


Abstract
Identification of primary microRNA (miRNA) gene targets is critical for developing miRNA-based therapeutics and understanding their mechanisms of action. However, disentangling primary target derepression induced by miRNA inhibition from secondary effects on the transcriptome remains a technical challenge. Here, we utilized RNA immunoprecipitation (RIP) combined with competitive binding assays to identify novel primary targets of miR-122. These transcripts physically dissociate from AGO2-miRNA complexes when anti-miR is spiked into liver lysates. mRNA target displacement strongly correlated with expression changes in these genes following in vivo anti-miR dosing, suggesting that derepression of these targets directly reflects changes in AGO2 target occupancy. Importantly, using a metric based on weighted miRNA expression, we found that the most responsive mRNA target candidates in both RIP competition assays and expression profiling experiments were those with fewer alternative seed sites for highly expressed non-inhibited miRNAs. These data strongly suggest that miRNA co-regulation modulates the transcriptomic response to anti-miR. We demonstrate the practical utility of this ‘miR-target impact’ model, and encourage its incorporation, together with the RIP competition assay, into existing target prediction and validation pipelines.
INTRODUCTION
microRNAs (miRNAs) are short non-coding RNAs that negatively regulate gene expression by destabilizing target transcripts through recruitment of RNA induced silencing complexes (RISCs) (1). Importantly, aberrant miRNA activity has been implicated in a number of diseases including diabetes, cancer and autoimmune diseases (2–4).
miRNA inhibition with chemically modified oligonucleotides that are complementary to miRNA (anti-miR) is a promising new area of therapeutic development (5–7). Because each miRNA can potentially regulate several hundred targets, pharmacological modulation of a single miRNA can affect numerous cellular pathways through both primary as well as secondary changes in the transcriptome (8–10). This far-reaching activity gives anti-miRs the potential to treat complex diseases that are otherwise difficult to remedy with traditional small molecule approaches whose actions are more limited in scope. It also creates, however, a technical challenge in deciphering direct from indirect effects. Direct targets serve as pharmacodynamic (PD) markers for assessing the potency and efficacy of anti-miR, which is essential for preclinical drug development.
Numerous computational methods are available to predict miRNA targets (11–14). The predominant target feature is the presence of a seven- or eight-nucleotide sequence within the 3′UTR of candidate genes that is complementary to the 5′end of a miRNA (the ‘seed region’). These seed sites tend to be highly conserved and a wealth of biochemical data supports that they comprise the majority of miRNA-target interactions (1). However, in part due to the low sequence specificity of seed sequences, computational predictions often suffer from high false-positive rates (15). Therefore, while upregulation of seed-matched genes in response to anti-miR treatment represents an on-target effect overall, some expression changes in seed-matched genes may be merely coincidental. Experimental validation is required to identify direct targets, yet to our knowledge no existing approaches have been developed specifically for target identification using anti-miRs in mammalian animal models.
We have now developed a RNA immunoprecipitation (RIP) based anti-miR competition assay for validating primary targets in situ using animal tissues with medium-to-high throughput. As a proof of concept study for this assay, we validated in mouse liver samples several novel direct targets of endogenous miR-122—a therapeutically relevant miRNA for liver disease (5,16–17). Most importantly, we discovered that genes predicted to be highly regulated by multiple miRNAs are less likely to respond to anti-miR directed against a single miRNA. This effect of alternative miRNAs has until now only been suggested, but not experimentally confirmed nor quantified. Based on these findings, we devise a ‘miR-Target Impact Model’ for use in directing and focusing efforts in miRNA validation and development of future anti-miR therapeutics.
MATERIALS AND METHODS
Animal care and treatments
All animal experiments were conducted according to the Institutional AAALAC Guidelines. Male C57BL/6 mice were housed four to five animals per cage with a 12 h light/dark cycle. For gene expression studies, oligonucleotides were dissolved in 1× phosphate buffered saline (PBS) and administered to mice by subcutaneous injection at 3 mg/kg on Day 1 and Day 3. On Day 7, livers were harvested and gene expression was measured by Nanostring or array profiling.
Array profiling
mRNA expression profiles were measured using Mouse Genome 430 2.0 or Human Genome U133 Plus 2.0 arrays (Affymetrix). mRNA microarrays were run in triplicate for anti-miR- or saline-treated mice, and processing was performed using Bioconductor for R (bioconductor.org) (18), specifically the limma package for differential expression analysis (19). Sylamer analysis (10) was performed using SylArray web server (http://www.ebi.ac.uk/enright-srv/sylarray) (20). For miR-221/222 expression studies, SKHep-1 cells were transfected in triplicate with 20 nM anti-221 using RNAiMax (Life Technologies), and cells were collected 48 h post-transfection.
Liver and SKHep-1 lysates
Liver lysates were prepared in lysis buffer (20 mM Tris, pH 7.4, 100 mM NaCl and 2.5 mM MgCl2) supplemented with ethylenediaminetetraacetic acid (EDTA)-free Halt Protease Inhibitor cocktail (ThermoFisher). Intact livers were homogenized with a glass Dounce in ice-cold lysis buffer by 20 strokes with each of the loose and tight plungers. The homogenate was centrifuged at 1000×g for 10 min at 4°C. The resulting supernatant was then centrifuged twice at 16 000×g for 10 min at 4°C to produce the final S16 lysate. Protein concentrations were measured by DS Protein Assay (BioRad) and adjusted to 10 mg/ml. Aliquots of lysate were flash frozen and stored at −80°C. SKHep-1 cultures were maintained in Eagle's Minimum Essential Medium with 10% fetal bovine serum. Cells were collected by scraping, resuspended in lysis buffer containing 0.05% NP-40, and homogenized by passing through a 27-gauge needle.
miR-target impact scoring (mTIS)
Sequence data for all mouse (GRCm38/mm10) and human (GRCh37/hg19) Refseq genes were downloaded from the UCSC Genome Bioinformatics Table Browser (http://genome.ucsc.edu/) (21). 3′UTR sequences were parsed based on annotated coding regions using Matlab (v7.12, Mathworks, Inc) scripts written in-house. For all 3′UTRs, miRNA binding sites were identified using seed sequences downloaded from TargetScan v6.2 (targetscan.org; ‘miR Family’ file). For all k genes containing a seed-match site to a given miRNA of interest (MOI), mTIScores were calculated by rewarding for number of MOI sites (Nmoi) while penalizing for other miRNA sites (Ni−j), such that
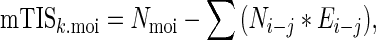 |
where E is the linear expression level of each alternative miRNA relative to the moi (Ei−j = ei−j/emoi). Summed expression levels were used for miRNAs within the same family, i.e. with identical seed sequences.
RIP competition assay
Hybridoma cells expressing monoclonal anti-AGO2 4F9 antibody were a generous gift from E. Chan (University of Florida, Gainesville) (22). Antibody-containing supernatant was dialyzed in binding buffer (0.1 M phosphate, 0.15 M NaCl; pH 7.2) and purified using protein-L agarose (Pierce), as instructed by the manufacturer. Purified antibody was conjugated to magnetic M-270 Epoxy Dynabeads (Life Technologies) as per manufacturer's instructions. For each IP, 6 µg antibody-conjugated beads were added to 250 µl lysate following pre-incubation with anti-miR oligonucleotides for 30 min at room temperature, at which point RNA remained stably intact (Supplementary Figure S1A and B). IP reactions were tumbled at 4°C for 2 h and subsequently washed ×3 in wash buffer (20 mM Tris, pH 8.0, 140 mM KCl, 5 mM EDTA, 1× Halt Protease Inhibitor (ThermoFisher), 40U/ml RnaseOut (Life Technologies) and 0.5 mM DTT). RNA was purified with either RNeasy micro or RNeasy 96-well kits (Qiagen). In both cases, 20 ng polyA+ carrier RNA (Qiagen) was added to aid RNA retention during purification. mRNA was quantified with reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) or Nanostring nCounter technology (Nanostring Technologies). Data were normalized using the geometric mean of the top two most stable non-target genes, as described elsewhere (23,24). RNA integrity was assessed with an Agilent 2100 Bioanalyzer.
nCounter expression analysis
A custom nCounter Gene Expression CodeSet was purchased from Nanostring Technologies. For IP samples, RNA was purified with RNeasy min-elute spin-columns and eluted with 16 µl ddH2O. For total RNA samples, input RNA concentrations were adjusted to 20 ng/µl. A volume of 5 µl was used for input and hybridized according to the manufacturer's protocol. For miRNA expression analysis, nCounter miRNA expression assays were used with total RNA samples purified using miRNeasy 96-well spin-plates (Qiagen).
RT-qPCR
Random cDNA was synthesized using a High Capacity cDNA Reverse Transcription (RT) kit (Applied Biosystems). In cases where RNA was purified with RNeasy min-elute spin-columns, 10 µl of the ∼14 µl eluant was added to RT master mix for a 20 µl final RT volume. Alternatively, when RNA was purified with the RNeasy 96-well spin-plates, 45 µl of the ∼50 µl eluant was added to RT master mix for a 90 µl final RT volume. After reverse transcription was complete, smaller volume RT reactions were diluted 1:5 with ddH2O while larger RT reactions were used as-is for input into qPCR. 2.0 µl cDNA was used for each 10 µl qPCR reaction prepared with Universal TaqMan Master Mix II without UNG (Applied Biosystems) and TaqMan primer/probesets (IDT).
RESULTS
RIP anti-miR competition assay
AGO2 is the central RISC component that binds miRNA complexes to target mRNA transcripts. Anti-miRs are synthetic oligonucleotides that bind miRNA and compete with target transcripts for AGO2-miRNA occupancy. To identify miRNA-target complexes that are sensitive to anti-miR (i.e. direct targets), we developed a simple RIP competition assay wherein anti-miR was titrated into cytosolic extracts prior to AGO2 immunoprecipitation (IP). Direct target transcripts displaced by anti-miR were then identified based on their depletion from IP fractions (Figure 1A).
Figure 1.

Development of AGO2-RNA Immunoprecipitation Competition Assay. (A) Schematic of the RIP-competition assay. Livers were extracted from mice and lysates were prepared ex vivo as described in the Materials and Methods section. In order to identify direct miRNA targets, lysates were pre-incubated with spiked-in anti-miR compound, causing miRNA inhibition and preventing dissociated target mRNA from re-associating with AGO2 complexes. Target mRNAs were identified as being under-enriched in AGO2 co-immunoprecipitated fractions in the presence of anti-miR (left side of panel) compared to PBS control (right side of panel). (B) Dose-dependent results for miR-122 target candidates Aldoa (blue), Dlat (red) and Gys1 (purple). mRNA levels were measured with RT-qPCR and fold-changes were calculated with the 2−ΔΔCt method relative to the geometric mean of genes Rnf167 and Ezh2 (gray), neither of which contain 3′UTR miR-122 seed sites. Data were fit with a competitive binding curve (for Aldoa: IC50 = 14.26 +/− 11.46 nM, bottom = 0.56, R2 = 0.64; for Dlat: IC50 = 477.1 +/− 391.3 nM, bottom = 0.71, R2 = 0.59; for Gys1: IC50 = 50.69 +/− 13.20 nM, bottom = 0.38, R2 = 0.94). (C) Western blots of AGO2 in input and IPs with anti-AGO2 or control (IgG) antibodies. (D) Quantification of Gys1 in AGO2-IPs or control by relative standard curve (compare to quantification method in panel (B)). (E) Comparison of Gys1 results obtained with different monoclonal antibodies recognizing AGO2 or TNRC6A, another RISC protein. (F) Comparison of Gys1 RIP competition results with various anti-miRs at 400 nM (top) or 4000 nM (bottom). Gys1 3′UTR contains three miR-122 sites but no sites for miR-22 or let-7.
To test whether our method could be used to identify direct targets in animal tissue, we used mouse liver extracts as a source of endogenous miR-122-AGO2 complex and benchmarked the assay using RT-qPCR to detect well-validated miR-122 targets Aldoa, Dlat and Gys1. After spiking in anti-miR-122 compound over a six-log concentration range, all three miR-122 targets showed dose-dependent depletion from AGO2-IPs (Figure 1B and C). In contrast, Ezh2 and Rnf167, which do not contain miR-122 3′UTR sites, remained stably bound in the IP fractions even at the highest doses, thus making them suitable as reference genes. Fitting these data with non-linear regression revealed differences between the miR-122 targets: while Aldoa and Gys1 exhibited similar IC50 values (∼10–50 nM), Dlat dissociated from AGO2 only at much higher anti-miR-122 concentrations (∼500 nM). The three targets also exhibited different maximum responses. Interestingly, a significant portion (∼38%) of Gys1, which showed the greatest response among the three targets, remained on the beads at even the highest anti-122 concentrations. Non-specific binding only accounted for a slim margin (∼4%) of this refractory population (Figure 1D), suggesting that under these conditions only certain portions of each AGO2-bound target are sensitive to anti-miR competition. Increasing pre-incubation time with anti-miR did not lead to enhanced target mRNA release (Supplementary Figure S1C). Furthermore, highly similar results were produced using different monoclonal antibodies against AGO2 or TNRC6A (GW182 homolog)—the effector protein of miRNA-mediated repression (25,26)— indicating that pulled-down complexes represented active RISC populations (Figure 1E). Currently, we are investigating the rationale of this phenomenon where only portions of total mRNA transcripts are seemingly susceptible to anti-miR-mediated competition.
Additional control experiments also showed that these responses were anti-miR-122 specific. Even at high concentrations, anti-miRs complementary to miR-22 and let-7 were ineffective by comparison, consistent with the lack of canonical 3′UTR sites for these alternative miRNAs (Figure1F).
We have thus developed a method that can directly identify target-miRNA complexes that are dose-sensitive to anti-miR under near-native conditions using relevant animal tissue.
Identification of novel anti-122 primary biomarkers
With the RIP competition assay in hand, we next sought to identify which target candidates from a shortlist of computational predictions best responded to anti-miR-122. A custom Nanostring nCounter codeset was designed with probes recognizing 37 genes predicted by the TargetScan algorithm (11) to be regulated by miR-122 (Table 1 (27–35)). An additional four genes without miR-122 sites were also included, among which the top two most stable genes were used as reference genes (Supplementary Figure S2). Similar to earlier results, these candidate transcripts were differentially displaced by anti-miR-122 from immunoprecipitated fractions (Figure 2A–C, Supplementary Figure S3). Based on the extent of displacement, we were able to classify candidate genes as either ‘RIP-responsive’ or ‘RIP-non-responsive’. Fourteen candidates (38%) showed statistically significant displacement with 400 nM anti-miR-122 compared to control (Figure 2A). An additional two candidates responded at the next highest tested anti-miR-122 concentration (4000 nM; Figure 2B), making for a total of 16 candidates (43%) that could be confirmed as being anti-miR-122 sensitive. These targets are consequently likely to be direct targets of miR-122. Consistently, all but one RIP-responsive gene (94%) showed significant derepression in vivo following anti-miR-122 treatment (Figure 2E). Notably, expression changes of RIP responsive genes were very strongly correlated (r = −0.91) with levels of RIP response (Figure 2D), indicating that changes in expression in vivo are proportional to the amount of target RNA released from RISC complexes by anti-miR. In contrast, a similar correlation did not exist with RIP-non-responsive candidates. Most non-responsive genes were also not significantly upregulated following in vivo anti-miR-122 treatment (Figure 2E). There were, however, a few exceptions: a total of nine genes were significantly upregulated, but RIP-non-responsive at any tested anti-miR-122 concentration (summarized in Table 1). Potentially, expression changes in these latter genes occur as a result of secondary effects of miR-122 inhibition, rather than as a direct consequence. Therefore, despite the fact that these genes contain a miR-122 seed-match and are upregulated in the presence of anti-122, they are less likely to be direct biomarkers of miRNA modulation. These results demonstrate how RIP competition assays can serve as a filter combined with expression analysis to assist in identifying high-confidence primary anti-miR PD markers.
Table 1. Summary of results, computational scores and properties of miR-122 target candidates assessed in this study.
| Gene symbol | RefSeq ID | #122 sites | TS context+ score prct | 3′UTR length (nt) | mTIScore | Avg RIP FC (400 nM) | Sig RIP | Avg exp FC | Sig exp. | Previously inferred | Previously validated | Validated here |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aldoa | NM_007438 | 1 | 98 | 195 | 0.86 | 0.44 | * | 4.97 | * | (30) | (31); (32) | Y |
| Ankrd13c | NM_001013806 | 1 | 89 | 992 | 0.79 | 0.59 | * | 2.81 | * | (11) | Y | |
| Brpf1 | NM_030178 | 1 | 96 | 552 | 0.68 | 0.87 | 1.52 | |||||
| Calm3 | NM_007590 | 1 | 77 | 1595 | 0.38 | 0.96 | 1.53 | * | ||||
| Ccng1 | NM_009831 | 1 | 97 | 2331 | 0.16 | 0.94 | 2.86 | * | (33,); (34) | |||
| Cd320 | NM_019421 | 2 | 67 | 1347 | 1.76 | 0.55 | * | 3.76 | * | (31) | Y | |
| Cdc42bpb | NM_183016 | 1 | 46 | 1130 | 0.37 | 1.00 | 1.29 | |||||
| Clic4 | NM_013885 | 2 | 82 | 3133 | 0.51 | 1.02 | 2.08 | * | (11) | (34) | ||
| Cs | NM_026444 | 1 | 83 | 1348 | 0.70 | 0.67 | * | 2.54 | * | (32) | Y | |
| Ctdnep1 | NM_026017 | 2 | 93 | 565 | 1.94 | 0.79 | 1.94 | * | ||||
| Dicer1 | NM_148948 | 1 | 84 | 3852 | −0.29 | 0.94 | 1.17 | |||||
| Dlat | NM_145614 | 2 | 90 | 1809 | 1.19 | 0.78 | 1.28 | |||||
| Efha2 | NM_030110 | 1 | 98 | 1697 | 0.65 | 0.81 | *,† | 2.49 | * | Y† | ||
| Eif2c1 | NM_153403 | 1 | 10 | 4492 | −1.10 | 0.72 | 1.24 | |||||
| Foxk2 | NM_001080932 | 1 | 73 | 2933 | 0.55 | 1.10 | 1.73 | * | (11) | |||
| Foxp2 | NM_212435 | 3 | 82 | 3957 | 1.35 | 1.04 | 2.37 | |||||
| Fundc2 | NM_026126 | 1 | 98 | 2772 | 0.43 | 0.89 | 1.24 | (32) | ||||
| G3bp2 | NM_001080794 | 1 | 17 | 2636 | 0.12 | 1.04 | 1.72 | * | ||||
| Git1 | NM_001004144 | 2 | 82 | 1187 | 1.33 | 0.68 | * | 1.61 | Y | |||
| Gpr172b | NM_029643 | 1 | 97 | 1005 | 0.87 | 0.69 | * | 2.16 | * | Y | ||
| Gys1 | NM_030678 | 3 | 92 | 1251 | 2.77 | 0.46 | * | 3.16 | * | (30); (11) | Y | |
| Hnrnpu | NM_016805 | 1 | 95 | 1055 | 0.75 | 0.84 | 1.27 | |||||
| Kif5b | NM_008448 | 1 | 41 | 2722 | 0.15 | 0.84 | *,† | 1.89 | * | Y† | ||
| Lcorl | NM_001163073 | 1 | 56 | 3125 | 0.43 | 1.19 | 1.31 | |||||
| Lrp10 | NM_022993 | 1 | 56 | 444 | 0.93 | 0.70 | * | 1.85 | * | Y | ||
| Maf1 | NM_026859 | 1 | 95 | 511 | 0.72 | 0.67 | * | 2.70 | * | Y | ||
| Mapre1 | NM_007896 | 1 | 79 | 6388 | −0.69 | 1.02 | 1.79 | * | (11) | |||
| Mcart1 | NM_001009949 | 2 | 29 | 3319 | 1.06 | 0.69 | * | 1.97 | * | Y | ||
| Mmgt1 | NM_146234 | 1 | 86 | 3633 | 0.10 | 1.14 | 1.26 | |||||
| Npepps | NM_008942 | 1 | 97 | 1281 | 0.85 | 0.85 | 1.70 | * | (11) | |||
| Ocln | NM_008756 | 1 | 98 | 1448 | 0.80 | 0.84 | 1.76 | * | ||||
| Orc2 | NM_008765 | 1 | 97 | 1434 | 0.66 | 0.66 | * | 2.24 | * | (11) | Y | |
| P4ha1 | NM_011030 | 2 | 98 | 2245 | 1.54 | 0.62 | * | 2.07 | * | Y | ||
| Rbm47 | NM_001127382 | 1 | 68 | 2650 | 0.60 | 1.06 | 1.48 | |||||
| Slc25a34 | NM_001013780 | 3 | 99 | 1589 | 2.67 | 0.36 | * | 6.68 | * | Y | ||
| Slc41a1 | NM_173865 | 3 | 87 | 2144 | 2.58 | 0.64 | * | 2.75 | * | Y | ||
| Zbtb41 | NM_172643 | 1 | 55 | 5478 | −0.20 | 0.91 | 1.78 | (11) |
Note that Nanostring probes were designed to hit all mRNA isoforms. For computing scores for genes with multiple isoforms, isoforms with the longest annotated 3′UTRs were used. TargetScan context+ score percentiles (TS Context+ Score Prct) were derived from the TargetScan database (11,45). Candidate genes that have been previously inferred (by gene expression analysis only) or previously validated (gene expression plus a secondary assay (e.g. luciferase, AGO-IP, etc.)) as miR-122 targets in mouse or human are marked by citation(s) to the original work(s). These citations were compiled by cross-referencing curated miRNA–mRNA target interactions in miRecords (27), miRWalk (28) and miRTarBase (29) databases.
*Statistical significance for RIP (Sig RIP) or expression (Sig Exp) changes are based on methods described in Figure 2.
†These genes responded significantly in RIP competition assays only at the highest (4000 nM) anti-122 concentration tested. Others marked as significant responded at ≤400 nM.
Figure 2.

Validation of novel miR-122 primary targets in mouse liver. AGO2-RIP competition responses of 37 miR-122 target candidates measured by Nanostring nCounter technology in the presence of (A) 400 nM or (B) 4000 nM anti-122. Fold-changes are relative to PBS. Green bars mark genes with statistically significant reduction of mRNA in IP with treatment (P < 0.05 by ANOVA with Dunnett correction for multiple comparisons, n = 3, error bars represent SEM). Results were normalized based on the geometric mean of Rnf167 and Nras, the most stable of all reference genes tested (Supplementary Figure S2). (C) Summary of genes significantly reduced in IP with varying doses of anti-122, represented as fraction of total (N = 37). (D) Correlation between RIP responses at 400 nM anti-122 and in vivo gene expression changes measured by Nanostring. Significantly upregulated genes resulting from anti-122 administration (unpaired t-test, FDR = 5%, n = 3) are shown in pink, others in gray. Statistical significance from RIP experiments is indicated by squares (significant) and circles (non-significant). Genes with significant RIP responses (grouped with a black outline) were fit by least-squares linear regression (r = −0.91, P (one-tailed) < 0.0001, Pearson correlation). In contrast, genes with non-significant RIP responses were poorly correlated with gene expression (r = −0.04, P = 0.4313). (E) Fraction of significantly upregulated genes with significant (pink; Ntotal = 16) or non-significant (gray; Ntotal = 21) RIP responses at any tested anti-122 concentration. Additional data are in Supplementary Figure S2-S3.
RIP target response is explained by miR-target impact model
RIP competition experiments were able to differentiate miR-122 seed-matched genes based directly on their sensitivity to anti-miR-122 competition. We next wanted to identify factors that influenced these outcomes. We hypothesized two influencing factors: first, we noticed that many of the top responding targets, such as Slc25a34 and Gys1, contained more than one miR-122 seed-match site (Table 1). The presence of multiple seed sites is well known to increase the extent of miRNA target regulation (11,36–37); presumably then, a greater magnitude of derepression or IP depletion may be expected with multi-seed targets. Second, since most gene transcripts contain many different seed-match sequences, they can potentially be regulated simultaneously by distinct miRNAs (38–40). Therefore, the presence of alternative, non-inhibited miRNAs may have retained target mRNA in IP fractions. This may at least partially explain why inhibition of only one miRNA, miR-122, was insufficient to release certain targets from the majority of bound RISCs (illustrated in Figure 3A).
Figure 3.

Co-regulation by alternative miRNAs strongly influences the effect of anti-miR. (A) Modified AGO2-RIP competition schematic illustrating the hypothesis that target mRNA is retained by alternative miRNAs. (B) Relative expression of miRNA profiled in mouse liver lysates. Expression levels were used for weighting the impact of alternative miRNAs. (C) Relationship between RIP responses at 400 nM anti-122 (y-axis) and miR-target impact score (mTIS; x-axis; see text for scoring details). Pearson coefficient and corresponding P-value are shown. (D) Receiver Operating Characteristic (ROC) curves for classification of responsive (IP fold-change ≤ 0.75) and non-responsive (IP fold-change > 0.75) targets based on mTIS, TargetScan, or 3′UTR length. As controls, mTIScores were computed using shuffled 3′UTR sequences (‘Scrambled’) or out-of-context miRNA expression levels (‘Hep3B’). Black dashed line on the diagonal represents expected result for indiscriminate, purely random classification. Areas under each curve (AUC) are plotted in the inset with a theoretical range from AUC = 0.5 (poor classifier) to AUC = 1.0 (perfect classifier). Only curves for mTIS and 3′UTR length, which are auto-correlated, were statistically significant as being non-random (mTIS: P = 0.0015; 3′UTR length: P = 0.017).
To assess both these factors in combination, we derived a quantitative model that ranked genes by rewarding for number of predicted miR-122 (MOI) seed sites while penalizing for number of predicted alternative sites (Materials and Methods). In addition, each alternative site was weighted based on expression level in liver tissue relative to miR-122 (Figure 3B), since an alternative miRNA's impact is likely a function of its abundance. We termed this metric ‘miR-target impact scoring’ (mTIScores) as it is an estimate of a given miRNA's repression potential on a target relative to all other miRNAs. Based on this concept, we hypothesized that genes with higher mTIScores for a particular miRNA are more likely to be sensitive to inhibition of that miRNA, whereas genes with lower mTIScores are less likely to be sensitive to miRNA inhibition. The presence of many alternative sites could in essence dilute the effects of any one miRNA.
Consistent with this hypothesis, a moderately strong correlation was observed between miR-122 target mTIScore and RIP response at 400 nM (r = −0.59, P < 0.0001; Figure 3C) and 4000 nM anti-miR-122 (r = −0.51, P = 0.0013; Supplementary Figure S4A). Furthermore, mTIScores were a strong classifier of RIP response, outperforming scrambled controls, TargetScan scoring (which does not take alternative miRNAs into account) and 3′UTR length alone (Figure 3D). Notably, all predictive power was lost when control mTIScores were derived using out-of-context miRNA expression data from the hepatocellular carcinoma Hep3B cell line, where miRNA expression is markedly different from normal liver (Figure 3D). Taken together with the strong correlation between RIP response and expression changes (Figure 2D), these data indicate that anti-miR has the greatest effect on mRNAs that are strongly regulated by a single miRNA, and presence of additional regulators mitigates these effects.
Additionally, we analyzed whether our model would benefit from including seed sites located within the coding sequence (CDS) of target genes. Previous efforts using cross-linking and immunoprecipitation followed by deep-sequencing (CLIP) have shown that AGO2 is capable of binding transcripts outside the 3′UTR in an otherwise canonical, seed-based fashion (41,42). We found that inclusion of CDS sites had little to no effect on classification performance and only very slightly improved the correlation between mTIScore and RIP response (r = −0.59 versus r = −0.62, at 400 nM anti-miR-122, Supplementary Figure S4B and C). As a result, for simplicity CDS sites were omitted from further analyses.
miR-target impact model is generalizable to transcriptome, other inhibited miRNAs
A moderately strong correlation was observed between mTIScores and expression changes for the tested panel of miR-122 target candidates (Supplementary Figure S4D). To gain a broader perspective, array profiling was performed on mice treated with either anti-miR-122 or anti-miR against the let-7 family. In both cases, a miRNA-specific signature in the transcriptomic response was observed with seed-matched transcripts being enriched among the most upregulated genes (Figure 4A and F). Cumulative distribution frequencies (CDFs) of seed-matched genes binned by mTIScores into equal sized groups indicated that the extent of derepression was commensurate with mTIScore: overall a greater proportion of higher scored bins were upregulated to a greater extent than lesser scored bins (Figure 4B and G). Importantly, similar results were found when comparing seed-matched genes containing the same number of seed sites (Figure 4C–D, H–I), indicating that the penalty for alternative miRNA binding sites was a significant contributing factor to these outcomes. In addition, higher mTIScored bins were more enriched for significantly derepressed targets, especially when only considering conserved seed and alternative sites (Figure 4E and J).
Figure 4.

Transcriptome-level assessment of miR-target impact model. Array profiling of liver tissue was performed following in vivo dosing of anti-122 (A–E) or anti-let-7 (F–J). (A) Sylamer analysis for anti-122 treatment. Genes were ranked from most upregulated (left-hand side of x-axis) to most downregulated (right-hand side), and hypergeometric enrichment scores were calculated for each possible short (7- or 8-mer) nucleic acid sequence based on their frequency of occurrence in the 3′UTRs of the ranked gene list (y-axis). miR-122 target sites (8mer, 7m8 and 7m1A) were highly enriched in 3′UTRs of the most upregulated genes, indicating an on-target effect of miRNA inhibition. Gray lines represent the background for all non-miR-122 hexamer sequences. (B) Cumulative distribution frequencies (CDFs) for miR-122 seed-matched genes binned by mTIScores into equal sized groups. Median mTIScores for each bin are indicated in parentheses. CDF for non-seed containing genes is shown in black. (C) CDFs for genes containing only one miR-122 site, binned by mTIScores into equal sized groups. (D) Same as (C) for genes containing exactly two miR-122 sites. (E) Fraction of miR-122 seed-matched genes significantly upregulated (FDR = 20%) in equal-sized ranked bins of mTIScores computed using all seed sequences (top) or only conserved seed sequences (bottom). (F–J) Analogous results for anti-let-7.
In addition, we also analyzed profiling data for miR-17 inhibition using inducible tough-acting decoy (TuD) (43) in Hep3B cells. Highly similar results compared to that of miR-122 and let-7 were observed (data not shown), thus demonstrating that our model is also applicable with non-synthetic miRNA inhibitors expressed in cell culture.
In mouse liver, miR-122 and let-7 are among the most highly expressed miRNA families, representing ∼33% and ∼10% of total miRNA, respectively (Supplementary Figure S5A). Likewise, the miR-17 family represents ∼25% of total miRNA in Hep3B cells. To test whether miR-target impact scoring could also be applied to a lesser expressed miRNA family, anti-miR against miR-221/222 was transfected into SKHep-1 cells, where miR-221/222 family expression accounts for <4% of total miRNA (Supplementary Figure S5A). Consistent with reduced expression (44,45), miR-221/222 inhibition did not show a strong transcriptomic signature (Supplementary Figure S5B), unlike other tested miRNAs. Successful inhibition, however, could be confirmed based on cumulative upregulation of seed-matched transcripts compared to those lacking seed sequences (Supplementary Figure S5C). Additionally, derepression of the previously well-validated miR-221/222 target CDKN1B (p27) was observed (Supplementary Figure S5D), and verified using RIP competition (Supplementary Figure S5E). Here, mTIScores were slightly less predictive: while the bottom three groups of genes binned by mTIScores had rankings commensurate with CDFs, the top scoring bin underperformed in terms of fold-change derepression (Supplementary Figure S5C); even though it was the bin most enriched with significantly upregulated targets (Supplementary Figure S5F). The observed underperformance may be partly due to improper penalization resulting from inaccurate alternative site predictions and/or miRNA expression measurements caused by ligation bias (46). These inaccuracies would have greater consequence when analyzing lower expressed MOI, where penalization contributes more overall to target scores due to expression weighted penalization.
Taken together, these results demonstrate that the miR-target impact model can be generalized to global transcriptomic responses of various miRNAs in both tissue and cultured cells following inhibition by either expressed TuD or synthetic anti-miR. In addition, they strongly indicate the widespread effects of alternative miRNAs on shaping the cellular response to miRNA inhibition.
DISCUSSION
AGO-based RIP is a valuable tool for biochemically assessing miRNA-target interactions (47–56). Unlike luciferase assays that use exogenously expressed reporters, RIP provides physical evidence for the interaction of miRNA complexes with endogenous target mRNAs. Despite these advantages, RIP is relatively less used compared to luciferase assays for validating direct downstream mRNA targets of miRNAs.
Limited use of RIP may in part be due to technical challenges inherent in the method. Because AGO2 binds a diverse range of miRNAs, RIP provides a global view of miRNA regulation. In order to identify direct targets specific to a particular miRNA, it is usually perturbed by transfection or inhibition in order to measure changes in the AGO-associated mRNAs. However, a complication arises from the fact that miRNA perturbation also induces changes in overall transcript abundance, thus necessitating another measurement in order to calculate a net IP (50). Accounting for these changes in total transcript abundance can confound interpretation, complicate analysis, and reduce sensitivity of IP data due to propagation and compounding of errors in all the necessary measurements.
Here, we expanded on the utility of AGO2-based RIP methods for target validation by coupling it with post-lysis, anti-miR competition assays. The simple alteration of treating lysates ex vivo over very short time periods—rather than obtaining lysates from pre-treated cells or tissues—eliminated gene expression changes, and as a result, improved sensitivity and interpretation. In addition, it improved both throughput and efficiency. Defined levels of anti-miR could be titrated into prepared lysates in 96-well format while only requiring very few animals.
The method's effectiveness was demonstrated by identifying several novel targets of miR-122 in mouse liver. Probing the interactions between miR-122 and its targets in their near-native context enabled direct correlations to be made with differential gene expression analysis, thereby reconnecting independent measurements of the two outcomes of miRNA inhibition: physical disruptions in RISC-target complexes and downstream changes in target mRNA abundance. Accordingly, very strong correlation between in vivo and ex vivo target responses suggests that the majority of AGO-target interactions observed by IP represent endogenous complexes rather than post-lysis artifacts (57).
Coordinated action of multiple miRNAs on common targets is a prevalent feature of miRNA regulatory networks (38–40). Several genes have been shown experimentally to be simultaneously repressed by multiple miRNAs (58,59). Yet the effects of co-regulation, if any, following miRNA inhibition were until now only inferred. Tan et al. (55) previously suggested that miRNA co-regulation may explain why four putative miR-17 targets (RAB12, E2F3, MYLIP and CDKN1A) showed less than expected changes in AGO-IP fractions following anti-miR-17 transfection in Hodgkin lymphoma (HL) cell lines. Our results now strongly indicate that miRNA co-regulation has a widespread impact in modulating the transcriptomic responses to anti-miR treatment, with more highly co-regulated targets being less sensitive to anti-miR. These results also refine our understanding of the mechanism of anti-miR drugs. Anti-miR potency is derived not only from its ability to bind and inhibit cognate miRNA, but ultimately through its ability to effectively alter RISC-mRNA target occupancy.
A priori knowledge of this modulatory effect of alternative miRNAs can assist target validation efforts. To this end, the miR-target impact model can potentially be used as a positive filter that is capable of enriching for targets most likely to be directly responsive to anti-miR treatment. We present this model, however, as more of a conceptual framework than an out-of-the-box solution. It has thus far only been tested on a handful of miRNAs, and out of these initial results, it appears to perform better with those that are more highly expressed. For miRNAs with lower expression, further optimization may be necessary both in terms of experimental measurements (more accurate miRNA expression levels) and bioinformatics (scaling of alternative site penalty, better understanding of relevant miRNA-binding sites). In this regard, the model's utility would likely be enhanced by combining its features with that of other target prediction methods to reduce false-positives in alternative predictions.
Another interesting aspect of the miR-target impact model is its reliance on cell-specific miRNA expression data. Indeed, use of out-of-context expression data derived from hepatocellular carcinoma cells diminished any predictive power for healthy mouse liver. These results emphasize the importance of having accurate expression data that reflect the cellular environment and are also consistent with widespread dysregulation of miRNA in cancer (60,61). Accordingly, changes in alternative miRNA abundance and target co-regulation may in part explain our previous finding that miR-21 inhibition leads to widely disparate transcriptome responses in cultured cancer cells compared to non-transformed tissue (44). Other instances of cell-specific attenuation of miRNA activity have also been reported. For instance, RNA-binding proteins (RBPs) HUR and PUM can strengthen or weaken miRNA-mediated repression of specific targets in a manner that is dependent on the expression level and/or phosphorylation state of the RBP (62,63). Incorporation of context-specific factors into next-generation computational tools may therefore be a critical element for improving sensitivity and specificity of miRNA target prediction.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank all of their colleagues at Regulus Therapeutics for their thoughtful insights and contributions, particularly B. Bhat, X. Huang, V. Kaimal, R. Pagarigan, A. Pavlicek and T. Vincent.
FUNDING
Regulus Therapeutics, Inc. Source of open access funding: Regulus Therapeutics Inc.
Conflict of interest statement. All authors are employees of Regulus Therapeutics, Inc.
REFERENCES
- 1.Bartel D. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar M., Nath S., Prasad H., Sharma G., Li Y. MicroRNAs: a new ray of hope for diabetes mellitus. Protein Cell. 2012;3:726–738. doi: 10.1007/s13238-012-2055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garzon R., Calin G., Croce C. MicroRNAs in cancer. Annu. Rev. Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 4.Pauley K., Cha S., Chan E. MicroRNA in autoimmunity and autoimmune diseases. J. Autoimmun. 2009;32:189–194. doi: 10.1016/j.jaut.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krützfeldt J., Rajewsky N., Braich R., Rajeev K., Tuschl T., Manoharan M., Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 6.Jackson A., Linsley P. The therapeutic potential of microRNA modulation. Discov. Med. 2010;9:311–318. [PubMed] [Google Scholar]
- 7.Stenvang J., Petri A., Lindow M., Obad S., Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1. doi: 10.1186/1758-907X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim L., Lau N., Garrett-Engele P., Grimson A., Schelter J., Castle J., Bartel D., Linsley P., Johnson J. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 9.Lewis B., Burge C., Bartel D. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 10.van Dongen S., Abreu-Goodger C., Enright A. Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods. 2008;5:1023–1025. doi: 10.1038/nmeth.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimson A., Farh K., Johnston W., Garrett-Engele P., Lim L., Bartel D. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.John B., Enright A., Aravin A., Tuschl T., Sander C., Marks D. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krek A., Grün D., Poy M., Wolf R., Rosenberg L., Epstein E., MacMenamin P., da Piedade I., Gunsalus K., Stoffel M., et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 14.Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 15.Witkos T., Koscianska E., Krzyzosiak W. Practical aspects of microRNA target prediction. Curr. Mol. Med. 2011;11:93–109. doi: 10.2174/156652411794859250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janssen H., Reesink H., Lawitz E., Zeuzem S., Rodriguez-Torres M., Patel K., van der Meer A., Patick A., Chen A., Zhou Y., et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 17.Tsai W.-C., Hsu S.-D., Hsu C.-S., Lai T.-C., Chen S.-J., Shen R., Huang Y., Chen H.-C., Lee C.-H., Tsai T.-F., et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 2012;122:2884–2897. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentleman R., Carey V., Bates D., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smyth G. In: Bioinformatics and computational biology solutions using R and Bioconductor. Gentleman R, Vince C, Huber W, Irizarry R, Dudoit S, editors. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 20.Bartonicek N., Enright A. SylArray: a web server for automated detection of miRNA effects from expression data. Bioinformatics. 2010;26:2900–2901. doi: 10.1093/bioinformatics/btq545. [DOI] [PubMed] [Google Scholar]
- 21.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The Human Genome Browser at UCSC. Genome Res. 2002;12 doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda K., Satoh M., Pauley K., Fritzler M., Reeves W., Chan E. Detection of the argonaute protein Ago2 and microRNAs in the RNA induced silencing complex (RISC) using a monoclonal antibody. J. Immunol. Methods. 2006;317:38–44. doi: 10.1016/j.jim.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fabian M., Cieplak M., Frank F., Morita M., Green J., Srikumar T., Nagar B., Yamamoto T., Raught B., Duchaine T., et al. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat. Struct. Mol. Biol. 2011;18:1211–1217. doi: 10.1038/nsmb.2149. [DOI] [PubMed] [Google Scholar]
- 26.Huntzinger E., Kuzuoglu-Öztürk D., Braun J., Eulalio A., Wohlbold L., Izaurralde E. The interactions of GW182 proteins with PABP and deadenylases are required for both translational repression and degradation of miRNA targets. Nucleic Acids Res. 2013;41:978–994. doi: 10.1093/nar/gks1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dweep H., Sticht C., Pandey P., Gretz N. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Hsu S.D., Lin F.M., Wu W.Y., Liang C., Huang W.C., Chan W.L., Tsai W.T., Chen G.Z., Lee C.J., Chiu C.M., et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fabani M., Gait M. miR-122 targeting with LNA/2’-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elmén J., Lindow M., Silahtaroglu A., Bak M., Christensen M., Lind-Thomsen A., Hedtjärn M., Hansen J., Hansen H., Straarup E., et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai W.-C., Hsu P., Lai T.-C., Chau G.-Y., Lin C.-W., Chen C.-M., Lin C.-D., Liao Y.-L., Wang J.-L., Chau Y.-P., et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- 33.Gramantieri L., Ferracin M., Fornari F., Veronese A., Sabbioni S., Liu C.-G., Calin G., Giovannini C., Ferrazzi E., Grazi G., et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 34.Lin C., Gong H.-Y., Tseng H.-C., Wang W.-L., Wu J.-L. miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem. Biophys. Res. Commun. 2008;375:315–320. doi: 10.1016/j.bbrc.2008.07.154. [DOI] [PubMed] [Google Scholar]
- 35.Xu H., He J.-H., Xiao Z.-D., Zhang Q.-Q., Chen Y.-Q., Zhou H., Qu L.-H. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52:1431–1442. doi: 10.1002/hep.23818. [DOI] [PubMed] [Google Scholar]
- 36.Doench J., Petersen C., Sharp P. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayr C., Hemann M., Bartel D. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedman Y., Balaga O., Linial M. Working together: combinatorial regulation by microRNAs. Adv. Exp. Med. Biol. 2013;774:317–337. doi: 10.1007/978-94-007-5590-1_16. [DOI] [PubMed] [Google Scholar]
- 39.Ivanovska I., Cleary M. Combinatorial microRNAs: working together to make a difference. Cell Cycle. 2008;7:3137–3142. doi: 10.4161/cc.7.20.6923. [DOI] [PubMed] [Google Scholar]
- 40.Hobert O. miRNAs play a tune. Cell. 2007;131:22–24. doi: 10.1016/j.cell.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 41.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jungkamp A.-C., Munschauer M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chi S., Zang J., Mele A., Darnell R. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haraguchi T., Ozaki Y., Iba H. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Androsavich J., Chau B., Bhat B., Linsley P., Walter N. Disease-linked microRNA-21 exhibits drastically reduced mRNA binding and silencing activity in healthy mouse liver. RNA. 2012;18:1510–1526. doi: 10.1261/rna.033308.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia D.M., Baek D., Shin C., Bell G.W., Grimson A., Bartel D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hafner M., Renwick N., Brown M., Mihailović A., Holoch D., Lin C., Pena J.T., Nusbaum J.D., Morozov P., Ludwig J., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beitzinger M., Peters L., Zhu J., Kremmer E., Meister G. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007;4:76–84. doi: 10.4161/rna.4.2.4640. [DOI] [PubMed] [Google Scholar]
- 48.Dölken L., Malterer G., Erhard F., Kothe S., Friedel C., Suffert G., Marcinowski L., Motsch N., Barth S., Beitzinger M., et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe. 2010;7:324–334. doi: 10.1016/j.chom.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 49.Hendrickson D., Hogan D., Herschlag D., Ferrell J., Brown P. Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PloS One. 2008;3 doi: 10.1371/journal.pone.0002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karginov F., Conaco C., Xuan Z., Schmidt B., Parker J., Mandel G., Hannon G. A biochemical approach to identifying microRNA targets. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19291–19296. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Landthaler M., Gaidatzis D., Rothballer A., Chen P., Soll S., Dinic L., Ojo T., Hafner M., Zavolan M., Tuschl T. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA. 2008;14:2580–2596. doi: 10.1261/rna.1351608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matkovich S., Van Booven D., Eschenbacher W., Dorn G. RISC RNA sequencing for context-specific identification of in vivo microRNA targets. Circ. Res. 2011;108:18–26. doi: 10.1161/CIRCRESAHA.110.233528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicolas F., Pais H., Schwach F., Lindow M., Kauppinen S., Moulton V., Dalmay T. Experimental identification of microRNA-140 targets by silencing and overexpressing miR-140. RNA. 2008;14:2513–2520. doi: 10.1261/rna.1221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nonne N., Ameyar-Zazoua M., Souidi M., Harel-Bellan A. Tandem affinity purification of miRNA target mRNAs (TAP-Tar) Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkp1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan L., Seinen E., Duns G., de Jong D., Sibon O., Poppema S., Kroesen B.-J., Kok K., van den Berg A. A high throughput experimental approach to identify miRNA targets in human cells. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang W.-X., Wilfred B., Hu Y., Stromberg A., Nelson P. Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA. 2010;16:394–404. doi: 10.1261/rna.1905910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mili S., Steitz J. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu S., Huang S., Ding J., Zhao Y., Liang L., Liu T., Zhan R., He X. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene. 2010;29:2302–2308. doi: 10.1038/onc.2010.34. [DOI] [PubMed] [Google Scholar]
- 59.Jiang Q., Feng M.-G., Mo Y.-Y. Systematic validation of predicted microRNAs for cyclin D1. BMC Cancer. 2009;9:194. doi: 10.1186/1471-2407-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Calin G., Croce C. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 61.Dvinge H., Git A., Gräf S., Salmon-Divon M., Curtis C., Sottoriva A., Zhao Y., Hirst M., Armisen J., Miska E., et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature. 2013;497:378–382. doi: 10.1038/nature12108. [DOI] [PubMed] [Google Scholar]
- 62.Bhattacharyya S., Habermacher R., Martine U., Closs E., Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 63.Kedde M., van Kouwenhove M., Zwart W., Oude Vrielink J., Elkon R., Agami R. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.