiple short regions that possess functional modules. The distinct modules of lncRNAs, which have a variety of secondary structure elements, interact with proteins, DNAs or RNAs to achieve specific regulatory outcomes (9–10,65–67). Thus, we captured the functional elements of lncRNAs in most possible ways by combining the power and advantages of various significant features: sequence, structure, robustness, coding potential and modular characteristics. RNA structural motifs are building blocks of the complex RNA architecture, and the recurrence of RNA structural motifs implies their high modularity and functional importance (10,68). On this basis, we hypothesized that lncRNAs containing various sequences and structural elements could be captured by scoring for these modular sequences and structural motifs. We generated the database containing various motifs, both structural and sequence-based (see the Materials and Methods section for more details). Furthermore, we hypothesized that certain combinations of significant features can reliably distinguish ncRNA elements from other sequences. To combine unrelated features with different scales, a unified statistical framework was needed. Therefore, we developed a scoring scheme based on the logistic regression function of the five significant features and used this scheme to discriminate lncRNA elements from other sequences. We generated several feature composition models based on LDA (Linear Discriminant Analysis), PCA (Principal Component Analysis) and LR (Logistic Regression) techniques and compared them by 10-fold cross validation. Based on their performances, we defined the logistic regression model as a composite feature, SCORE, because the method has a higher AUC compared to other techniques (R-squared = 0.7258, AUC = 0.9015). Moreover, logistic regression is relatively robust and easily updated, and allows meaningful interpretation. It is a hypothesis-driven model that can provide more useful information for ncRNA identification.
To examine the spread of SCORE values in the training data and compare to the well-known MFE feature, we plotted boxplots and histograms using rattle (69) in R. As shown in Figure 5, a significant difference was observed between ncRNAs and other classes in the SCORE feature. To explore whether the SCORE feature is able to capture lncRNA elements and improve the performance of the classifier, we compared the lncRNA prediction results obtained with our model framework with and without the SCORE feature. We randomly selected 10 lncRNA families from the Rfam database and used them as testing data. The performances of the model with and without the SCORE feature are compared in Table 4. These results provide preliminary evidence to support our hypothesis that the SCORE feature facilitates the identification of lncRNA regions. Table 4 shows that both of our RF frameworks successfully recovered most of the lncRNAs. In terms of sensitivity, the results suggest that our RF model can be applied generally to lncRNA elements. Moreover, these results indicate that our features can be used effectively in ML models to detect lncRNAs.
Figure 5.
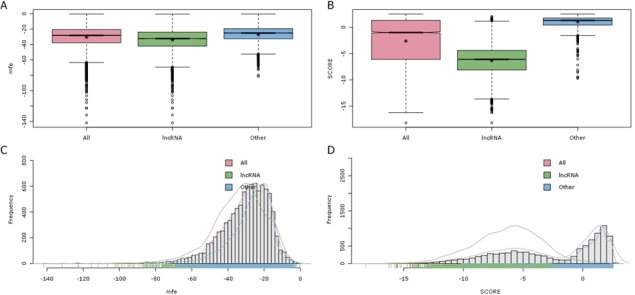
(A) and (B) The boxplots illustrate the spread of the training 2 dataset for the MFE feature (left) and the SCORE feature (right). The y-axis shows the values of the feature, and the x-axis shows the class of data. (C) and (D) Histogram plots of the MFE and SCORE features.
Table 4. Performance comparison of the model with and without the SCORE feature for detecting various Rfam lncRNA regions.
| Rfam | Description | No. of sequences | No. of correctly predicted sequences (%) | |
|---|---|---|---|---|
| with SCORE | without SCORE | |||
| RF01890 | LincRNA-p21 conserved region 2 | 10 | 9 (90) | 6 (60) |
| RF01905 | HOTAIR intron conserved region2 | 68 | 65 (95.5) | 62 (91.2) |
| RF01909 | CDKN2B antisense RNA1 intronic conserved region | 59 | 59 (100.0) | 59 (100) |
| RF01977 | HOX antisense intergenic RNA myeloid conserved 3 | 76 | 74 (97.3) | 68 (89.5) |
| RF02090 | DAOA antisense RNA1 conserved region 1 | 66 | 58 (87.8) | 50 (75.7) |
| RF02124 | JPX transcript, XIST activator conserved region 1 | 78 | 71 (91.0) | 63 (80.8) |
| RF02132 | HOXB13 antisense RNA 1 conserved region 1 | 53 | 50 (94.3) | 49 (92.5) |
| RF02138 | HOXA11 antisense RNA 1 conserved region 2 | 83 | 81 (97.5) | 78 (93.9) |
| RF02143 | Hydatidiform mole associated and imprint region | 56 | 53 (94.6) | 48 (85.7) |
| RF02148 | MEST intronic transcript 1 | 51 | 49 (96.1) | 47 (92.2) |
Genome-wide screen performance
We used our framework to scan for ncRNAs in a wide range of genomes (Table 5). To identify known annotated ncRNAs in bacterial genomes, we used M. tuberculosis H37Rv and P. aeruginosa PA14 as testing genomes. Our framework was able to identify known ncRNAs with a sensitivity of >90%.
Table 5. Performance of our model in a genome-wide screen.
| Genome | Strand | Sensitivity accuracy |
|---|---|---|
| Mycobacterium tuberculosis H37Rv (AL123456.2) | + | 90.7% |
| Mycobacterium tuberculosis H37Rv (AL123456.2) | − | 91.9% |
| Pseudomonas aeruginosa PA14 (CP000438.1) | + | 97.5% |
| Pseudomonas aeruginosa PA14 (CP000438.1) | − | 94.3% |
| Oryza sativa, Japonica, chromosome 1 (CM000138.1) | + | 92.3% |
| Oryza sativa, Japonica, chromosome 1 (CM000138.1) | − | 86.5% |
| Homo sapiens 1 (Containing five known lncRNAs: GNAS_AS-1-5) | + | 80% (4/5) |
| Homo sapiens 2 (Containing nine known miRNAs) | + | 77.7% (7/9) |
| Homo sapiens 3 (Containing six known lncRNAs: MAT2A_A-F) | + | 83.3% (5/6) |
| Homo sapiens 4 (Containing five known lncRNAs: HOTAIRM1_1-5) | + | 100% (5/5) |
| Homo sapiens 5 (Containing six known lncRNAs: HOXA11_AS1_1-6) | + | 83.3% (5/6) |
| Arthrospira platensis NIES-39 (AP011615.1, region: 1–1 200 000) | + | 413a |
| Penaeus monodon mitochondria (AF217843) | + | 39a |
aNeed to be experimentally verified.
Earlier methods have been applied to various prokaryotic genomes, but we wanted to test the ability of our method to identify lncRNAs and miRNAs, which are usually found in higher eukaryotes. Therefore, we obtained the following eukaryotic sequences from GenBank: O. sativa (chr.1) genomic sequence and five H. sapiens genomic regions (Region1: the 57 417 000–57 426 000 bp of chr. 20; Region2: the 49 767 700–49 779 500 bp of chr. x; Region3: the 85 770 900–85 772 300 bp of chr.2; Region4: the 27 135 000–27 139 900 bp of chr. 7; Region5: the 27 225 000–27 228 900 bp of chr. 7). Our framework also correctly identified most of the known ncRNAs in the eukaryotic genomic regions, with a sensitivity of >77%.
The tool was also evaluated for its ability to identify putative ncRNA candidates in unannotated genomes based on known ncRNA databases (33,70,71). The P. monodon mitochondria and the A. platensis genomes were used for testing. The model framework was applied to the A. platensis genome (NC_016640.1, genomic region: 1–1 200 000 nt; strand +) and 413 putative ncRNA candidates were identified and reported (results are shown in the Supplementary data 1). A total of 49 putative ncRNA candidates were identified as ncRNAs with high confidence (>0.85 Confidence, highlighted in bold in the Supplementary data 1). We also clustered the putative ncRNA candidates with Rfam sequences using RNAClust (72) and compared them to Rfam CM to suggest their putative functions via sequence-structure homology. The RNAClust (72) and Locarna (73) pipelines were used to detect similarity to Rfam sequences based on sequence-structure similarity (clustering trees provided upon request). To achieve maximal sensitivity with our method, the screening should be repeated with different window sizes. Consistent with a previous study (74), we also suggest using three different sizes of sliding windows (one short, 55–100 nt; one long, 125–200 nt; and one intermediate, 120 nt) to increase sensitivity because ncRNAs vary in size and structure.
Performance with unknown ncRNAs
To verify that our method has the ability to detect novel unknown ncRNAs in addition to known ncRNAs in the database, 52 sRNA candidates (in bacteria, the regulatory RNAs are called ncRNAs or sRNAs) validated by northern blot (37) were used. These candidate sequences have not been submitted to the current Rfam (ver.11). These test data are completely independent from the training data and are not part of our training dataset. To test the ability of our method to detect unknown ncRNAs, we obtained the genome sequence of PA14 from GenBank (accession number CP000438.1) and used our method to identify sRNAs in its genome. The prediction results were compared with the experimental sRNA identification data (37), as summarized in Table 6. The sRNA candidates were classified according to functional/structural categories for regulatory RNA in bacteria (75): Class I represents trans-encoded or intergenic RNAs; Class II represents 5′-untranslated region (5′-UTR) sense RNAs; Class III represents antisense RNAs (asRNAs) or cis-encoded; Class IV represents intergenic containing CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-like array; and Class V represents 3′-untranslated region (3′-UTR) sense RNAs.
Table 6. Performance of our model in detecting unknown verified ncRNAs in P. aeruginosa PA14.
| Regulatory class of sRNAs | No. of sRNAs | |
|---|---|---|
| Tested candidates | Detected by our method | |
| I intergenic sRNAs | 19 | 17 |
| II 5′-UTR sRNAs | 10 | 9 |
| III asRNAs | 19 | 14 |
| IV intergenic contains CRISPR | 1 | 1 |
| V 3′-UTR sRNAs | 3 | 3 |
| Total | 52 | 44 (84.6%) |
Our approach was able to predict various types of sRNA transcripts derived from 3′-UTR or 5′-UTR sense regions, antisense regions and intergenic regions. Most of the missed predictions were in the ‘antisense sRNA’ class (five of eight missed). This result is consistent with an earlier study (76) that addressed the observation that computational approaches are likely to focus on identifying ncRNAs in intergenic regions and are likely to miss small RNAs expressed from the non-coding strands of known genes and small RNAs of <50 nt. In addition, asRNAs may function mainly by complementing the coding sequences rather than being expressed as a specific sequence and/or structure-based feature (77). Moreover, our method depends on the fixed size of a sliding window (120 nt). As discussed earlier, to increase the overall sensitivity of the framework, multiple sizes of sliding windows are desirable. However, the high sensitivity of multiple sliding windows comes at the expense of computational time. We demonstrate here that our method can also detect novel ncRNA candidates. In contrast to the sequences used in the preceding section, these sRNA candidates appear to represent unannotated novel ncRNA genes identified by our tool. Taken together, we have demonstrated that our framework, in combination with the selected discriminating features, can identify both known and unknown ncRNAs.
CONCLUSION
We describe a hybrid method that uses a logistic regression model as a composite feature in an RF-based classifier model to detect various ncRNAs. The RF model has the advantage of robustness due to a bagging process and random selection of features to build the ensemble of trees. Moreover, the RF method can cope with the heterogeneous characteristics of diverse ncRNAs because its algorithm combines multiple decision trees with multiple classification rules. In addition to the robustness of the model, the composite feature incorporated as a statistical model in the RF-based classification method enhances the performance of the model for identifying lncRNA elements. The lncRNA is a challenging set of ncRNAs because of limited knowledge of lncRNA characteristics. The ncRNA identification framework proposed here exhibits high performance not only in recognizing known ncRNAs in a wide range of genomes but also in identifying novel ncRNAs in unannotated genomes. Both known and unknown ncRNAs can be identified with high accuracy. However, our scheme has some limitations. First, the size of the sliding window affects the performance of the classifier. Second, because ncRNA knowledge is limited, the framework may not be completely accurate and may need to be further refined to incorporate new ncRNA families. The logistic regression can be easily updated to incorporate new data. This composite feature may be improved in the future by using a non-linear combination of features based on a GA. We are also interested in building various logistic regression models, including separate logistic regression models for long, intermediate and short ncRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [77–86].
FUNDING
National Research University Project of Thailand's Office of the Higher Education Commission [54000318]; King Mongkut's University of Technology Thonburi. Funding for open access charge: King Mongkut's University of Technology Thonburi; National Research University Project of Thailand's Office of the Higher Education Commission [54000318].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Mattick J.S., Makunin I.V. Non-coding RNA. Hum. Mol. Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 2.Weinberg Z., Ruzzo W.L. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- 3.Brosnan C.A., Voinnet O. The long and the short of noncoding RNAs. Curr. Opin. Cell Biol. 2009;21:416–425. doi: 10.1016/j.ceb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 5.Costa F.F. Non-coding RNAs: Meet thy masters. BioEssays. 2010;32:599–608. doi: 10.1002/bies.200900112. [DOI] [PubMed] [Google Scholar]
- 6.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pauli A., Rinn J.L., Schier A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011;12:136–149. doi: 10.1038/nrg2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Managadze D., Rogozin I.B., Chernikova D., Shabalina S.A., Koonin E.V. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol. Evol. 2011;3:1390–1404. doi: 10.1093/gbe/evr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 10.Guttman M., Rinn J.L. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorodkin J., Hofacker I.L. From structure Prediction to genomic screens for novel non-coding RNAs. PLoS Comput. Biol. 2011;7:e1002100. doi: 10.1371/journal.pcbi.1002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponjavic J., Ponting C.P., Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17:556–565. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sati S., Ghosh S., Jain V., Scaria V., Sengupta S. Genome-wide analysis reveals distinct patterns of epigenetic features in long non-coding RNA loci. Nucleic Acids Res. 2012;40:10018–10031. doi: 10.1093/nar/gks776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen G., Wang Z., Wang D., Qiu C., Liu M., Chen X., Zhang Q., Yan G., Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41:D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moran V.A., Perera R.J., Khalil A.M. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012;40:6391–6400. doi: 10.1093/nar/gks296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Chen Z., Wang X., Huang Z., He Z., Chen Y. Long non-coding RNA: a new player in cancer. J. Hematol. Oncol. 2013;6:37. doi: 10.1186/1756-8722-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marques A.C., Ponting C.P. Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol. 2009;10:R124. doi: 10.1186/gb-2009-10-11-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernhart S.H., Hofacker I.L. From consensus structure prediction to RNA gene finding. Brief Funct. Genomic Proteomic. 2009;8:461–471. doi: 10.1093/bfgp/elp043. [DOI] [PubMed] [Google Scholar]
- 19.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G., et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivas E., Eddy S.R. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2001;2:8. doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Washietl S., Hofacker I.L., Stadler P.F. Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coventry A., Kleitman D.J., Berger B. MSARI: multiple sequence alignments for statistical detection of RNA secondary structure. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12102–12107. doi: 10.1073/pnas.0404193101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedersen J.S., Bejerano G., Siepel A., Rosenbloom K., Lindblad-Toh K., Lander E.S., Kent J., Miller W., Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran T., Zhou F., Marshburn S., Stead M., Kushner S., Xu Y. De novo computational prediction of non-coding RNA genes in prokaryotic genomes. Bioinformatics. 2009;25:2897–2905. doi: 10.1093/bioinformatics/btp537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saetrom P., Sneve R., Kristiansen K.I., Snøve O., Grünfeld T., Rognes T., Seeberg E. Predicting non-coding RNA genes in Escherichia coli with boosted genetic programming. Nucleic Acids Res. 2005;33:3263–3270. doi: 10.1093/nar/gki644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C., Ding C., Meraz R.F., Holbrook S.R. PSoL: a positive sample only learning algorithm for finding non-coding RNA genes. Bioinformatics. 2006;22:2590–2596. doi: 10.1093/bioinformatics/btl441. [DOI] [PubMed] [Google Scholar]
- 27.Washietl S., Findeiß S., Müller S.A., Kalkhof S., Bergen M.V., Hofacker I.L., Stadler P.F., Goldman N. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011;17:578–594. doi: 10.1261/rna.2536111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raasch P., Schmitz U., Patenge N., Vera J., Kreikemeyer B., Wolkenhauer O. Non-coding RNA detection methods combined to improve usability, reproducibility and precision. BMC Bioinformatics. 2010;11:491. doi: 10.1186/1471-2105-11-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salari R., Aksay C., Karakoc E., Unrau P.J., Hajirasouliha I., Sahinalp S.C. smyRNA: A Novel Ab Initio ncRNA Gene Finder. PLoS ONE. 2009;4:e5433. doi: 10.1371/journal.pone.0005433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao M., Cervantes-Cervantes M., Zhong L., Wang J.T.L. Searching for non-coding RNAs in genomic sequences using ncRNAscout. Genomics, Proteomics Bioinformatics. 2012;10:114–121. doi: 10.1016/j.gpb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lertampaiporn S., Thammarongtham C., Nukoolkit C., Kaewkamnerdpong B., Ruengjitchatchawalya M. Heterogeneous ensemble approach with discriminative features and modified-SMOTEbagging for pre-miRNA classification. Nucleic Acids Res. 2013;41:e21. doi: 10.1093/nar/gks878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivas E., Eddy S.R. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- 33.Burge S., Daub J., Eberhardt R., Tate J., Barquist L., Nawrocki E., Eddy S., Gardner P., Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pruitt K.D., Tatusova T., Brown G.R., Maglott D.R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amaral P.P., Clark M.B., Gascoigne D.K., Dinger M.E., Mattick J.S. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2011;39:D146–D151. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrara S., Brugnoli M., De Bonis A., Righetti F., Delvillani F., Deho G., Horner D., Briani F., Bertoni G. Comparative profiling of Pseudomonas aeruginosa strains reveals differential expression of novel unique and conserved small RNAs. PLoS One. 2012;7:e36553. doi: 10.1371/journal.pone.0036553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall M.A. Proceedings of the Seventeenth International Conference on Machine Learning. San Francisco, CA: Morgan Kaufmann; 2000. Correlation-based feature selection for discrete and numeric class machine learning; pp. 359–366. [Google Scholar]
- 39.Bishop C.M. Pattern Recognition and Machine Learning. New York: Springer; 2006. [Google Scholar]
- 40.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Austria: Vienna; 2006. [Google Scholar]
- 41.Altschul S.F., Gish W., Miller W., Myers E.W., Libman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 42.Nawrocki E.P., Kolbe D.L., Eddy S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eddy S.R., Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao Z., Weinberg Z., Ruzzo W.L. CMfinder—a covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22:445–452. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- 45.Lee M.T., Kim J. Self containment, a property of modular RNA structures, distinguishes microRNAs. PLoS Comput. Biol. 2008;4:e1000150. doi: 10.1371/journal.pcbi.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freyhult E., Gardner P.P., Moulton V. A comparison of RNA folding measures. BMC Bioinformatics. 2005;6:241. doi: 10.1186/1471-2105-6-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong L., Zhang Y., Ye Z., Liu X., Zhao S., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slater G.C. Algorithms for the Analysis of Expressed Sequence Tags. Cambridge: University of Cambridge; 2000. [Google Scholar]
- 49.Breiman L. Random forests. Mach. Learning. 2001;45:5–32. [Google Scholar]
- 50.Larrañaga P., Calvo B., Santana R., Bielza C., Galdiano J., Inza I., Lozano J.A., Armañanzas R., Santafé G., Pérez A., et al. Machine learning in bioinformatics. Brief. Bioinform. 2006;7:86–112. doi: 10.1093/bib/bbk007. [DOI] [PubMed] [Google Scholar]
- 51.Liaw A., Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 52.Yang P., Hwa Yang Y., Zhou B., Zomaya A. A review of ensemble methods in bioinformatics. Curr. Bioinformatics. 2010;5:296–308. [Google Scholar]
- 53.Frank E., Hall M., Trigg l. E., Holmes G., Witten I.H. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- 54.Strobl C., Boulesteix A., Zeileis A., Hothorn T. Bias in random forest variable importance measures: illustrations, sources and a solution. BMC Bioinformatics. 2007;8:25. doi: 10.1186/1471-2105-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strobl C., Boulesteix A., Kneib T., Augustin T., Zeileis A. Conditional variable importance for random forests. BMC Bioinformatics. 2008;9:307. doi: 10.1186/1471-2105-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hothorn T., Hornik K., Zeileis A. party: a laboratory for recursive part(y)tioning. 2006 [R package version 0.9-0] [Google Scholar]
- 57.Lu Z.J., Yip K.Y., Wang G., Shou C., Hillier L.W., Khurana E., Agarwal A., Auerbach R., Rozowsky J., Cheng C., et al. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res. 2011;21:276–285. doi: 10.1101/gr.110189.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Babak T., Blencowe B.J., Hughes T.R. Considerations in the identification of functional RNA structural elements in genomic alignments. BMC Bioinformatics. 2007;8:33. doi: 10.1186/1471-2105-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clote P., Ferré F., Kranakis E., Krizanc D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005;11:578–591. doi: 10.1261/rna.7220505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klein R.J., Misulovin Z., Eddy S.R. Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7542–7547. doi: 10.1073/pnas.112063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schattner P. Searching for RNA genes using base-composition statistics. Nucleic Acids Res. 2002;30:2076–2082. doi: 10.1093/nar/30.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsson P., Hinas A., Ardell D.H., Kirsebom L.A., Virtanen A., Söderbom F. De novo search for non-coding RNA genes in the AT-rich genome of Dictyostelium discoideum: performance of Markov-dependent genome feature scoring. Genome Res. 2008;18:888–899. doi: 10.1101/gr.069104.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gardner P.P. The use of covariance models to annotate RNAs in whole genomes. Brief. Funct. Genomic Proteomic. 2009;8:444–450. doi: 10.1093/bfgp/elp042. [DOI] [PubMed] [Google Scholar]
- 64.Menzel P., Gorodkin J., Stadler P.F. The tedious task of finding homologous noncoding RNA genes. RNA. 2009;15:2075–2082. doi: 10.1261/rna.1556009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong C., Zhang S. Clustering RNA structural motifs in ribosomal RNAs using secondary structural alignment. Nucleic Acids Res. 2012;40:1307–1317. doi: 10.1093/nar/gkr804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miler T.L. Modular organization and composability of RNA. Publicly Accessible Penn Dissertations. 2009 Paper 244. [Google Scholar]
- 67.Bhartiya D., Pal K., Ghosh S., Kapoor S., Jalali S., Panwar B., Jain S., Sati S., Sengupta S., Sachidanandan C., et al. lncRNome: a comprehensive knowledgebase of human long noncoding RNAs. Database. 2013;11 doi: 10.1093/database/bat034. doi: 10.1093/database/bat034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Novikova I.V., Hennelly S.P., Sanbonmatsu K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 2012;40:5034–5051. doi: 10.1093/nar/gks071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.William G.J. Rattle: a data mining GUI for R. R J. 2009;1:45–55. [Google Scholar]
- 70.Bu D., Yu K., Sun S., Xie C., Skogerbø G., Miao R., Xiao H., Liao Q., Luo H., Zhao G., et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012;40:D210–D215. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kin T., Yamada K., Terai G, Okida H., Yoshinari Y., Ono Y., Kojima A., Kimura Y., Komori T., Asai K. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res. 2007;35:D145–D148. doi: 10.1093/nar/gkl837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaczkowski B., Torarinsson E., Reiche K., Havgaard J., Stadler P., Gorodkin J. Structural profiles of human miRNA families from pairwise clustering. Bioinformatics. 2009;25:291–294. doi: 10.1093/bioinformatics/btn628. [DOI] [PubMed] [Google Scholar]
- 73.Will S., Joshi T., Hofacker I. locaARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA. 2012;18:900–914. doi: 10.1261/rna.029041.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kavanaugh L.A., Dietrich F.S. Non-coding RNA prediction and verification in Saccharomyces cerevisiae. PLoS Genet. 2009;5:e1000321. doi: 10.1371/journal.pgen.1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waters L.S., Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kawano M., Reynolds A.A., Miranda-Rios J., Storz G. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 2005;33:1040–1050. doi: 10.1093/nar/gki256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Georg J., Voss B., Scholz I., Mitschke J., Wilde A., Hess W.R. Evidence for a major role of antisense RNAs in cyanobacterial gene regulation. Mol. Syst. Biol. 2009;5:305. doi: 10.1038/msb.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.