


Abstract
Many enhancers regulate their target genes via long-distance interactions. High-throughput experiments like ChIA-PET have been developed to map such largely cell-type-specific interactions between cis-regulatory elements genome-widely. In this study, we integrated multiple types of data in order to reveal the general hidden patterns embedded in the ChIA-PET data. We found characteristic distance features related to promoter–promoter, enhancer–enhancer and insulator–insulator interactions. Although a protein may have many binding sites along the genome, our hypothesis is that those sites that share certain open chromatin structure can accommodate relatively larger protein complex consisting of specific regulatory and ‘bridging’ factors, and may be more likely to form robust long-range deoxyribonucleic acid (DNA) loops. This hypothesis was validated in the estrogen receptor alpha (ERα) ChIA-PET data. An efficient classifier was built to predict ERα-associated long-range interactions solely from the related ChIP-seq data, hence linking distal ERα-dependent enhancers to their target genes. We further applied the classifier to generate additional novel interactions, which were undetected in the original ChIA-PET paper but were validated by other independent experiments. Our work provides a new insight into the long-range chromatin interactions through deeper and integrative ChIA-PET data analysis and demonstrates DNA looping predictability from ordinary ChIP-seq data.
INTRODUCTION
Many distant enhancer elements in the human genome regulate their target genes through long-range deoxyribonucleic acid (DNA) looping interactions (1–4). Such long-range interactions are often related to 3D chromatin conformations that are important for gene regulation in specific cell types (5–7). In general, these chromatin interactions can be roughly grouped into two different types: one is closely related to gene regulation and dynamically changes during development or in response to external stimuli (8,9) and the other plays more of a structural role, forming non-tissue-specific chromosome conformations (10).
To biochemically detect how and where these long-range interactions occur, chromatin conformation capture (3C) (11) and related methods, such as 4C (12) and 5C (13), have been developed. These methods are well suited for studying targeted local chromatin regions. Recently, genome-wide high-throughput techniques have been applied to detect large numbers of multiple interacting regions at the same time, which can delineate a global landscape of long-range chromatin interactions. Two of the best known methods are Hi-C (14) and ChIA-PET (15). Hi-C is directly derived from 3C, which sequences all the interacting DNA fragments with biotin-marked ligation junctions. It can detect substantial interactions simultaneously and is not restricted to the type of protein that ‘bridges’ these interactions. However, Hi-C can only provide a relatively low-resolution interaction map (∼1 Mbp), which is unsuitable for studying interactions between specific cis-regulatory elements, such as enhancer–promoter loops. ChIA-PET combines ChIP and 3C together, which enriches crosslinked DNA–protein complexes using an antibody against the protein of interest and uses proximity ligation to collect interacting DNA fragments tethered by the ChIP-ed protein. It has a higher resolution (similar to ChIP-seq) than Hi-C, but is limited to detecting interactions mediated by only one type of protein per experiment. Thus, Hi-C and ChIA-PET provide us with global view of different aspects of the chromosomal contact structure. However, they both require very extensive sequencing depth and are often affected by noise from random contact of DNA fragments in solution.
We decided to integrate genome-wide transcription factor (TF) binding and histone modification profiles in order to better understand ChIA-PET data for two reasons. First, it has been reported that active enhancers are most often bound by multiple TFs, which form large protein complexes to link enhancers to promoters (16,17). This may generate a different epigenetic pattern compared with simple protein binding sites. Second, if two distant TF binding sites are interacting with each other, ChIP-seq experiments will likely detect both peaks in these two regions (18), and this phenomenon can partially explain the fact that some TF binding sites do not contain the canonical motif. We can infer that those regions pulled down by the same antibody are more likely to form interactions. These facts imply that aligning multiple TF binding sites (e.g. according to ChIP-seq and DNase-seq data) will be informative for predicting physical interactions between functional cis-regulatory elements.
We started from MCF7 estrogen receptor alpha (ERα) ChIA-PET data and combined gene expression, TF binding, histone modification profiles and open chromatin conformation data to uncover hidden features buried beneath the long-range interactions. Firstly, invariant distance features from different ChIA-PET data sets were extracted to characterize promoter–promoter (PP), enhancer–enhancer (EE) and insulator–insulator (II) interactions. Secondly, we determined the genetic and epigenetic features that could discriminate loop-associated and non-loop-associated ERα binding sites (ERBSs). Our analysis demonstrates that ERBSs associated with loop formation, especially those that contain an estrogen response element (ERE), are more likely to be nucleosome depleted, which is a surprise as some previous study (19) reported that ERα could bind to nucleosomes directly. Assembly of co-factors, such as FoxA1, GATA3 and AP2γ, and general co-activator p300 and ERα complex appear to require such chromosome conformation with higher DNA accessibility. Lastly, we used these features to predict loop-associated ERBSs (laERBSs) and to develop a DNA looping prediction algorithm. This allowed us to recover novel ERα-mediated interactions that were missed from the original ERα ChIA-PET paper (15). Many known ERα-regulated genes that were not found in the original ChIA-PET paper were identified by our method, and some are supported by other independent 3C data or Pol2 ChIA-PET data. To our knowledge, this is the first successful attempt to use multiple ChIP-seq data to predict long-range chromatin interactions, thus could serve as a complement to the complicated and costly ChIA-PET experiments.
MATERIALS AND METHODS
Data sources
ChIA-PET data of ERα from E2 (17β-Estradiol) induced MCF7 cell was obtained from (15), where the P-value of each Paired-End Tag (PET) cluster was given; Pol2 and CTCF data from MCF7 and K562 cells were obtained from ENCODE project (20), and the coordinates of data were changed to hg18 with the lift-over tool. The inter-chromosomal interactions were not considered in our analysis as they were only composed of a very small proportion of the interactions (a few tens) and were not enough for statistical analysis. ChIP-seq data of ERα, Pol2, H3K4me1, H3K4me3, H3K9ac, H3K27me3 and Input control from E2-induced MCF7 cells were from (21); ChIP-seq data of H3K4me2 and DNase-seq data from E2-induced MCF7 cells were from (22); FoxA1 and AP2γ data from E2-induced MCF7 cells were from (23); GATA3 and p300 data from E2-induced MCF7 cell lines were from (24). GRO-seq data for E2-induced MCF7 cells after 40 min were from (25). E2-induced differential expressed genes were obtained from the supplementary data of (26). The related Gene Expression Omnibus (GEO) accession numbers are GSE11352, GSE18046, GSE23701, GSE23852, GSE29073, GSE33216, GSE39495, GSM678539 and GSM678540.
Binding sites detection
Binding sites from ChIP-seq were called by Model-based Analysis for ChIP-Seq (MACS) (27) with default parameters for ERα, FoxA1, GATA3, AP2γ, Pol2 and p300. Enrichment regions for histone modifications were called by broad peak options with parameter setting ‘–nomodel –nolambda’ as suggested by Feng et al. (28).
EE, PP and enhancer–promoter interactions classification
Distance between two ChIP-seq binding peaks was calculated as the genomic distance between their summits called by MACS (27). Distance between two genomic regions was defined as the genomic distance between their mid-points. Density estimation was called by the R density function with default parameters. Peak position of a uni-modal distribution was chosen as the point with the highest density value. For multi-modal distribution, we first fitted the data with a Mixture Gaussian Distribution by R package mixtools (29) and chose the estimated expectation of each component as peak position.
We used log2-transformed read-count ratio between H3K4me3 and H3K4me1 ChIP-seq data in the Pol2 binding sites to classify Pol2 ChIA-PET interaction clusters into three types: EE interactions, PP interactions and enhancer–promoter (EP) interactions. Windows for computing read counts were selected as ±1.5-kb region around the peak summits. Binding sites whose summits were closer than 1.5 kb were merged and the mid-point of merged regions was chosen as the new peaks’ ‘pseudo summit’. The data were then fitted with Mixture Gaussian Model and a Bayesian posterior probability of 0.5 was set to determine whether a Pol2 binding site was promoter-like or enhancer-like. PET clusters with both ends classified as enhancer-like were called EE interactions, both promoter-like were called PP interactions and the rest were called EP interactions. For ERα ChIA-PET data, we applied this classification in those interactions that overlapped with H3K4me1 or H3K4me3 peaks at both ends. The subsequent steps were similar to those of Pol2 ChIA-PET data.
Histone modification, DNase-seq and GRO-seq profiles surrounding ChIP-seq binding peaks
Each tag was extended by 200 bp along the read direction. We selected peak summit as the center for signal alignment. Each region was divided into 25-bp sub-regions and its read coverage was computed as the read counts covering this sub-region. The difference of read coverage between ChIP data and input data was computed as the log2-transformed read-coverage ratio at each sub-region. DNase-seq data were not extended and only the 5′ end was used as aggregation inputs. GRO-seq data were divided into two groups according to the strand of the reads and then processed in the same manner as histone modification data.
To classify whether an ERBS was an ERE-containing one, we used STORM (30) to scan ±200-bp region around the summit of ERBS for ERE sequence motif, with P-value 1e−4.
Model to predict laERBSs
ERBSs were extracted from ChIP-seq data. To predict laERBSs, three types of features were computed:
Fi,j: loge-transformed read counts for ChIP-seq data of protein j (or DNase-seq data) in ERBS i, with a window size of 400 bp centered around ERBS peak summit;
Di: loge-distance between the neighboring ERBS of ERBS i and ERBS i;
Hi,j: the differences between log2-transformed ratio of read-coverage against input in central region (±100-bp region relative to the peak summit) to average of read-coverage against input in the two flanking regions (−400 bp to −200 bp and +200 bp to +400 bp relative to peak summit) of ERBS i for histone modification j.
We selected the 1822 ERBSs that related to 903 interactions identified in both two ChIA-PET experimental replicates as foreground training set. An equal number of ERBSs that did not overlap with any interactions in either replicate were randomly selected as background training set. We used a logistic classifier to perform the classification, i.e.
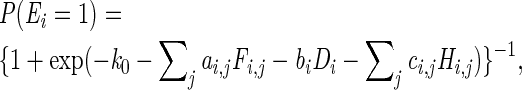 |
in which Ei is the indicator whether ERBS i is a laERBS, P is a probability function and k0,ai,j,bi,ci,j are the model parameters.
After training, we chose the most significant features that showed improvement over ERα ChIP-seq read counts in ERBSs to fix the final classifier. Then ERBSs were filtered by setting the threshold of the logistic classifier to be 0.2 to find putative laERBSs. Summits of these laERBSs closer than 3 kb were merged and the mid-point was selected as the new ‘pseudo summit’. This resulted in ∼15 000 candidate anchors.
Model to predict ERBS interaction clusters
To predict ER-associated interactions, the candidate anchors were paired with each other to form candidate ERBS pairs. Those pairs that crossed topological domain boundaries in h1-ESC were excluded since domain boundaries were roughly invariant across cell lines, and long-range interactions were largely restricted within topological domains (31). To predict interactions, two types of features were computed for each candidate pair:
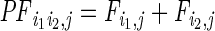
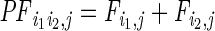 :
the sum of loge-transformed jth ChIP-seq (or DNase-seq) read counts for
each candidate ERBS pair
(i1,i2), with a window
size of 3 kb for each end (±1.5-kb region relative to the peak
summit);
:
the sum of loge-transformed jth ChIP-seq (or DNase-seq) read counts for
each candidate ERBS pair
(i1,i2), with a window
size of 3 kb for each end (±1.5-kb region relative to the peak
summit);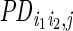
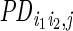 :
the loge-distance and inverse distance between each ERBS pair
(i1,i2), since previous
study (32) suggested that probability of
long-range interaction was not monotonically related to genomic
distance.
:
the loge-distance and inverse distance between each ERBS pair
(i1,i2), since previous
study (32) suggested that probability of
long-range interaction was not monotonically related to genomic
distance.
Eight hundred ERBS interactions (out of 903 interactions identified in both two ChIA-PET experimental replicates) with both ends restricted within the same topological domain were selected as foreground training set. Equal number of candidate ERBS pairs that did not overlap with any ERα interactions identified in either experimental replicate was randomly selected background training set. We still used a logistic classifier to perform the classification, i.e.
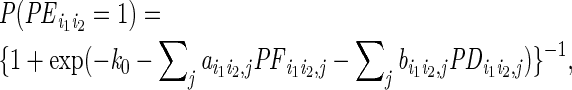 |
in which 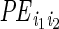
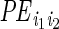 is the
indicator whether ERBS pair
(i1,i2) formed an interaction,
P is a probability function and
is the
indicator whether ERBS pair
(i1,i2) formed an interaction,
P is a probability function and 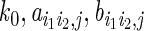
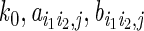 are the model parameters.
are the model parameters.
Performance of the classifier was evaluated by a 5-fold cross-validation; AUC (area under curve) and ROC (receiver operating characteristic) were plotted as the average of each 5-fold cross-validation.
To predict novel ERBS–ERBS interactions, we trained the model parameters on the whole training set and applied it to the remaining candidate ERBS pairs. Predicted interactions that were not reported in either replicate were further analyzed.
Model to predict promoter–ERBS interaction clusters
To predict new promoter–ERBS interactions, we first found out those promoters containing at least one of the FoxA1, GATA3 or AP2γ binding sites but not ERBSs from RefSeq-annotated promoters (±1500-bp region surrounding the transcription start site (TSS) and alternative TSSs within 1.5 kb were merged). Those features used for predicting ERBS–ERBS interactions were computed by replacing distance between two ERBSs with distance between promoter and ERBS.
RESULTS
Invariant distance distributions are associated with different types of interactions
We collected the ChIA-PET data for ERα (15), Pol2 (33) and CTCF from the GEO data sets (GEO accession numbers GSE18046 and GSE39495) and ENCODE project (20) for this study. We first analyzed the distribution of the distance between the two ends of chimeric and non-chrimeric PETs (34). Raw PETs of ERα ChIA-PET data from MCF7 cells presented a trimodal distribution (Figure 1A). The third peak at the right, which was mainly composed by single PET, was similar to that of chimeric PETs that represented randomly paired interaction sites. This suggested that PETs with a long span (>1 Mbps) were more likely to be derived from random DNA contact noise in solution or some non-specific interactions. Thus, they should be filtered more carefully. In the following analysis, we removed all PETs that had a span larger than 1 Mbps and only considered the interactions that could be supported by no less than two PETs (PETs 2+ clusters) instead of threshold (no less than three PETs without distance restriction) used in (15). This is because that majority of PETs 2+ clusters have a span less than 1 Mbps, which is well separated from the chimeric PETs. Setting a higher threshold will lose many true positives and make the selection of background less reliable. Interaction clusters from both ERα and Pol2 data presented bimodal distribution patterns (Figure 1B and C). For the Pol2 data, we used H3K4me3/H3K4me1 log2 read-count ratio to classify the PETs 2+ clusters into three groups (33), i.e. EE interactions where both ends of the clusters showed higher H3K4me1 signals, PP interactions where both ends of the clusters showed higher H3K4me3 signals and EP interactions for the rest (see details in the Materials and Methods section). It was clear that the two peaks from Pol2 data corresponded to the characteristic distances ∼5 kb and ∼60 kb for EE interactions and PP interactions, respectively (Figure 1D). And we found that a great percentage of EE interactions are indeed within the flanking regions (±10 kb) of the same gene (Supplementary Figure S1). We removed common interactions between MCF7 and K562 cells to study the cell-type specificity of EE and PP interactions. Both of the two peaks were present in MCF7 unique and K562 unique interactions (Figure 1C). ERα data displayed a similar, albeit less pronounced, character. The bimodal pattern of distance distributions of chromatin interaction clusters was largely conserved across different cell types (in both MCF7 and K562 cells; Figure 1C), and thus appeared to be an invariant feature embedded in the typical promoter interacting TF ChIA-PET data.
Figure 1.
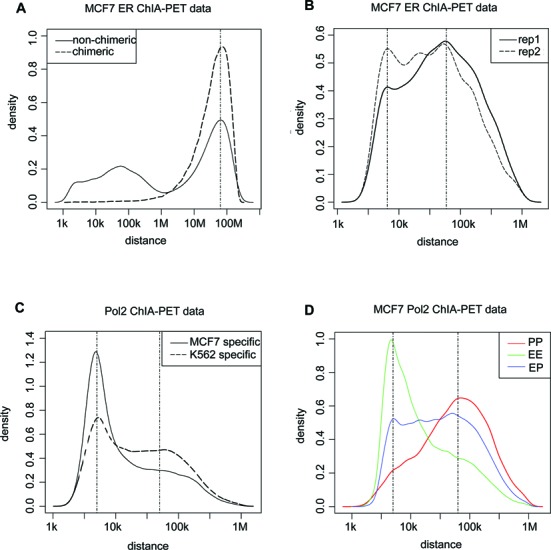
Characteristic distance features of ChIA-PET data. (A) Log10-distance distribution between the two ends of chimeric and non-chimeric PETs, where non-chimeric PETs presented a trimodal distribution. The third peak at right of non-chimeric PETs was close to that of chimeric PETs. (B) Log10-distance distribution between the two ends of PETs 2+ clusters of MCF7 ERα ChIA-PET data for two replicates. Interactions with span more than 1 Mbps were excluded. Peak positions located at 3.8 and 4.77. (C) Log10-distance distribution between the two ends of PETs 2+ clusters from MCF7-specific and K562-specific Pol2 ChIA-PET data. Peak positions located at 3.7 and 4.64. (D) Log10-distance distribution of EE, PP and EP interactions from MCF7 Pol2 ChIA-PET data. Positions of peaks for EE and PP interactions were 3.7 and 4.8, respectively.
We also checked II interactions using CTCF ChIA-PET data. CTCF binding sites and interactions that overlapped with H3K4me1 or H3K4me3 peaks at either end were filtered out to avoid promoter- or enhancer-associated interactions. The log10-distance distribution of the rest CTCF interaction clusters had a unimodal pattern (Supplementary Figure S2A). The peak position was roughly invariant between MCF7 (160 kb) and K562 (200 kb) cells (Supplementary Figure S2A), and was roughly 5-fold as large as the median distance of the neighboring CTCF binding sites (Supplementary Figure S2B). This was consistent with the notion that CTCF regulation is largely conserved between different cell types (35). Thus, II interactions share a different distance preference, which is also largely invariant across different cell lines.
laERBSs are more likely to be nucleosome depleted
To identify genetic or epigenetic features that could discriminate ERBSs involved in loop formation from those solo ones, we carried out integrative analysis of multiple (TF and histone modification) ChIP-seq data sets from the same cell line (MCF7). Firstly, we analyzed histone modification patterns around laERBSs and non-loop-associated ERBSs (nlaERBSs). Here laERBSs were selected as ERBSs overlapping with PETs 2+ clusters identified in both two ChIA-PET experimental replicates (903 clusters). To align histone modification signals around ERBSs, we selected the strongest summit of ERBS as the center if more than one ERBS was contained in the same end of an interaction cluster, resulting 1203 ERBSs as the foreground set. We randomly selected an equal number of ERBSs that were not associated with any interaction clusters identified in either ChIA-PET experimental replicate as the background set. It was noticed that laERBSs showed subtle depletion of histone modification signal in the center compared with nlaERBSs (Supplementary Figure S3A–C). laERBSs and nlaERBSs were further divided into two groups by the presence or absence of an ERE. Surprisingly, we found that a significantly more proportion of laERBSs with an ERE shown depleted histone modification signals in the center (Figure 2A–C, Supplementary Figure S4 and Supplementary Table S1) than the other three groups. DNase-seq data confirmed that laERBSs were more frequently located in open chromatin regions (Supplementary Figure S3D), especially those that contained an ERE (Figure 2D and Supplementary Figure S5). Since only about 10% laERBSs located in promoter regions, our observation could not be explained by nucleosome depletion in active promoter regions (Supplementary Figure S6). Therefore, although to the best of our knowledge, there were no direct genome-wide nucleosome positioning data available in E2-induced MCF7 cells, our integrative analysis of multiple histone modifications and DNase-seq data suggested that nucleosomes were more likely to be depleted in laERBS. In fact, a previous paper (22) reported that ERα binding to DNA was not sensitive to nucleosomes; while on the contrary, we found that those laERBSs with an ERE showed significant nucleosome depletion in the center. This indicated that there might be some other co-factors that can bind to these laERBSs cooperatively to facilitate long-range interactions and be responsible for nucleosome eviction.
Figure 2.
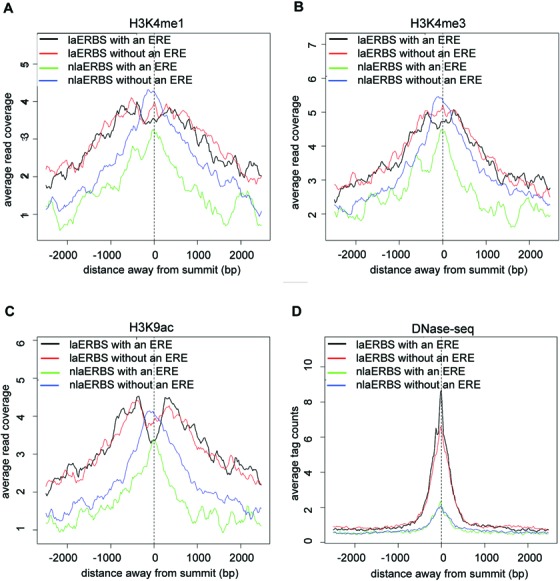
Histone modification and DNase-seq signal profile for four groups of ERBSs. Average H3K4me1 (A), H3K4me3 (B) and H3K9ac (C) read-coverage against input surrounding laERBSs with and without an ERE and nlaERBSs with and without an ERE. Average DNase-seq tag counts surrounding laERBSs with and without an ERE and nlaERBSs with and without an ERE (D).
To test this hypothesis, we conducted an enrichment analysis against the ChIP-seq data of three known ERα's co-factors, and the general co-activator p300. More than 80% laERBSs (either with an ERE or not) overlapped with FoxA1, GATA3 and AP2γ binding sites (Figure 3). The percentages were significantly higher than those of nlaERBSs (P < 2.2e−16; Supplementary Table S2), indicating that these three TFs are likely in the complex mediating ERα-associated long-range interactions. Reported RNAi knock-down experiments also support the notion that these three co-factors are important for ERα loop formation (23,24). Thus they might be associated with nucleosome eviction in laERBSs. For p300, the percentage of overlapping with laERBSs was not as high as that of those three co-factors. However, interestingly, p300 was significantly enriched in laERBSs with an ERE in comparison with laERBSs without an ERE (P = 7.59e−4), but showed no difference between nlaERBSs with an ERE and nlaERBSs without an ERE (P = 0.72), while none of the three co-factors have a similar pattern. A previous study (36) showed that p300 is a component of an estrogen receptor co-activator complex and the formation of the p300 complex is associated with nucleosome eviction (37). This indicated that p300 might be more frequently recruited to laERBSs with an ERE for higher level of nucleosome depletion. In addition, with the GRO-seq data (GEO accession numbers GSM678539 and GSM678540) we found that laERBSs showed stronger bidirectional transcription of small RNAs (smRNAs) compared to nlaERBSs (Supplementary Figure S7), with the highest transcription level occurring in laERBSs with an ERE (Supplementary Figure S7A). This is consistent with the notion that the bidirectional smRNAs transcription may help maintain the open chromatin structure in these regions (38). Taken together, these observations suggest that laERBSs, especially those with canonical EREs, are often associated with an open chromatin structure, which is likely resulted by the formation of the looping-specific protein complex of ERα, its co-factors and p300, and associated with the bidirectional transcription of smRNAs.
Figure 3.
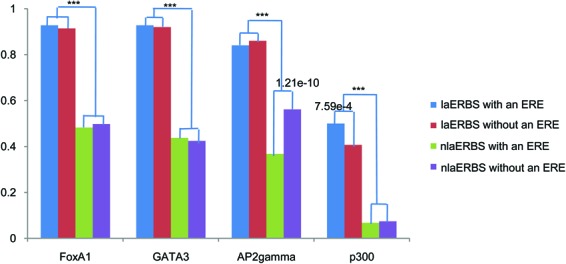
Proportion of FoxA1, GATA3, AP2γ and p300 binding peaks that overlapped with four groups of ERBSs. P-values smaller than 0.05 were listed above the bars, which were given by one-tailed Fisher test in R. ***P < 2.2e−16.
Clustered ERBSs are more likely to be associated with loops
Of all the 1299 regions that formed PETs 2+ interaction clusters identified in both two ChIA-PET experimental replicates, 30% contained more than one ERBS. Statistical tests showed the clustered ERBSs within 3 kb were more likely to associate with DNA loops (P < 2.2e−16, 45% versus 20%).
A logistic classifier can predict laERBSs and associated long-range interactions
We tested the prediction power of the above features extracted from previous sections by building a classifier to predict ERα-associated long-range interactions based on TF and histone modification ChIP-seq data. Given a pair of ERα ChIP-seq binding peaks, a powerful classifier should be able to judge how likely they are actually forming an interacting DNA loop. Our predictor was built in two steps. First, we used the above features to distinguish laERBSs and nlaERBSs. We used 903 PETs 2+ clusters confirmed in both two replicates as the positive training samples, since they were more reliable than those non-reproducible ones. We evaluated the performance of the classifier with 5-fold cross-validation and got the average true positive rate (TPR) at 74% and average false positive rate (FPR) at 21% at the Bayesian threshold (0.5) with only three features, i.e. ERα ChIP-seq signals, neighboring ERBS distances and DNase-seq signals. AUC for predictor with single feature and ROC comparisons for different feature combinations were shown in Figure 4A and C, respectively. Although transcriptional rate of bidirectional smRNA and the binding intensity of p300 showed good predictive power, they are redundant with ERα ChIP-seq signals, neighboring ERBS distances and DNase-seq signals. And unexpectedly, histone modification features did not provide additional information over DNase-seq signals for the prediction. We finally set the Bayesian threshold 0.2 to contain as many laERBSs (in the training set) as possible (97%) while the relatively high FPR (61%) can be further dealt with in the next filtering step.
Figure 4.

AUC and ROC curves for the predictors. (A) AUC for each feature to predict laERBSs. Here AUC was computed as the average area under ROC curve for predictors with each single feature for 5-fold cross-validation. (B) AUC for each feature to predict interactions between predicted laERBSs. AUC was computed in the same manner as in (A). "dis" is distance between two ERBSs and "idis" is the inverse distance. (C) ROC trained for ERα alone, ERα+distance of neighboring ERBSs, ERα+distance of neighboring ERBSs+DNase-seq and all the features. (D) ROC trained for ERα alone, ERα+distance and ERα+distance+6 significant features and all the features.
In the second step, we tried to predict long-range interactions between the putative laERBSs obtained from the first step. Recently, Hi-C data analysis suggests that chromatins are organized as large topological domains and interactions between regulatory elements are largely restricted within these domains (31). As shown in (31), although strengths of chromatin interactions between and within these domains may vary across different cell lines, such domains are roughly invariant across different cell types. Thus, we further restricted the two ends of the same interaction cluster within the same Hi-C-determined topological domain. This criterion only excluded 69 (∼8%) PETs clusters from our positive set, but reduced more than one half of ERBS pair-wise combinations, a large percentage of which are likely to be noise (there are 97 202 candidate ERBS pairs by using constrain of topological domain while there exist 239 016 and 6 377 706 possible intra-chromosome ERBS pairs with and without restriction of 1-Mb genomic distance, respectively). With the above restrictions, we revealed 800 PETs 2+ interactions between filtered laERBSs within the topological domains, which served as the foreground training set. Classifiers with different feature combinations on the training set were evaluated by 5-fold cross-validation. The best performer achieved a TPR of 93% and an FPR of 8% at the Bayesian threshold 0.5. The intensity of ERBSs and the distance between candidate pairs acted as the top two significant features to distinguish foreground and background (Figure 4B). The binding intensity of FoxA1 and AP2γ had a similar predictive power, but was somewhat redundant with that of ERα. Beyond these features, DNase-seq and histone modification signals that were able to describe chromatin accessibility could also improve the performance. Finally, eight features (Supplementary Tables S3 and S4) that showed significant improvement over the ERα+distance combination (Figure 4D) were selected to build the final classifier.
We applied the classifier to predict interactions among the 97 202 candidate ERBS pairs. Originally, Fullwood et al. (15) reported 3527 high confidential interactions that share ERα ChIP-seq binding peaks at both ends for the combined two replicates. Among them, 3113 were restricted in the topological domains, 76% (2356) of which could be recalled, while only 9% (8805) of the remaining 94 089 unreported pairs were predicted to be ERα-associated interaction clusters. Therefore we conclude that our classifier is highly effective in predicting known ChIA-PET interactions from multiple ChIP-seq data.
Many novo predicted interactions are supported by other data sources
Next, we compared our newly predicted 8805 interactions with other sources of data to confirm their biological significance. They are significantly overlapping with known Pol2 PETs clusters (33) (P < 2.2e−16) (Figure 5A and Supplementary Table S5). And the overlapping proportion was comparable with that of 3113 high-confidential ERBS–ERBS pairs reported in (15) (Figure 5A and Supplementary Table S5). This observation suggested that our predicted novel ERBS interactions might contain a considerate proportion of true positives that were not captured by the published ChIA-PET analysis (15), probably due to the fact that the original ERα experiments were not saturated and many interactions were filtered out to reduce false positives.
Figure 5.
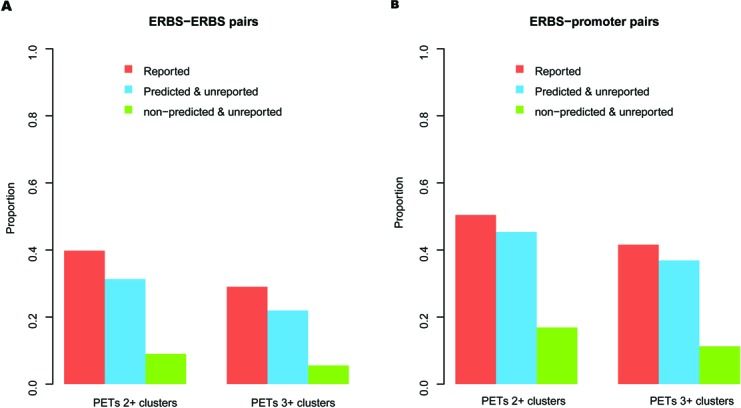
Evaluation of novo predicted interactions by other independent data sets. (A) Proportion of reported, predicted and unreported and rest of the candidate (non-predicted and unreported) ERBS–ERBS pairs that overlapped with Pol2 PETs 2+ and PETs 3+ clusters, respectively. (B) Proportion of reported, predicted and unreported and rest of the candidate (non-predicted and unreported) ERBS–promoter pairs that overlapped with Pol2 PETs 2+ and PETs 3+ clusters, respectively.
New ERα target genes are predicted
There are thousands of ERBSs in the genome (21), but only a small fraction of them (10–15% in different experiments) are located in regions proximal to gene TSS (±1500 bp). ChIA-PET experiments validated that a considerate percentage of ERBSs (∼10%) function via long-range interactions to their target promoters (15). Here we further extended our classifier to predict potential target genes regulated by the distal ERα-bound enhancers but without direct ERα binding at the promoters, which were hard to detect with traditional ChIP-seq analysis methods. Candidate promoters were selected as those that contained at least one of the FoxA1, GATA3 or AP2γ ChIP-seq binding peaks, but not ERBSs. Again, we restricted all those promoter–ERBS pairs within the topological domains. The classifier identified 507 pairs of ERBS–promoter interactions, associated with 374 genes. These predicted interactions were significantly overlapping with Pol2 PETs clusters (P < 2.2e−16) as expected (Figure 5B and Supplementary Table S6), and similar to the 113 ERBS–promoter pairs (without ERBS at promoters and restricted in topological domains) reported in (15) (Figure 5B and Supplementary Table S6). From the gene expression data (GSE11352), we found those genes up-regulated at 4 h, 12 h and 24 h were all significantly enriched in the predicted genes than in rest of the candidate genes, while the down-regulated genes were not (Supplementary Table S7). Interestingly, some well-known ERα target genes that have been reported in the literature but not detected in the original ERα ChIA-PET study appeared in our prediction. For example, 3C experiments confirmed that CTSD's promoter (Figure 6A) can form a loop with the −9 kb upstream ERBS containing enhancer (39). CTSD is important for tumor progression and in metastasis and is used as a specific biomarker in breast cancer diagnosis (40). XBP1 (Figure 6B), a TF involved in the unfolded protein response (41), is also an estrogen-regulated gene and its expression is strongly correlated with ERα expression in breast cancer. Sengupta et al. (41) have reported that the −13-kb enhancer upstream of the XBP1 promoter is an E2-response regulatory element that can functionally regulate XBP1 gene expression as we predicted. These observations suggest that our classifier can reliably predict ERα target genes regulated by distal elements while the common ChIP-seq data analysis has failed.
Figure 6.

Genome browser view by IGV tools (42, 43) of predicted loops around CTSD and XBP1 gene. (A) predicted interaction between promoter of CTSD gene and a -9kb upstream ERBS. (B) predicted interaction between promoter of XBP1 gene and a -13kb upstream ERBS.
DISCUSSION
Long-range interaction is an important and complex mechanism that regulates gene expression in space and time. With 3C-based technologies, many functional long-range interactions can be detected genome-widely. Among them, ChIA-PET can provide a more detailed view of whole genome interactions for a given TF. Our comprehensive reanalysis of ChIA-PET data revealed invariant characteristic distance features between different regulatory elements. Such distance features are largely unchanged across different cell types for EE interactions (5–6 kb), PP interactions (60–80 kb) and II interactions (160–200 kb). These characteristic distance features may reflect the underlying invariant properties of the structural organization of these regulatory elements in 3D chromatin DNA.
Our integrative analysis of histone modification and DNase-seq data showed that, although some ERα DNA binding sites may not be sensitive to nucleosomes, laERBSs are often found in open chromatin regions. This implies those ERBSs that are involved in specific long-range regulatory interactions with their target genes may be strongly dependent on local open chromatin structure in order to accommodate sophisticated protein complexes through 3D DNA looping. We expect that this insight may be generally applicable to many other different TFs, where nucleosome eviction can act as a predictive mark to distinguish loop-associated transcription factor binding sites (TFBSs) from solo TFBSs.
Another sex hormonal receptor, the androgen receptor (AR), is a TF that is very similar to ERα, as they both cooperate with FoxA1 for binding. A previous paper (22) reports that their binding sites have different chromatin accessibility patterns: AR favors pre-defined nucleosome-depleted regions, while ERα does not. However, here we showed that laERBSs, especially those with an ERE, have a similar static open chromatin structure just like most AR binding sites, indicating a more general long-range regulation mechanism by nuclear hormone receptors.
By extracting features from multiple ChIP-seq data sets of histone modification and TF binding profiles, we have developed classifiers to predict laERBSs and significantly interacting ERBS pairs. The ERα ChIP-seq signal, distance of the neighboring ERBS and DNase-seq signals are the most predictive features for laERBS, while other features are more or less redundant with these three ones. The restrictions of interactions between predicted laERBSs and within Hi-C-defined topological domains have proved to be very effective filters. Our final trained logistic classifier can not only recover a large percentage (76%) of reported ERα interactions but also predict many novel ERBS pairs that are validated by independent 3C or ChIA-PET experiments. We also applied our model to predict ERBS–promoter interactions. Some newly predicted ERα target genes, whose promoters were not directly bound by ERα and hence undetected by common ChIP-seq analysis, can also be linked with the E2-induction process or breast cancer pathways through other experimental supporting evidence. This in turn validates our model. The method we have described here should be applicable to other TFs, such as AR.
There are still two main potential limitations of our model. One is that we used topological domains in H1 cells, not in MCF7 cells due to lack of the data, to filter ERBS pairs before interaction prediction. Although it is reported that topological domains are largely conserved between different cell types and only a small proportion of ERα ChIA-PET-detected interactions in MCF7 cells were filtered out. Using topological domains detected in the same cell type would be able to further improve the performance. The other is that our predictions depend on multiple ChIP-seq data, especially those of ERα's co-factors, which is not always available in other types of cells. Overall, our work indicates that integrative analysis of ChIP-seq, Hi-C, ChIA-PET data, etc. could overcome the limits of each single method and provide a more comprehensive understanding of the chromatin interaction landscape at multi-scales.
CONCLUSION
In this work, we carried out a comprehensive analysis of ERα ChIA-PET data by combining gene expression, TF binding, histone modification profiles and open chromatin conformation data together. We showed that laERBSs were more likely to be nucleosome depleted compared with nlaERBSs. They were also significantly overlapping with FoxA1, GATA3, AP2γ, and p300 ChIP-seq binding peaks. An efficient classifier was developed to predict laERBSs and chromatin interactions between these laERBSs. Among all the features, the ERα ChIP-seq signal, distance of the neighboring ERBS and DNase-seq signals are the most predictive features for laERBSs. When predicting ERBS interactions, the restriction within Hi-C determined topological domains is very effective to filter many potential false positives. The logistic classifier we trained can recover a large percentage (76%) of ChIA-PET experiment identified ERα interactions. Besides, many of our predicted novo ERBS interactions could be validated by independent 3C or other ChIA-PET data sets. The model was applied to predict the interactions between distal ERBS and promoter. We found that some newly predicted ERα target genes whose promoters did not overlap with ERBSs were associated with breast cancer related gene ontology items. Comparing with traditional analysis of ChIP-seq and ChIA-PET data, our integrative analysis and predictive model can provide a better understanding of the chromatin long range interactions.
ACCESSION NUMBERS
Data used in this study are available in NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession numberGSE11352, GSE18046, GSE23701, GSE23852, GSE29073, GSE33216, GSE39495, GSM678539 and GSM678540.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Monica Sleumer for helpful discussion and checking the proofreading.
FUNDING
National Basic Research Program of China [2012CB316503]; National Natural Science Foundation of China [91019016, 61322310, 31371341, 31361163004]; Foundation for the Author of National Excellent Doctoral Dissertation of PR China [201158]; National Institutes of Health [HG001696, ES017166 to M.Q.Z.]. Funding for open access charge: National Basic Research Program of China [2012CB316503].
Conflict of interest statement. None declared.
REFERENCES
- 1.Barton M., Emerson B. Regulated expression of the beta-globin gene locus in synthetic nuclei. Genes Dev. 1994;8:2453–2465. doi: 10.1101/gad.8.20.2453. [DOI] [PubMed] [Google Scholar]
- 2.Forrester W., Fernández L., Grosschedl R. Nuclear matrix attachment regions antagonize methylation-dependent repression of long-range enhancer–promoter interactions. Genes Dev. 1999;13:3003–3014. doi: 10.1101/gad.13.22.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter D., Chakalova L., Osborne C., Dai Y., Fraser P. Long-range chromatin regulatory interactions in vivo. Nat. Genet. 2002;32:623–626. doi: 10.1038/ng1051. [DOI] [PubMed] [Google Scholar]
- 4.Tolhuis B., Palstra R., Splinter E., Grosveld F., de Laat W. Looping and interaction between hypersensitive sites in the active β-globin locus. Mol. Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 5.Jing H., Vakoc C., Ying L., Mandat S., Wang H., Zheng X., Blobel G. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol. Cell. 2008;29:232–242. doi: 10.1016/j.molcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majumder P., Boss J. CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol. Cell. Biol. 2010;30:4211–4223. doi: 10.1128/MCB.00327-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dean A. In the loop: long range chromatin interactions and gene regulation. Brief. Funct. Genomic. 2011;10:3–10. doi: 10.1093/bfgp/elq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chien R., Zeng W., Kawauchi S., Bender M., Santos R., Gregson H., Schmiesing J., Newkirk D., Kong X., Ball A., et al. Cohesin mediates chromatin interactions that regulate mammalian β-globin expression. J. Biol. Chem. 2011;286:17870-17878. doi: 10.1074/jbc.M110.207365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chepelev I., Wei G., Wangsa D., Tang Q., Zhao K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012;22:490–503. doi: 10.1038/cr.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Liang J., Li Y., Xuan C., Wang F., Wang D., Shi L., Zhang D., Shang Y. CCCTC-binding factor acts upstream of FOXA1 and demarcates the genomic response to estrogen. J. Biol. Chem. 2010;285:28604–28613. doi: 10.1074/jbc.M110.149658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dekker J., Rippe K., Dekker M., Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 12.Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., De Wit E., Van Steensel B., De Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 13.Dostie J., Richmond T., Arnaout R., Selzer R., Lee W., Honan T., Rubio E., Krumm A., Lamb J., Nusbaum C., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieberman-Aiden E., Van Berkum N., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B., Sabo P., Dorschner M., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289-293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fullwood M., Liu M., Pan Y., Liu J., Xu H., Mohamed Y., Orlov Y., Velkov S., Ho A., Mei P., et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borggrefe T., Yue X. Interactions between subunits of the Mediator complex with gene-specific transcription factors. 2011;22:759–768. doi: 10.1016/j.semcdb.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Young R. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson K., Bresnick E. Dissecting long-range transcriptional mechanisms by chromatin immunoprecipitation. Methods. 2002;26:27–36. doi: 10.1016/S1046-2023(02)00005-1. [DOI] [PubMed] [Google Scholar]
- 19.Ruh M., Chrivia J., Cox L., Ruh T. The interaction of the estrogen receptor with mononucleosomes. Mol. Cell. Endocrinol. 2004;214:71–79. doi: 10.1016/j.mce.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 20.Feingold E., Good P., Guyer M., Kamholz S., Liefer L., Wetterstrand K., Collins F., Gingeras T., Kampa D., Sekinger E., et al. The ENCODE (ENCyclopedia of DNA elements) project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 21.Joseph R., Orlov Y., Huss M., Sun W., Kong S., Ukil L., Pan Y., Li G., Lim M., Thomsen J., et al. Integrative model of genomic factors for determining binding site selection by estrogen receptor- Mol. Syst. Biol. 2010;6:456. doi: 10.1038/msb.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He H., Meyer C., Chen M., Jordan V., Brown M., Liu X. Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res. 2012;22:1015–1025. doi: 10.1101/gr.133280.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan S., Lin Z., Chang C., Varang V., Chng K., Pan Y., Yong E., Sung W., Cheung E. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J. 2011;30:2569–2581. doi: 10.1038/emboj.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong S., Li G., Loh S., Sung W., Liu E. Cellular reprogramming by the conjoint action of ERα, FOXA1, and GATA3 to a ligand-inducible growth state. Mol. Syst. Biol. 2011;7:526. doi: 10.1038/msb.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hah N., Murakami S., Nagari A., Danko C., Kraus W.L. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223. doi: 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hua S., Kallen C., Dhar R., Baquero M., Mason C., Russell B., Shah P., Liu J., Khramtsov A., Tretiakova M., et al. Genomic analysis of estrogen cascade reveals histone variant H2A. Z associated with breast cancer progression. Mol. Syst. Biol. 2008;4:188. doi: 10.1038/msb.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y., Liu T., Meyer C., Eeckhoute J., Johnson D., Bernstein B., Nussbaum C., Myers R., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng J., Liu T., Zhang Y. Using MACS to identify peaks from ChIP-Seq data. 2011;Chapter 2:2–14. doi: 10.1002/0471250953.bi0214s34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benaglia T., Chauveau D., Hunter D.R., Young D. mixtools: an R package for analyzing finite mixture models. J. Stat. Softw. 2009;32:1–29. [Google Scholar]
- 30.Schones D.E., Smith A.D., Zhang M.Q. Statistical significance of cis-regulatory modules. BMC Bioinformatics. 2007;8:19. doi: 10.1186/1471-2105-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon J., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ringrose L., Chabanis S., Angrand P., Woodroofe C., Stewart A. Quantitative comparison of DNA looping in vitro and in vivo: chromatin increases effective DNA flexibility at short distances. EMBO J. 1999;18:6630–6641. doi: 10.1093/emboj/18.23.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G., Ruan X., Auerbach R., Sandhu K., Zheng M., Wang P., Poh H., Goh Y., Lim J., Zhang J., et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li G., Fullwood M., Xu H., Mulawadi F., Velkov S., Vega V., Ariyaratne P., Mohamed Y., Ooi H., Tennakoon C., et al. Software ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11:R22. doi: 10.1186/gb-2010-11-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim T.H., Abdullaev Z.K., Smith A.D., Ching K.A., Loukinov D.I., Green R.D., Zhang M.Q., Lobanenkov V.V., Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanstein B., Eckner R., DiRenzo J., Halachmi S., Liu H., Searcy B., Kurokawa R., Brown M. p300 is a component of an estrogen receptor coactivator complex. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11540–11545. doi: 10.1073/pnas.93.21.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luebben W.R., Sharma N., Nyborg J.K. Nucleosome eviction and activated transcription require p300 acetylation of histone H3 lysine 14. Proc. Natl. Acad. Sci. U.S.A. 2010;107:19254–19259. doi: 10.1073/pnas.1009650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D., Garcia-Bassets I., Benner C., Li W., Su X., Zhou Y., Qiu J., Liu W., Kaikkonen M., Ohgi K., et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bretschneider N., Kangaspeska S., Seifert M., Reid G., Gannon F., Denger S. E2-mediated cathepsin D (CTSD) activation involves looping of distal enhancer elements. Mol. Oncol. 2008;2:182–190. doi: 10.1016/j.molonc.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Byun H., Han N., Lee H., Kim K., Ko Y., Yoon G., Lee Y., Hong S., Lee J. Cathepsin D and eukaryotic translation elongation factor 1 as promising markers of cellular senescence. Cancer Res. 2009;69:4638–4647. doi: 10.1158/0008-5472.CAN-08-4042. [DOI] [PubMed] [Google Scholar]
- 41.Sengupta S., Sharma C.G., Jordan V.C. Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Horm. Mol. Biol. Clin. Invest. 2010;2:235–243. doi: 10.1515/HMBCI.2010.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.T Robinson James, Thorvaldsdóttir Helga, Winckler Wendy, Guttman Mitchell, S Lander Eric, Getz Gad, P Mesirov Jill. Integrative genomics viewer. Nature biotechnology. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorvaldsdóttir Helga, T Robinson James, P Mesirov Jill. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.