


Abstract
TET1 is a 5-methylcytosine dioxygenase and its DNA demethylating activity has been implicated in pluripotency and reprogramming. However, the precise role of TET1 in DNA methylation regulation outside of developmental reprogramming is still unclear. Here, we show that overexpression of the TET1 catalytic domain but not full length TET1 (TET1-FL) induces massive global DNA demethylation in differentiated cells. Genome-wide mapping reveals that 5-hydroxymethylcytosine production by TET1-FL is inhibited as DNA methylation increases, which can be explained by the preferential binding of TET1-FL to unmethylated CpG islands (CGIs) through its CXXC domain. TET1-FL specifically accumulates 5-hydroxymethylcytosine at the edges of hypomethylated CGIs, while knockdown of endogenous TET1 induces methylation spreading from methylated edges into hypomethylated CGIs. We also found that gene expression changes after TET1-FL overexpression are relatively small and independent of its dioxygenase function. Thus, our results identify TET1 as a maintenance DNA demethylase that does not purposely decrease methylation levels, but specifically prevents aberrant methylation spreading into CGIs in differentiated cells.
INTRODUCTION
DNA methylation at the C5 position of cytosine (5-methylcytosine, 5mC) is a crucial epigenetic modification that has been implicated in numerous cellular processes in mammals, including embryonic development, transcription, X chromosome inactivation, genomic imprinting and chromatin structure (1,2). The methylation pattern of the genome is dynamic during normal development, starting from fertilization through embryogenesis and postnatal growth, and abnormal methylation changes are involved in various human diseases, such as cancer (3,4). The patterns of DNA methylation in cells are initially established by de novo DNA methyltransferases DNMT3a and DNMT3b, and then faithfully maintained during DNA replication by the maintenance methyltransferase DNMT1 (5–8). In contrast to the well-defined DNA methyltransferases, the potential enzymes that erase DNA methylation are only beginning to be understood (9,10).
The ten–eleven translocation (TET) family proteins were recently identified as 5mC dioxygenases which can consecutively convert 5mC into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine and 5-carboxylcytosine and further induce passive or active DNA demethylation in genomic DNA (11–20). Tet1, the founding member of the TET family, has been intensely studied since its dioxygenase catalytic function was demonstrated (19,20). Depletion of Tet1 in mouse embryonic stem cells (mESCs) causes decreased 5hmC levels and increased DNA methylation at its target regions, and also affects gene transcription and cell lineage specification (19,21–25). Tet1 was also reported to induce locus-specific demethylation in mouse primordial germ cells and activate some meiotic genes (26–28). Moreover, Tet1-mediated demethylation was observed in the reprogramming processes for generation of induced pluripotent stem cells (29,30). However, outside of embryonic development and reprogramming, little is known about the role of TET1 in DNA methylation regulation in differentiated cells where it is commonly expressed (19,31). Previous studies, which reported that overexpression of TET1 in HEK293 cells can induce DNA demethylation in exogenous non-replicable DNA reporters and endogenous genomic loci, used overexpression of the TET1 catalytic domain (TET1-CD) but not full length TET1 (TET1-FL) (17,32). Given the possibility that the residual domains in TET1 may regulate the accessibility to its catalytic domain, those results on TET1-CD do not precisely reveal the function of TET1 in physiologic DNA methylation regulation.
In this report, we systematically investigated the effect of TET1 on DNA methylation in HEK293T cells by overexpression of TET1-FL and knockdown of endogenous TET1. Our results demonstrate that TET1 works as a maintenance DNA demethylase, which does not change DNA methylation globally, but specifically maintains the DNA hypomethylation state of CpG islands (CGIs) by preventing methylation spreading from methylated edges.
MATERIALS AND METHODS
DNA construct and overexpression of TET1
To clone the open reading frame (ORF) of human TET1-FL (1–2136 amino acid), we extracted total RNA from SY5Y cells using TRIzol® Reagent (Invitrogen). Reverse transcription was performed with a gene-specific primer (5′-TATATACTGCAAGTTGCTAATACTTGAATG-3′) and AccuScript PfuUltra II RT-PCR Kit (Stratagene) according to the manufacturer's instructions. After PCR amplification with AccuPrime™ Taq DNA Polymerase High Fidelity (Invitrogen), TET1-FL ORF was cloned into pCR®-XL-TOPO® vector (Invitrogen). Finally, the fragment of TET1-FL ORF was transferred into pIRES-hrGFP II vector (Stratagene) which contains a green fluorescent protein (GFP) reporter and a 3× FLAG tag. The ORF of TET1-CD (1418–2136 amino acid) was amplified from the above TET1-FL ORF clone, and it was also inserted into pIRES-hrGFP II vector. Catalytically mutant mTET1-FL and mTET1-CD (H1671D, Y1673A) (20) and CXXC domain-mutated TET1-FL-C594A (22) were generated by site-directed mutagenesis. The sequences of all plasmids were validated by Sanger DNA sequencing. Transfection of those plasmids into HEK293T cells were carried out with FuGene HD transfection reagent (Roche) according to manufacturer's instructions. GFP-positive cells were collected by fluorescent activated cell sorting (FACS) at indicated time points using Becton Dickinson FACS Calibur Flow Cytometer. HEK293T cells were obtained from ATCC, cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, and tested negative for mycoplasma contamination.
Western blot and DNA dot blot assays
For western blot assays, protein extraction was performed using the radio-immunoprecipitation assay (RIPA) buffer (Fisher) supplemented with 1× protease inhibitor cocktail solution (Roche). The primary antibodies used included anti-FLAG (Cat. #200471, Stratagene), anti-TET1 (GTX124207, GeneTex), anti-Lamin B (AB16048, Abcam) and anti-β-actin (GTX109639, GeneTex). For DNA dot blot assays, different amounts of genomic DNA samples diluted in 0.4 mM NaOH/10 mM ethylenediaminetetraacetic acid (EDTA) were denatured at 100°C for 10 min, followed by rapid chilling on ice. Two microliters of each denatured DNA was then spotted onto the positively charged nylon membrane (Roche), and the diameter of each dot was kept to <4 mm. After the membrane became dry, it was rinsed in 2× SSC buffer (0.3 M NaCl, 30 mM sodium citrate) followed by complete air dry. The dry membrane was wrapped in UV-transparent plastic wrap, and then placed DNA-side-down on a UV transilluminator for 3 min to immobilize the DNA. After blocking with 5% non-fat dry milk in PBS, the membrane was immunoblotted using 5hmC antibody (Cat. #39769, Active Motif) and HRP-conjugated anti-rabbit secondary antibody (NA934, GE Healthcare), and finally developed with enhanced chemiluminescence reagents and exposed to X-ray imaging film.
Bisulfite-pyrosequencing and bisulfite-cloning-sequencing
Bisulfite conversion of genomic DNA was done with EpiTect bisulfite kits (Qiagen) according to the manufacturer's instructions. For bisulfite-pyrosequencing, a two-step polymerase chain reaction (PCR) for amplification was generally used as previously described (33,34). The results were analyzed with Pyro Q-CpG Software (Qiagen) software. For bisulfite-cloning-sequencing, a similar two-step PCR as that in bisulfite-pyrosequencing was used but no biotinylated primers were included in the second step PCR. The final PCR product was then cloned into pCR4-TOPO vector (Invitrogen) and transformed TOP10 chemical competent cells (Invitrogen). After ∼14 h of incubation at 37°C, individual clones were picked and amplified with PCR using pCR4 forward primer (5′-TCTGGAATTGTGAGCGGATA-3′) and reverse primer (5′-GTTTTCCCAGTCACGACGTT-3′). Those PCR products were then sequenced with M13-RV primer. The primers used are listed in Supplementary Table S1.
HpaII-PCR DNA methylation assay
HpaII-PCR DNA methylation assay was performed as previously described (17). In brief, 500 ng genomic DNA were incubated with 10 units HpaII (NEB) or in a mock reaction without HpaII at 37°C for 8 h or overnight, followed by 80°C inactivation for 20 min. The DNA from HpaII digestion or mock treatment was tested by qPCR (Applied Biosystems 7500) using Power SYBR® Green PCR Master Mix (Applied Biosystems) and primers flanking specific HpaII digestion sites (CCGG). PCR reaction comprised a 10 min activation step at 95°C, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min. DNA methylation of a CCGG site was calculated by 2Ct(mock) − Ct(HpaII) × 100%. The primers used are listed in Supplementary Table S1.
Chromatin immunoprecipitation and quantitative PCR (ChIP-qPCR)
Cells were fixed with fresh 1% formaldehyde at room temperature for 10 min and quenched with 125 mM glycine. The cells were then washed with cold phosphate buffered saline (PBS) and resuspended in sodium dodecyl sulphate (SDS) lysis buffer (50 mM Tris pH 8.1, 10 mM EDTA, 1% SDS), followed by sonication with Bioruptor (Diagenode) to get an average fragment size of 200–500 bp. The resultant chromatin samples were 10-fold diluted in ChIP dilution buffer (16.7 mM Tris pH 8.1, 1.2 mM EDTA, 167 mM NaCl, 1.1% Triton X-100, 0.01% SDS) and incubated with Dynal®Protein G magnetic beads (Invitrogen) at 4°C overnight for pre-clearing. At the same time, anti-FLAG or anti-TET1 antibodies described above or control IgG were pre-crosslinked with Dynal®Protein G magnetic beads at 4°C overnight. The pre-cleared chromatin samples were then incubated with the antibody-bead complex at 4°C overnight. The immunoprecipitated chromatin–antibody–bead complexes were extensively washed with RIPA washing buffer (0.5 M EDTA, 5 M LiCl, 1 M HEPES–KOH pH 7.6, 10% NP-40, 10% Na-deoxycholate) and TE buffer (pH 8.0) containing 50 nM NaCl, resuspended in elution buffer (50 mM Tris pH 8.1, 10 mM EDTA, 1% SDS), and heated at 65°C for 15 min to separate chromatin from beads. For reversing crosslinks, the isolated chromatin was incubated at 65°C overnight, followed by digestion with RNase A and Proteinase K. Finally, the resultant DNA was purified with Qiaquick PCR Purification Kit (Qiagen). Enrichment of target regions was determined by qPCR under similar conditions described in HpaII-PCR DNA methylation assay. The primers used are listed in Supplementary Table S1.
Lentiviral shRNA-mediated TET1 knockdown
Two different TET1 shRNAs in the pTRIPZ vectors (shTET1#1: V2THS_141063 and shTET1#2: V2THS_203196, Open Biosystems) were transferred into MulI and XhoI sites of the pGIPZ vectors (Open Biosystems). A non-targeting shRNAmir-pGIPZ vector was used as a negative control (RHS4743, Open Biosystems). To produce lentiviral particles, pGIPZ-shTET1 and package plasmids psPAX2 and pMD2.G (Addgene) were transfected into HEK293FT cells (Invitrogen) at a ratio of 1:1:1 using Lipofectamine®2000 Transfection Reagents (Invitrogen). Two days after transfection, the viral supernatant was collected and filtered with 0.45 μm filters (Millipore). HEK293T cells were then infected with each lentivirus supernatant in the presence of 8 μg/ml of polybrene (Sigma). Puromycin selection (1.5 μg/ml, Sigma) began 2 days after infection. Stable knockdown cells were cloned by limiting dilution and selected by western blot assay based on TET1 protein level. These knockdown cells were subjected to further analyses 3–4 months after transfection.
Digital restriction enzyme analysis of methylation (DREAM)
DREAM was performed as previously described (35). Five micrograms of genomic DNA from (m)TET1-FL-or (m)TET1-CD-overexpressing HEK293T cells were first spiked with 0.05 ng of a set of specific calibrators with different methylation levels. The DNA mixture was then sequentially digested by 5 μl SmaI (3 h at 37°C, Fermentas) and 50 U XmaI (∼16 h at 37°C, NEB), resulting in distinct DNA signatures at unmethylated or methylated SmaI sites (CCCGGG). After purification with a QIAquick PCR purification kit, the digested DNA was heated at 65°C for 3 min followed by snap cooling to create free concatenated CCGG overhangs. Klenow fragment (3′→5′ exo-) (NEB) and CGA mix (dCTP, dGFP, dATP, 10 mM each) were then added to fill the overhangs and added ‘A’ tail to 3′ end. The resultant DNA was purified again and then ligated with Illumina paired ends adapters (PEA1: 5′-phosphate-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG-3′, PEA2: 5′-ACACTCTTTCCCTACACGACGCTCTTC-CGATCT-3′) using Quick T4 DNA ligase (Enzymatics). Subsequently, the DNA ligated with adapters was separated through 2% agarose gel. The gel slice with the size of 250–375 bp was cut off and purified with QIAquick Gel Extraction Kit (Qiagen). PCR amplification (18 cycles) of the gel-extracted DNA was performed using Illumina paired end PCR primers and iProof HF master mix (Bio-Rad). Resulting sequencing libraries were purified with Agencourt AMPure PCR Purification Kit (Beckman Coulter). Then the libraries were sequenced by paired-end 36 nt sequencing on Illumina Genome Analyzer II. The sequencing reads were mapped to SmaI sites in the human genome hg18 and signatures corresponding to methylated and unmethylated CpG were enumerated for each SmaI site. The minimum coverage was set at ≥20 reads unless otherwise indicated. The methylation value was calculated as the ratio of the number of methylated tags over total number of tags mapped to a given SmaI/XmaI site. Data have been deposited in GEO with accession number: GSE44038.
As for the genomic location of SmaI sites, each SmaI site was assigned to the gene that has the closest transcription start site (TSS). Then the region was classified by its location to the gene: upstream (−5 to −1 kb from TSS), promoter (−1 to 0.5 kb from TSS), exon, intron, downstream (−0.5 to 1 kb from transcription end site (TES)) and intergenic (1 kb from TES to −5 kb of downstream gene). The gene list used to annotate the enriched regions is the RefSeq gene list downloaded from UCSC genome browser (http://genome.ucsc.edu/) in April 2010. The CGI annotation was also obtained from the UCSC website.
To analyze the distribution of DNA methylation on gene bodies, each SmaI site was classified into bins according to its relative location to the closest gene. Five kilobase upstream of TSS and 5 kb downstream of TES were subdivided into 20 bins, with 500 bp for each bin. The gene body was subdivided evenly into 20 bins. The average percentage of methylation for all the SmaI sites in each bin was calculated.
Hydroxymethylated DNA immunoprecipitation combined with next generation DNA sequencing (hMeDIP-seq)
Three micrograms of genomic DNA from (m)TET1-FL or (m)TET1-CD-overexpressing HEK293T cells were diluted in TE buffer (pH 7.6) and sonicated with Bioruptor. The desirable fragment size was 100–500 bp. The resultant DNA was purified with QIAquick PCR purification kit. Five hundred nanograms purified sonicated DNA was used and spiked with 20 pg of a set of specific calibrators containing different 5hmC levels. End repair, addition with ‘A’ bases to the 3′ end of the DNA fragments and ligation with Illumina paired ends adapters were then performed similar to the procedure described in DREAM. The resultant DNA was purified again, diluted in 450 μl TE buffer and denatured at 95°C for 10 min, followed by snap chilling in ice. The denatured DNA was subsequently mixed with 50 μl 10× IP buffer (1.4 M NaCl, 100 mM Na-phosphate, pH 7.0, 0.5% Triton X-100), followed by incubation with 1 μl anti-5hmC antibody described above at 4°C overnight. Then Dynal®Protein G magnetic beads were added and incubated at 4°C for 2 h. After extensive washing with 1× IP buffer (140 mM NaCl, 10 mM Na-phosphate, pH 7.0, 0.05% Triton X-100), DNA was separated from beads at 65°C for 15 min in elution buffer (10 mM Tris pH 7.6, 1 mM EDTA, 1% SDS). The eluted products were size selected by electrophoresis in a 2% agarose gel. A slice corresponding to 300 ± 25 bp size window based on DNA ladder was cut out. The DNA extracted from agarose was amplified with PCR (10–15 cycles) by using Illumina paired end PCR primers and Phusion™ High-Fidelity DNA Polymerase (NEB). The resultant sequencing libraries were purified with Agencourt AMPure PCR Purification Kit and sent for single-read sequencing on Illumina Genome Analyzer II. Sequenced DNA tags were mapped to human genome hg18 and uniquely mapped tags were kept. For multiple tags that were mapped to the same genomic location, only one was considered in the analysis to avoid PCR bias. CCAT (version 3.0) (36) was used to detect 5hmC peaks in hMeDIP-Seq samples. The window size was set as 500 bp. Peaks with FDR ≤0.05 and ≥5 fold enrichment to input were deemed as significant. Each peak was assigned to the gene that has the closest TSS as SmaI sites described above. As for landscape of the data, each tag was extended by 250 bp to its 3′ end. Then the number of overlapped tags in each genome position was rescaled to normalize the number of background tags to 10 M and averaged over 10 bp resolution. The averaged values were displayed using UCSC genome browser (http://genome.ucsc.edu/). Data have been deposited in GEO with accession number: GSE44036.
The distribution of hMeDIP signal on different genomic features was analyzed as follows. Gene body: for each gene, 5 kb upstream of TSS and 5 kb downstream of TES were subdivided into 1 kb bins, and the gene body was subdivided evenly into 20 bins. TSS: 10 kb upstream to 10 kb downstream of the TSS of each gene was subdivided into 250 bp bins. Exon: 200% upstream to 200% downstream of each exon was subdivided into 25 bins, with each bin 20% of the exon length. Exon–intron boundary: at each exon–intron boundary, 100 bp into exon and 100 bp into intron were subdivided into 25 bp bins. CGI: 10 kb upstream to 10 kb downstream of each CGI was subdivided into 50 bins, consisting of 20 bins for upstream or downstream (500 bp each) and 10 bins for CGI (10% of the length of CGIs each). In each bin, the number of tags was normalized by the length of the bin and the total number of background tags in the genome (the number of background tags was calculated as total number of tags multiplied by the noise rate from CCAT). The normalized tag density of the hMeDIP sample in each bin was then subtracted by that of the corresponding input and averaged over all the respective features in the genome.
RNA isolation and reverse transcription-qPCR analysis
Total RNA was isolated using TRIzol Reagent as per manufacturer's specifications. cDNA was synthesized from 1 μg of DNase-treated total RNA using High-Capacity cDNA reverse transcription kit (Applied Biosystems). qPCR was performed under similar conditions described in HpaII-PCR DNA methylation assay. The average threshold (Ct) was determined for each gene and normalized to β-actin as an internal normalization control. The primers used are listed in Supplementary Table S1.
Whole-genome gene expression microarray analysis
Affymetrix GeneChip® Whole-Transcript Human Gene 2.0 ST Arrays were used for global gene expression analysis. RNA samples were labeled and hybridized according to the manufacturer's instructions (Affymetrix, Santa Clara, CA, USA). Scanned microarray images were analyzed using the Affymetrix Gene Expression Console with RMA (Robust Multi-array Average) normalization algorithm. A criterion of 1.5-fold expression change was used to identify differentially regulated genes. Hierarchical clustering was performed on significant genes using signal intensities after RMA normalization by hclust function in R. Signal intensity values were rescaled to z-scores by row before clustering. Heatmap was generated by heatmap.2 function in R. Data have been deposited in GEO with accession number: GSE50016.
RNA-seq
RNA-seq sequencing libraries were made from 100 ng of DNase-treated total RNA samples using Encore® Complete RNA-Seq Library System (NuGEN, San Carlos, CA, USA) following the manufacturer's protocol. The libraries were sequenced using a 2 × 100 bases paired end protocol on the Illumina HiSeq 2000 instrument at the Fox Chase Cancer Center (Philadelphia, PA, USA). Each library was sequenced in a single lane, generating 188–241 million reads per sample. The reads were mapped to human genome (hg19) by TopHat (V2.0.5) (37). The number of fragments in each known gene from the RefSeq database (downloaded from UCSC Genome Browser on 9 March 2012) was enumerated using htseq-count from HTSeq package (V0.5.3p9). The differential expression between samples was statistically accessed by R/Bioconductor package edgeR (V3.0.8) (38) and DESeq (V1.10.1) (39), using the two most similar samples (mTET1-FL and TET1-FL) to estimate the biological variation. Genes with FDR ≥0.05 by both edgeR and DESeq and fold change ≥2 were called significant.
For the fragments that have both ends mapped, the first reads were kept. Together with the reads from the fragments that have only one end mapped, every read was extended to its 3′ end by 200 bp in exon regions. For each read, a weight of 1/n was assigned, where n is the number of positions the read was mapped to. The sum of weights for all the reads that cover each genomic position was rescaled to normalize the total number of fragments to 1 M and averaged over 10 bp resolution. The averaged values were displayed using UCSC genome browser (http://genome.ucsc.edu/). Data have been deposited in GEO with accession number: GSE49833.
Hierarchical clustering was performed on significant genes from any of the four comparisons (mTET1-CD, TET1-CD, mTET1-FL, TET1-FL versus control) using their FPKM values (fragments per kilobase of exon per million fragments mapped) by hclust function in R. FPKM values were rescaled to z-scores by row before clustering. Heatmap was generated by heatmap.2 function in R.
RESULTS
TET1-FL and TET1-CD display differential ability for 5hmC production and DNA demethylation
Overexpression of TET1-CD induces significant DNA demethylation in selected genomic loci in HEK293 cells (17,32) but it is not known if this is true for TET1-FL. To test this, HEK293T cells were transiently transfected with TET1-FL or TET1-CD expression plasmids and harvested for expressing cells by GFP sorting (Figure 1A and B and Supplementary Figure S1A–C). DNA dot-blot assays confirmed a dramatic production of 5hmC in cells transfected with wild type TET1-FL or TET1-CD but not those transfected with catalytically mutant TET1-FL or TET1-CD (referred to as mTET1-FL and mTET1-CD, respectively) 3 days after transfection (Supplementary Figure S1D). However, compared with TET1-CD, TET1-FL showed a much lower 5hmC production (∼1/8 of that by TET1-CD) (Supplementary Figure S1D), which may be attributable to its lower expression level (Figure 1B) and/or its possible lower inherent efficiency for 5hmC production in the genome. We thus increased the expression time of TET1-FL as a compensation for its low expression level, and found >2-fold increase of 5hmC content from 3 to 5 days after transfection but a slight increase from 5 to 7 days after transfection (Figure 1C). Moreover, given that some 5hmC would be further converted to other types of cytosine (including unmodified cytosine) during the prolonged expression time, the actual 5hmC production by TET1-FL 7 days after transfection should be higher than what we have seen in DNA dot-blot assay results (Figure 1C). Thus, to compare the effects of TET1-CD and TET1-FL on 5hmC production and potential DNA demethylation in subsequent experiments, we always used the TET1-CD or TET1-FL-overexpressing cells which were collected 3 or 7 days after transfection, respectively, unless indicated.
Figure 1.

5hmC production and DNA demethylation induced by TET1 overexpression. (A) Western blot analysis of TET1-FL overexpression in HEK293T cells with anti-TET1 antibody. The cells were collected by FACS 3 days after transfection. (B) Western blot analysis of overexpression of TET1-FL, TET1-CD and their catalytically mutant controls in HEK293T cells with anti-FLAG antibody. The cells were collected by FACS 3 days after transfection. (C) DNA dot blot analysis of genomic 5hmC level in HEK293T cells overexpressing (m)TET1-FL or (m)TET1-CD. The cells were collected by FACS at the indicated time points after transfection. (D) Bisulfite-pyrosequencing analysis of four methylated endogenous genomic loci in HEK293T cells overexpressing (m)TET1-CD or (m)TET1-FL collected by FACS 3 or 7 days after transfection, respectively. Data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 by Student's t-test compared to mTET1-CD-overexpressing cells. (E) HpaII-PCR based DNA methylation assay of four other endogenous genomic loci in HEK293T cells overexpressing (m)TET1-CD or (m)TET1-FL collected by FACS 3 or 7 days after transfection, respectively. The hypomethylated UBE2B gene was used as a control for complete digestion of HpaII. Data represent mean ± SD (n = 2). *P < 0.05, **P < 0.01 by Student's t-test compared to mTET1-CD-overexpressing cells.
We next examined whether TET1-mediated 5mC oxidation leads to DNA demethylation of endogenous genomic DNA. We initially used bisulfite-pyrosequencing for quantitative analysis of DNA methylation. Although the oxidated products of TET1 can interfere with bisulfite analysis in that 5hmC reacts the same as 5mC while 5fC and 5caC react as unmodified cytosine (15,40,41), the abundance of these modified bases is typically one to several orders of magnitude lower than that of 5mC (14), suggesting that their impact on the final results would be relatively low. We found that overexpression of TET1-CD induced significant demethylation at randomly selected methylated genomic loci, including long interspersed nucleotide element-1 (LINE-1) and promoters of RASSF1a, OCT4 and PGR genes (Figure 1D). In marked contrast, TET1-FL had no measurable effect on DNA methylation in this assay. Since HpaII digestion is blocked by 5mC and its oxidative derivatives (15,17), we also used HpaII-PCR based DNA methylation assays to confirm these results. Here again, only TET1-CD overexpression induced a significant decrease of DNA methylation at the promoters of INVS1ABP, NPAS3 and PARES1 (Figure 1E). Taken together, the above results at least suggest that TET1-FL and TET1-CD have differential ability for 5hmC production and DNA demethylation.
Overexpression of TET1-CD but not TET1-FL induces global DNA demethylation
To further characterize the effects of TET1-FL and TET1-CD on DNA methylation, we examined global DNA methylation changes in transfected HEK293T cells by using DREAM. This method provides quantitative analysis of DNA methylation with high accuracy (35). DNA methylation levels of 34,322; 33,395; 43,936 and 42,988 CpG sites were quantified in cells transfected with TET1-CD, mTET1-CD, TET1-FL and mTET1-FL, respectively. Pair-wise comparison of 32,803 common CpGs between TET1-CD and mTET1-CD transfections revealed a massive DNA demethylation induced by overexpression of TET1-CD (Figure 2A and B), with 2,957 CpGs (∼8.6%) demethylated by >20% (Figure 2A). These DNA demethylation events were the same in CGI and non-CGI DNA, upstream, promoter, exons, introns, downstream and intergenic regions, indicating that TET1-CD induces a genome-wide DNA demethylation without distribution bias (Figure 2C and D and Supplementary Figure S2A). Very different results were seen for TET1-FL. Pair-wise comparison of 40,937 common CpGs between TET1-FL and mTET1-FL transfections showed no significant DNA demethylation induced by TET1-FL overexpression (Figure 2E and F). Given that Tet1 specifically binds CpG-rich regions in mESCs (21,22,25), we then asked whether TET1-FL selectively induces DNA demethylation in CGIs. However, neither CGIs nor non-CGIs showed significant DNA demethylation induced by TET1-FL overexpression (Figure 2G and H). Similarly, further analyses of upstream, promoter, exons, introns, downstream and intergenic regions also did not find any significant demethylation after TET1-FL overexpression (Supplementary Figure S2B). Therefore, unlike TET1-CD, overexpression of TET1-FL in HEK293T cells cannot induce significant DNA demethylation genome-wide, indicating that TET1 is not as efficient a DNA demethylase as previously predicted (17,32).
Figure 2.
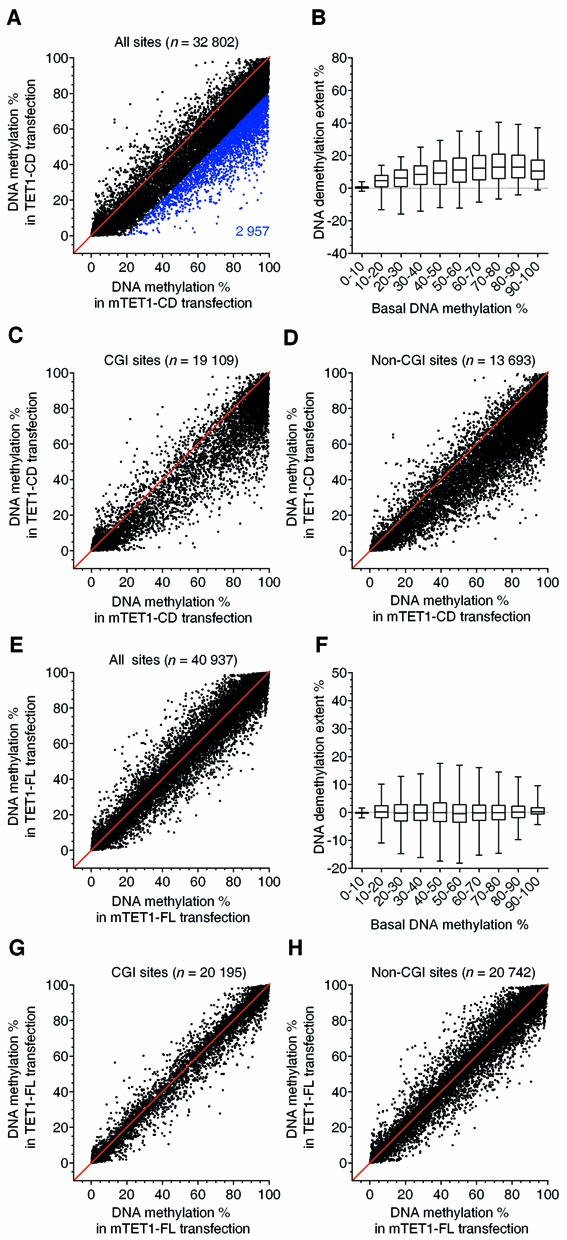
Overexpression of TET1-CD but not TET1-FL induces global DNA demethylation. (A, C–E, G and H) Shown are comparisons of % methylation in HEK293T cells overexpressing TET1-CD versus mTET1-CD (A, C and D) and TET1-FL versus mTET1-FL (E, G and H) for thousands of CpG sites quantitatively analyzed by DREAM (exact number of sites indicated in each graph). (A) and (E) plot all detected sites, (C) and (G) plot sites in CGIs, (D) and (H) plot non-CGI sites. In (A), blue dots and number represent CpG sites with ≥20% demethylation. The CGI annotation was obtained from the UCSC website. (B and F) Boxplots of DNA demethylation extent as a function of basal methylation level in HEK293T cells overexpressing TET1-CD (B) or TET1-FL (F). The basal methylation levels (% methylation in corresponding mutant control cells) are grouped in 10% intervals from 0 to 100% methylation. Boxes mark the interquartile range, and whiskers the 2.5th and 97.5th percentiles. The cells overexpressing (m)TET1-CD or (m)TET1-FL were collected by FACS 3 or 7 days after transfection, respectively.
TET1-FL and TET1-CD differentially regulate 5hmC distribution patterns
As the primary product of TET-catalyzed 5mC oxidation reaction, 5hmC also serves as a critical intermediate for TET-induced DNA demethylation (13,15,17,18). We therefore asked whether the differences in demethylation induction between TET1-CD and TET1-FL were due to their different regulation of 5hmC distribution. We performed genome-wide mapping of 5hmC in HEK293T cells with 5hmC antibody-based hMeDIP-seq (Figure 3A and Supplementary Figure S3). Once again, we used as controls mTET1-CD and mTET1-FL, which cannot catalyze 5mC oxidation. In these two control transfections, a similar number of 5hmC peaks and an almost identical distribution pattern were detected (Figure 3B) with 5hmC enrichment around promoters but even distribution in exons and introns at a relatively low level (Figure 3C and Supplementary Figure S4A and B). The enrichment of 5hmC around TSSs is similar to what was previously observed in mESCs (25,42). Interestingly, 5hmC density showed a dip in CGI-overlapped TSSs but peaked at non-CGI-overlapped TSSs (Figure 3D and E).
Figure 3.
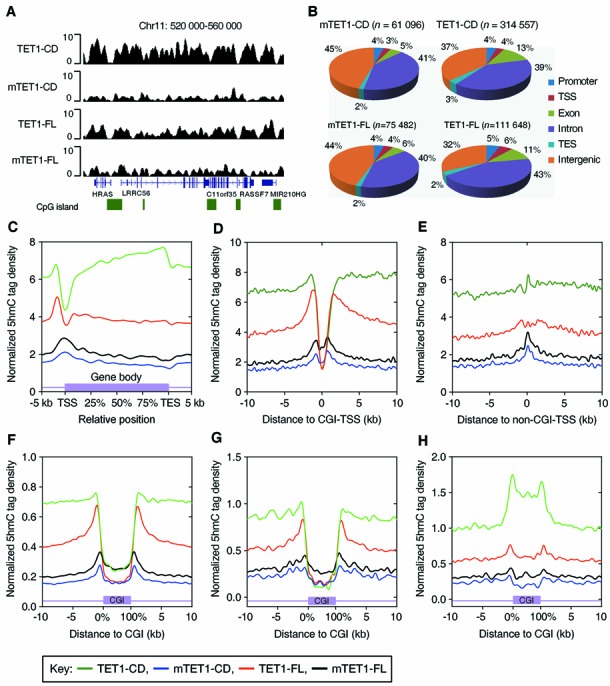
TET1-FL induces specific accumulation of 5hmC at the edges of hypomethylated CGIs. (A) Examples of hMeDIP-Seq profiles in HEK293T cells overexpressing (m)TET1-CD or (m)TET1-FL. Gene distribution (exons) and CGIs are indicated below the graph. (B) The genomic distributions of 5hmC peaks detected in HEK293T cells overexpressing (m)TET1-CD or (m)TET1-FL. Upstream: −5 to −1 kb relative to TSS; promoter: −1 to 0.5 kb relative to TSS; downstream: −0.5 to 1 kb relative to TES; intergenic: 1 kb from TES to −5 kb of downstream gene. (C) Distributions of 5hmC tag across gene bodies in HEK293T cells overexpressing (m)TET1-CD or (m)TET1-FL. Each gene body was normalized to 0–100%. (D and E) Distributions of 5hmC tag around TSSs that overlap CGIs (D) or non-CGIs (E). (F) Distributions of 5hmC tag across CGIs which locates in gene promoters. Total 13 913 out of 27 639 CGIs reside in promoter in human genome. (G and H) Distributions of 5hmC tag across the non-promoter CGIs which were either hypomethylated (methylation < 10%, G) or hypermethylated (methylation > 50%, H) based on DREAM data. Total 739 hypomethylated and 1059 hypermethylated non-promoter CGIs were covered by DREAM results and analyzed for 5hmC distribution. The cells overexpressing (m)TET1-CD or (m)TET1-FL were collected by FACS 3 or 7 days after transfection, respectively. Each CGI in (F–H) was normalized to 0–100%. The key to (C)–(H) is at the bottom.
We next examined the 5hmC distributions in TET1-CD- and TET1-FL-overexpressing cells. Consistent with the DNA dot blot assays (Figure 1C), TET1-FL markedly increased the number of 5hmC peaks (from 75 482 to 111 648), with an even greater increase in TET1-CD-overexpressing cells (from 61 095 to 314 557) (Figure 3B). Importantly, TET1-FL and TET1-CD showed very different 5hmC distribution patterns across gene bodies and around TSSs. While TET1-CD markedly increased 5hmC in gene bodies with higher enrichment in exons than in introns, TET1-FL resembled the distribution of 5hmC in control cells with the highest levels just upstream to TSSs (Figure 3C and Supplementary Figure S4A and B). The contrast between TET1-CD and TET1-FL was more apparent around CGI-overlapped TSSs. In both cases, 5hmC was depleted just at TSSs (Figure 3D). However, TET1-CD induced equal accumulation of 5hmC outside of TSSs, while TET1-FL formed two dramatic 5hmC peaks flanking TSSs (Figure 3D). By contrast, the 5hmC increase induced by TET1-FL was much lower around non-CGI-overlapped TSSs (Figure 3E), suggesting that TET1-FL preferentially functions in CGIs.
We next directly examined the 5hmC distribution across CGIs. Interestingly, the TET1-FL-induced 5hmC peaks described above (Figure 3D) precisely resided at the edges of promoter CGIs, and the increased 5hmC by TET1-CD also spread evenly outside of those CGIs (Figure 3F). However, other CGIs (in upstream, exons, introns, downstream and intergenic regions) showed much less 5hmC accumulation by TET1-FL at their edges and significantly increased 5hmC by TET1-CD over their bodies (Supplementary Figure S5A–E). Contrary to promoter CGIs which were predominantly unmethylated, most non-promoter CGIs were moderately or highly methylated (Supplementary Figure S6A and B). We thus hypothesized that those different 5hmC distribution patterns across promoter and non-promoter CGIs are associated with their different basal methylation levels. As expected, hypomethylated (methylation < 10%) non-promoter CGIs exhibited highly similar 5hmC distribution patterns as promoter CGIs (Figure 3F and G), while methylated (methylation > 50%) non-promoter CGIs showed extremely increased 5hmC by TET1-CD but much less 5hmC enrichment at CGI edges by TET1-FL (Figure 3H). Thus, these results indicate that TET1-FL and TET1-CD differentially regulate 5hmC distribution in HEK293T cells, with a marked preference of TET1-FL toward the edges of hypomethylated CGIs, while TET1-CD appears to monotonously increase 5hmC levels.
DNA hypermethylation inhibits 5hmC production by TET1-FL
The requirement for pre-existing 5mC in TET-catalyzed 5hmC production implies a positive correlation between 5hmC and 5mC distributions in genomic DNA. However, 5hmC and 5mC actually have distinct genomic distributions in mESCs (22,23,25,42). For example, contrary to 5mC, 5hmC is significantly enriched around TSSs but generally not detectable at repetitive elements and minor satellite repeats in mESCs (25). To study this issue, we combined our global DNA methylation and 5hmC distribution data and analyzed the correlation between 5mC and 5hmC levels in TET1-CD- and TET1-FL-overexpressing cells. The 5hmC tag density in TET1-CD-overexpressing cells positively correlated with basal DNA methylation (Figure 4A; Pearson r = 0.89, P = 0.0005). By contrast, 5hmC tag density remained at a relatively constant level regardless of basal methylation in control cells and also in TET1-FL-overexpressing cells, suggesting that high levels of 5mC actually inhibit TET1-FL catalytic function (Figure 4A). We next profiled 5hmC tag density together with DNA methylation levels across gene bodies. The basal DNA methylation levels in control cells were lowest at TSSs and gradually increased along gene bodies, followed by a dramatic drop around TESs (Figure 4B and C). The 5hmC distribution profile in TET1-CD-overexpressing cells highly resembled the basal DNA methylation patterns, further confirming the positive correlation between 5mC and 5hmC in the setting of TET1-CD overexpression (Figure 4B). Indeed, as the 5hmC density increased toward TESs, the extent of TET1-CD-induced DNA demethylation also increased, demonstrating the requirement of high 5hmC production for significant DNA demethylation by TET-CD. By contrast, TET1-FL overexpression failed to produce more 5hmC as basal DNA methylation increased along gene bodies and also failed to induce DNA demethylation (Figure 4C). Therefore, this divergent distribution of 5hmC by TET1-CD and TET1-FL likely explains their differences in inducing DNA demethylation.
Figure 4.
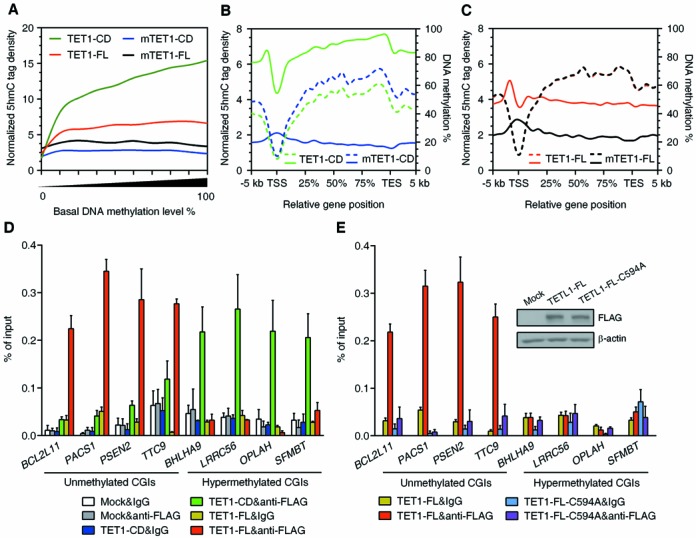
The preferential binding of TET1-FL to unmethylated CGIs through its CXXC domain contributes to its limited 5hmC production and failure to induce significant DNA demethylation. (A) Normalized 5hmC tag density as a function of basal DNA methylation level. The basal methylation levels (% methylation in corresponding mutant control cells) were grouped in 10% intervals from 0 to 100% methylation. (B and C) Normalized 5hmC tag density (left axis) and DNA methylation levels (right axis) plotted across gene bodies in HEK293T cells overexpressing (m)TET1-CD (B) or (m)TET1-FL (C). Solid line represents 5hmC tag density and dotted line DNA methylation. (D) FLAG ChIP-qPCR analysis of TET1-CD and TET1-FL occupancy at differentially methylated CGIs. Data represent mean ± SD (n = 3). The cells were collected by FACS 3 days after transfection. (E) FLAG ChIP-qPCR analysis of TET1-FL and the CXXC domain-mutated TET1-FL-C594A occupancy at differentially methylated CGIs. Data represent mean ± SD (n = 3). The cells were collected by FACS 3 days after transfection. Western blot analysis of TET1-FL and TET1-FL-C594A overexpression is also shown in the graph.
TET1 specifically binds unmethylated CGIs through its CXXC domain
Considering that a high 5hmC yield depends not only on a high level of substrate 5mC but also on an enriched amount of TET1 binding, we hypothesized that the gradual loss of TET1-FL binding as DNA methylation levels increase may underlie its inability to demethylate. Indeed, Tet1-bound CGIs have been reported to be associated with lower 5mC levels compared to the CGIs not bound by Tet1 in mESCs (21). We next compared TET1-FL occupancy at eight unmethylated (BCL2L11, PACS1, PSEN2 and TTC9) or hypermethylated (BHLHA9, LRRC56, OPALH and SFMBT1) promoter CGIs (methylation levels shown in Supplementary Figure S7A) by ChIP-qPCR. TET1-FL was highly enriched at unmethylated CGI promoters but dramatically excluded from hypermethylated ones (Figure 4D). This was also true for endogenous TET1 in HEK293T cells (Supplementary Figure S7B). By contrast, through an unknown mechanism, TET1-CD, which lacks the CXXC domain previously linked to specific binding to CGIs (22), was extensively bound to both kinds of CGIs, with a preference for hypermethylated CGIs (Figure 4D). The CXXC domain-mutated TET1-FL-C594A completely lost enrichment at unmethylated CGI promoters, confirming the CXXC domain-dependent binding of TET1 to genomic DNA (Figure 4E). As a result of the loss of DNA binding, a much lower 5hmC yield was detected by overexpression of TET1-FL-C594A compared to TET1-FL (Supplementary Figure S8). Taken together, our data suggest that the preferential binding of TET1-FL to unmethylated CGIs through its CXXC domain essentially limits its 5hmC production (compared to TET1-CD, seen in Figure 1C) and consequently leads to its failure to induce significant DNA demethylation (Figure 2).
TET1-FL decreases DNA methylation levels in sparsely methylated CGIs
The preferential binding of TET1-FL to unmethylated CGIs further triggered us to ask whether TET1-FL selectively induces demethylation in hypomethylated CGIs. Although generally referred to as ‘unmethylated’, many CGIs actually have low levels of DNA methylation when carefully examined by quantitative (and sensitive) methods. We therefore re-analyzed the DREAM results of (m)TET1-FL transfections with a focus on hypomethylated CGI sites for which very precise methylation data were available by virtue of having a high level of sequence coverage (≥100 tags). Around 1,100 CpG sites were identified with measurable low methylation levels (1–20%) and divided into four groups with basal methylation of 1–5, 5–10, 10–15 and 15–20%, respectively. Using pair-wise analysis, we found that TET1-FL overexpression significantly decreased methylation in sites with 1–5% (P < 0.0001) and 5–10% (P < 0.0001) basal methylation, but not those with 10–15 or 15–20% basal methylation (Figure 5A–D). Therefore, TET1-FL exhibits a specific demethylating activity in sparsely methylated CGIs.
Figure 5.
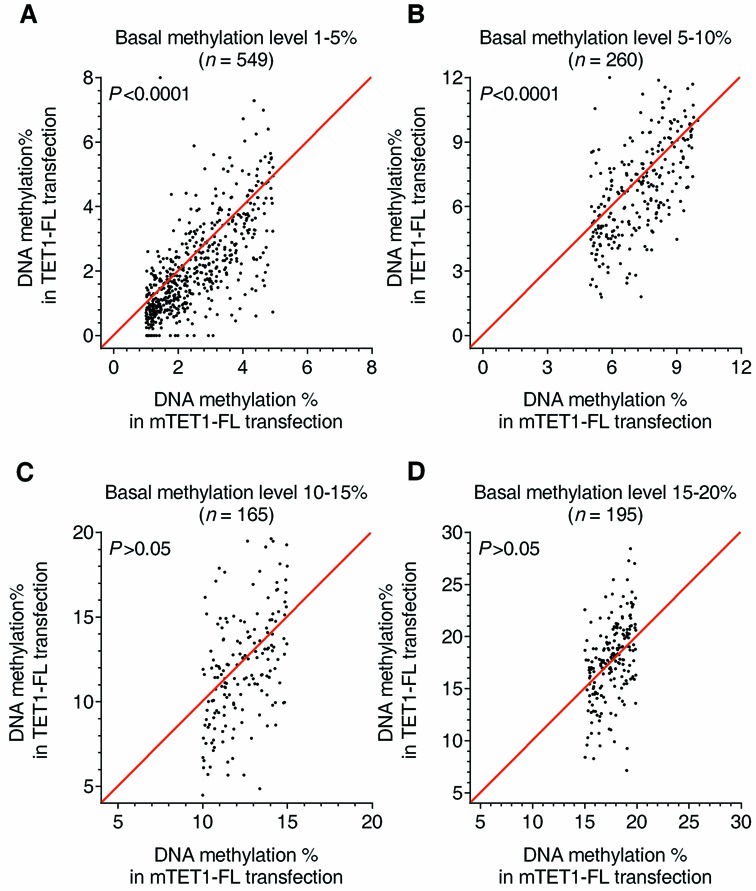
Decreased DNA methylation in sparsely methylated CGIs after TET1-FL overexpression. (A–D) Shown are comparisons of % methylation in HEK293T cells overexpressing TET1-FL versus mTET1-FL for CGI CpG sites with basal DNA methylation levels of 1–5% (A), 5–10% (B), 10–15% (C) or 15–20% (D). The cells were collected by FACS 7 days after transfection. DNA methylation was quantitatively measured by DREAM and only CpG sites with ≥100 tags sequenced were included to ensure accuracy (exact number of sites indicated in each graph). P values were obtained from Wilcoxon paired signed-rank test.
TET1 prevents DNA methylation spreading into CGIs
In contrast to the sporadically distributed CpGs that are heavily methylated, CGIs are typically unmethylated in mammalian genome (2). The above results that TET1-FL induces striking 5hmC accumulation at the edges of hypomethylated CGIs and specific demethylation in sparsely methylated CGIs in HEK293T cells, suggested that TET1 functions primarily to prevent methylation of CGIs rather than dynamically switch methylation states in differentiated cells. To test this directly, we next established shRNA-mediated TET1 knockdown also in HEK293T cells because of their relatively high endogenous TET1 expression (Supplementary Figure S9). Knockdown of TET1 in two independent clones did not change cell morphology but significantly inhibited cell growth (Figure 6A and Supplementary Figure S10) as reported in NIH3T3 cells (43). It also expectedly decreased genomic 5hmC content (Figure 6B), as well as the enrichment of TET1 at the unmethylated promoter CGIs of BCL2L11, PACS1, PSEN2 and TTC9 (Figure 6C and Supplementary Figure S11A). We then studied DNA methylation at both upstream edges and central regions of these four CGIs by bisulfite-cloning-sequencing. Among the eight tested regions in control cells, the edge of BCL2L11 was completely methylated, that of PACS1 showed partial methylation and the other regions were almost unmethylated (Figure 6D and Supplementary Figure S11B–D). TET1 knockdown induced a significant increase of methylation only at the edge of the PACS1 CGI with no measurable changes in the other tested regions (Figure 6D and Supplementary Figure S11B–D), suggesting that TET1 mainly regulates DNA methylation at the boundary between methylated and unmethylated CpG sites close to CGIs. Given that pre-existing DNA methylation could serve as a seed for methylation spreading into nearby unmethylated regions (44,45) and the specific accumulation of 5hmC at CGI edges by TET1-FL, it seems plausible that by its 5mC dioxygenase function TET1 blocks DNA methylation spreading in this context.
Figure 6.

TET1 knockdown leads to DNA methylation spreading at methylated CGI edges. (A) Western blot analysis of shRNA-mediated TET1 knockdown in two independent HEK293T clones transfected with different TET1 shRNA. (B) DNA dot blot shows decreased genomic 5hmC level after TET1 knockdown. (C) ChIP-qPCR shows decreased occupancy of TET1 at unmethylated CGIs after TET1 knockdown. Data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 by Student's t-test compared to shControl clone cells. (D) Bisulfite-sequencing analysis shows increased DNA methylation at the methylated edges of unmethylated CGIs after TET1 knockdown. The top panel contains diagrams of PACS1, KAZN, MUM1, RFX6 and VAX2 CGI promoters. Horizontal green bars represent CGIs. Red bars show the location of boundary amplicons studied by bisulfite-sequencing. In the lower panel, each line represents a different cloned sequence, with black squares representing methylated CpG sites. The average methylation is shown below each panel as mean ± SD (n = 3). In all cases, boundary methylation increased after TET1 knockdown.
To confirm these data, we randomly selected four other unmethylated promoter CGIs (KAZN, MUM1, RFX6 and VAX2) that had methylated edges based on DREAM results (Supplementary Table S2). The specific binding of TET1 and the methylated edges were further validated for each CGI (Figure 6C and D). Consistent with the findings in the PACS1 CGI, the methylated edges of these CGIs also showed significant methylation spreading in both TET1 knockdown clones (Figure 6D). Thus, TET1 knockdown resulted in DNA methylation spreading in all five CGIs specifically at their methylated edges, consistent with the hypothesis that TET1 binds to hypomethylated CGIs (through its CXXC domain) and functions as a ‘maintenance’ demethylase that inhibits the spreading of de novo DNA methylation from methylated CGI edges.
Effects of TET1 on gene transcription
DNA methylation of CGI promoters is associated with repressed gene transcription (2,46), and TET1 has previously been reported to affect gene expression (21,22,25,28). We therefore asked whether TET1 is required for the active transcription of target genes by preventing de novo DNA methylation spreading into the CGI promoters. We first analyzed the effect of TET1 knockdown on the expression of target genes previously analyzed. TET1 knockdown reduced the expression of only three out of five genes for which we confirmed DNA methylation spreading into their promoter CGIs (Figure 7A), and no or inconsistent expression changes of other three genes that had unchanged DNA methylation at their promoter CGIs (Figure 7B). We next used cDNA microarrays to analyze whole genome gene expression changes after TET1 knockdown. Using a criterion of 1.5-fold expression change, 89 upregulated and 97 downregulated genes were identified in both TET1 knockdown cell clones (Supplementary Figure S12A), and some of them were further validated by RT-qPCR (Supplementary Figure S12B and C). Three of these validated downregulated genes were further proved to be TET1 target genes but did not consistently gain DNA methylation spreading in their CGI promoters after TET1 knockdown (Supplementary Figure S12D and E). Thus, consistent with previous results indicating that depletion of Tet1 induces similar gene expression changes in wild type and Dnmt TKO mESCs (25), our data suggest relatively minor effects of TET1 on gene transcription in HEK293T cells that are both DNA methylation-dependent and -independent.
Figure 7.
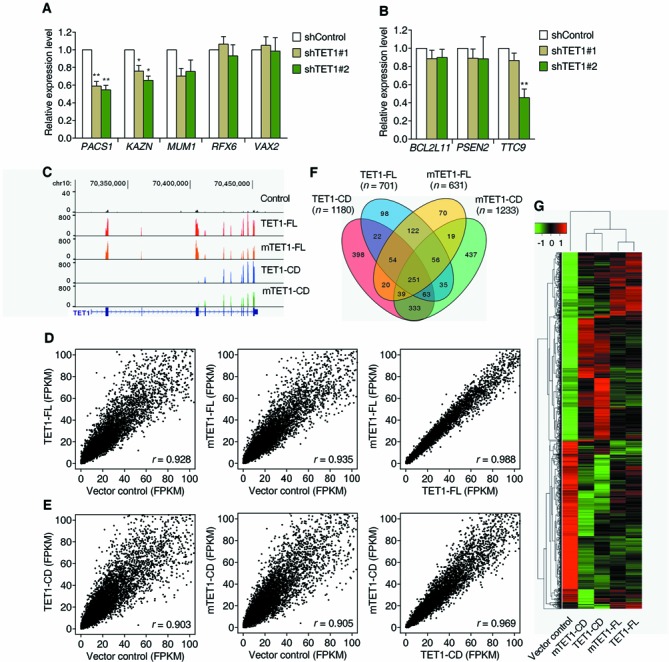
TET1 regulates gene transcription independent of its demethylating activity. (A and B) Effect of TET1 knockdown on the expressions of its target genes with (A) or without (B) increased DNA methylation spreading into their CGI promotes. Data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 by Student's t-test compared to shControl cells. (C) RNA-seq transcriptional landscapes of the TET1 gene in the vector control and (m)TET1-FL- or (m)TET1-CD-overexpressing HEK293T cells. The two introduced substitution mutations in mTET1-FL and mTET1-CD were also validated by RNA-seq. (D and E) Scatter plots comparing the RNA-seq-derived gene expression profiles among the vector control, TET1-FL and mTET1-FL overexpressions (D), and among the vector control, TET1-CD and mTET1-CD overexpressions (E). Pearson correlation coefficients, r, are listed in each graph. (F) Venn diagram showing the overlap of differentially expressed genes (>2-fold change, FDR < 0.05) of TET1-FL, mTET1-FL, TET1-CD and mTET1-CD overexpressions. (G) Heatmap showing hierarchical clustering of differentially expressed genes of TET1-FL, mTET1-FL, TET1-CD and mTET1-CD overexpressions. All cells were collected by FACS 3 days after transfection.
To more directly determine whether TET1 functions in gene transcription regulation, we undertook RNA-seq analysis of HEK293T cells after overexpression of TET1-FL or mTET1-FL. As expected, TET1-FL- and mTET1-FL-overexpressing cells showed an extremely high mRNA level of TET1 (∼150-fold increase compared with the vector control, Figure 7C). Strikingly, TET1-FL and mTET1-FL had a nearly identical gene expression profile, but both were significantly different from the control transfections (Figure 7D). Consistent with this, when we computed differentially expressed genes (>2-fold change, FDR < 0.05) in TET1-FL and mTET1-FL compared with control, we found a significant overlap and a strong positive correlation between the gene sets (Figure 7F and G). Thus, overexpression of TET1-FL induces significant expression changes despite its limited ability to increase 5hmC, and these changes are also seen with an enzymatically dead mutant TET1 vector (mTET1-FL). Our data do not imply a direct contribution of TET1 to gene expression and suggest that the effects observed are independent of its catalytic activity and of its demethylating activity.
Very similar results were seen with TET1-CD. Both TET1-CD and mTET1-CD overexpression changed gene expression profiles compared with the control, even though only TET1-CD induced massive DNA demethylation (Figure 7C and E). TET1-CD and mTET1-CD also had very similar gene expression profiles (Figure 7E) despite the mutation in the catalytic domain of mTET1-CD, and their differentially expressed genes compared to controls also significantly overlapped and positively correlated (Figure 7F and G). The differentially expressed genes of (m)TET1-CD overexpression substantially overlapped and positively correlated with those of (m)TET1-FL (Figure 7F and G), suggesting that the catalytic domain is still important to the observed gene expression differences, even though this effect is independent of its ability to catalyze 5hmC formation and induce demethylation. The above RNA-seq data were also confirmed by gene expression microarrays with similar results (Supplementary Figure S13A and B). Thus, these results conclusively establish that the gene expression changes seen after TET1 overexpression or depletion are separate from and independent of its demethylating activity.
DISCUSSION
In this paper, we demonstrate that TET1-FL overexpression has minimal effects on global DNA methylation. Rather, TET1 is a critical component of methylation boundaries and serves as a maintenance DNA demethylase that prevents aberrant methylation spreading into hypomethylated CGIs. We also provide evidence that the effects of TET1 on gene transcription observed after overexpression are unrelated to its 5mC dioxygenase function.
Our results are different from previous studies which investigated the DNA demethylating effects of TET1 using overexpression of a truncated catalytic domain-only version of TET1 (TET1-CD) (17,32). We found that TET1-CD but not TET1-FL induces genome-wide demethylation. This difference cannot be simply the result of the lower TET1-FL expression because TET1-FL and TET1-CD had similar mRNA levels by RNA-seq (Figure 7C) and the apparent difference in protein expression is likely overestimated due to their differing membrane transfer efficiency due to the large difference in protein size. Moreover, increasing expression time significantly compensated for TET1-FL low expression level in terms of 5hmC production (Figure 1C), and finally, the different regulation patterns of 5hmC distribution (Figure 3C–H) cannot be accounted for by differing expression levels. Conversely, the failure of TET1-FL to induce genome-wide demethylation can be essentially explained by its DNA CpG motif binding domain, CXXC, which appears to strongly limit the 5hmC production capacity and demethylating potential of TET1 by guiding it toward hypomethylated DNA.
Previous studies have also shown that Tet1 is predominantly enriched at CpG-rich genomic regions (21,22,25), where most CpGs are unmethylated (2,47). We found that TET1 binds unmethylated but not hypermethylated CGIs (Figure 4E and F). Thus, the CXXC domain and the catalytic domain of TET1 form an interesting but conflicting domain combination: the CXXC domain specifically targets TET1 to hypomethylated regions where its substrate (5mC) is almost depleted and consequently limits 5hmC yield and demethylating activity of the catalytic domain in hypermethylated genomic regions. Therefore, at least in HEK293T cells, TET1-mediated 5mC oxidation is not used for global DNA demethylation but rather prevents methylation spreading into CGIs. By contrast, the absence of the CXXC domain and the binding to both unmethylated and hypermethylated CGIs (with a preference for the latter, Figure 4D) adequately explain why TET1-CD overexpression is able to induce massive global DNA demethylation. It is interesting to note that TET1-FL increases 5hmC by DNA dot blots and hMeDIP-Seq yet fails to affect DNA methylation genome wide. This paradox may be technical in part—it is impossible to relate dot blot 5-hmC levels to actual 5-hmC amount/methylated CpG site. Considering that the basal 5hmC level is extremely low in HEK293 cells (only ∼0.35% of 5mC) (14), doubling or tripling this amount may be detectable by an enrichment method such as hMeDIP-seq but may not be enough to significantly change global DNA methylation level. Also, the hMeDIP-seq data show that 5hmC increase with TET-FL is highly concentrated at the edges of hypomethylated CpG islands thus affecting <1% of all CpG sites, which might not result in significant global demethylation.
We note that the TET1 CXXC domain was recently reported to bind both unmethylated and methylated DNA probes in in vitro GST pull-down assays (22,32). Still, even in these studies, the binding of the TET1 CXXC domain to methylated DNA was much lower than that to unmethylated DNA probes. Furthermore, this detected binding to a methylated DNA probe may disappear in vivo, in the complex chromatin environment in cells. For example, the recruitment of endogenous methyl-CpG binding proteins to methylated CpGs and the densely packed heterochromatin state in hypermethylated genomic regions both could dramatically inhibit the access of TET1 to methylated CpGs. Consistent with this, we were unable to detect binding of transfected or endogenous TET1 to methylated CGIs in our ChIP-qPCR studies.
Our finding that TET1-FL is unable to induce global DNA demethylation in differentiated cells may provide some mechanistic insight into the recent studies on the role of TET1 in embryonic development. Although the conversion of 5mC to 5hmC underlies global demethylation in mouse primordial germ cells where Tet1 and Tet2 is highly expressed, loss of Tet1 and Tet2 does not greatly affect this epigenome remodeling (26,28). Moreover, both Tet1-knockout and Tet1/Tet2 double-knockout mice are viable, fertile and grossly normal, though abnormal imprinting of some genes can be detected in a fraction of Tet1/Tet2 double-knockout embryos (48,49). Thus, the conflicting combination of CXXC and catalytic domains may explain why Tet1 has a limited role in global demethylation in primordial germ cells. The subtle changes in DNA methylation induced by TET1 would suggest that this protein may play a greater role in situations where there is a stress on the system and a pressure to methylate CpG islands, such as during aging, in inflammatory conditions or in cancer (50–52).
Our observation that TET1-FL induces specific accumulation of 5hmC at the edges of hypomethylated CGIs, while TET1 knockdown induces methylation spreading into hypomethylated CGIs also uncovers a DNA demethylase-based mechanism for the immunity of CGIs to DNA methylation, which is one of the most striking feature of DNA methylation patterns in mammals (2). By contrast, all the other known methylation-protecting factors such as binding of transcription factors, high transcription activity, and the active chromatin mark H3K4me3, protect CGIs from de novo DNA methylation by excluding DNMTs (53). Thus, the demethylase-based mechanism may reasonably cooperate with the DNMT-exclusion mechanism to provide a more solid protection for hypomethylated CGIs against methylation attack. This finding also suggests a possible involvement of TET1 in the yet unexplained occurrence of aberrant CGI hypermethylation in aging and cancer, which provides cancer cells with an advantage in cell growth and invasion (1,54). Indeed, methylation boundaries around CGIs become much less well-defined during cancer formation (55), and it would be interesting to see if this is caused in part by the frequently detected TET1 mutations and downregulation in neoplastic cells (31,56–59).
One of the paradoxes in the TET protein field has been the disconnection between effects on DNA methylation and effects on gene expression. Recent studies have suggested a complex function of Tet1 in gene transcription regulation in mESCs: Tet1 knockdown induces similar upregulation and downregulation of gene transcription in wild type and Dnmt KO mESCs (25) and, by recruiting PRC2 and Sin3a to chromatin, Tet1 has been proposed to repress gene transcription independent of its effects on DNA methylation (21,25). More recent studies also suggested that Tet1 regulates gene transcription through its interaction with O-linked N-acetylglucosamine transferase (60–62). However, the lack of a major phenotype in the Tet1-knockout and Tet1/Tet2 double-knockout mice suggest that the effects of Tet1 on transcription may be relatively minor. Here, by comparing the wild type TET1-FL and its catalytically mutant control, we directly demonstrate that the gene expression changes observed after TET1 overexpression are independent of its enzymatic and demethylating activity. It remains to be seen whether these effects are direct or indirect, and whether they are physiologically relevant. These data also have implications for understanding the function of TET2 and TET3 as well. In particular, TET2 mutations that are common in myeloid leukemias seem to have little effects on CGI DNA methylation (63). Dissecting DNA methylation-dependent and -independent effects of TET2 will be important to understand its role in cancer development.
In summary, our study demonstrates that in post-development cells TET1 works as a maintenance DNA demethylase which does not purposely decrease DNA methylation levels, but rather specifically prevents de novo DNA methylation spreading from methylated CGI edges into CGIs using its 5mC dioxygenase catalytic function. These findings support a role for DNA demethylation in maintaining normal DNA methylation patterns post-development and have implications for understanding methylation deregulation in aging and cancer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACCESSION NUMBERS
GEO accession numbers: GSE44038, GSE44036, GSE50016 and GSE49833.
FUNDING
National Institutes of Health [CA158112 to J.P.I.]; Ellison Medical Foundation (to J.P.I.); F.M. Kirby Foundation (to J.P.I.). Funding for open access charge: Fels Institute, Temple University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 2.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh C.L. Dynamics of DNA methylation pattern. Curr. Opin. Genet. Dev. 2000;10:224–228. doi: 10.1016/s0959-437x(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 4.Bergman Y., Cedar H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013;20:274–281. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- 5.Okano M., Xie S., Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 6.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 7.Bestor T., Laudano A., Mattaliano R., Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988;203:971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 8.Hermann A., Goyal R., Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 9.Wu S.C., Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ooi S.K., Bestor T.H. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Inoue A., Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue A., Shen L., Dai Q., He C., Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21:1670–1676. doi: 10.1038/cr.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valinluck V., Sowers L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 14.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maiti A., Drohat A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo J.U., Su Y., Zhong C., Ming G.L., Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H., Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S., D'Alessio A.C., Taranova O.V., Hong K., Sowers L.C., Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H., D'Alessio A.C., Ito S., Xia K., Wang Z., Cui K., Zhao K., Sun Y.E., Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y., Wu F., Tan L., Kong L., Xiong L., Deng J., Barbera A.J., Zheng L., Zhang H., Huang S., et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ficz G., Branco M.R., Seisenberger S., Santos F., Krueger F., Hore T.A., Marques C.J., Andrews S., Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 24.Koh K.P., Yabuuchi A., Rao S., Huang Y., Cunniff K., Nardone J., Laiho A., Tahiliani M., Sommer C.A., Mostoslavsky G., et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams K., Christensen J., Pedersen M.T., Johansen J.V., Cloos P.A., Rappsilber J., Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vincent J.J., Huang Y., Chen P.Y., Feng S., Calvopina J.H., Nee K., Lee S.A., Le T., Yoon A.J., Faull K., et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hackett J.A., Sengupta R., Zylicz J.J., Murakami K., Lee C., Down T.A., Surani M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaguchi S., Hong K., Liu R., Shen L., Inoue A., Diep D., Zhang K., Zhang Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature. 2012;492:443–447. doi: 10.1038/nature11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piccolo F.M., Bagci H., Brown K.E., Landeira D., Soza-Ried J., Feytout A., Mooijman D., Hajkova P., Leitch H.G., Tada T., et al. Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol. Cell. 2013;49:1023–1033. doi: 10.1016/j.molcel.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y., Chen J., Li K., Wu T., Huang B., Liu W., Kou X., Zhang Y., Huang H., Jiang Y., et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12:453–469. doi: 10.1016/j.stem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Lorsbach R.B., Moore J., Mathew S., Raimondi S.C., Mukatira S.T., Downing J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H., Zhang X., Clark E., Mulcahey M., Huang S., Shi Y.G. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390–1393. doi: 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- 33.Kroeger H., Jelinek J., Estecio M.R., He R., Kondo K., Chung W., Zhang L., Shen L., Kantarjian H.M., Bueso-Ramos C.E., et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood. 2008;112:1366–1373. doi: 10.1182/blood-2007-11-126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colella S., Shen L., Baggerly K.A., Issa J.P., Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques. 2003;35:146–150. doi: 10.2144/03351md01. [DOI] [PubMed] [Google Scholar]
- 35.Jelinek J., Liang S., Lu Y., He R., Ramagli L.S., Shpall E.J., Estecio M.R., Issa J.P. Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics. 2012;7:1368–1378. doi: 10.4161/epi.22552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H., Handoko L., Wei X., Ye C., Sheng J., Wei C.L., Lin F., Sung W.K. A signal-noise model for significance analysis of ChIP-seq with negative control. Bioinformatics. 2010;26:1199–1204. doi: 10.1093/bioinformatics/btq128. [DOI] [PubMed] [Google Scholar]
- 37.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y., Pastor W.A., Shen Y., Tahiliani M., Liu D.R., Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth M.J., Branco M.R., Ficz G., Oxley D., Krueger F., Reik W., Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 42.Wu H., D'Alessio A.C., Ito S., Wang Z., Cui K., Zhao K., Sun Y.E., Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang S., Zhu Z., Wang Y., Xu L., Chen X., Xu Q., Zhang Q., Zhao X., Yu Y., Wu D. Tet1 is required for Rb phosphorylation during G1/S phase transition. Biochem. Biophys. Res. Commun. 2013;434:241–244. doi: 10.1016/j.bbrc.2013.02.110. [DOI] [PubMed] [Google Scholar]
- 44.Song J.Z., Stirzaker C., Harrison J., Melki J.R., Clark S.J. Hypermethylation trigger of the glutathione-S-transferase gene (GSTP1) in prostate cancer cells. Oncogene. 2002;21:1048–1061. doi: 10.1038/sj.onc.1205153. [DOI] [PubMed] [Google Scholar]
- 45.Turker M.S. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene. 2002;21:5388–5393. doi: 10.1038/sj.onc.1205599. [DOI] [PubMed] [Google Scholar]
- 46.Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 47.Bird A.P. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 48.Dawlaty M.M., Ganz K., Powell B.E., Hu Y.C., Markoulaki S., Cheng A.W., Gao Q., Kim J., Choi S.W., Page D.C., et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dawlaty M.M., Breiling A., Le T., Raddatz G., Barrasa M.I., Cheng A.W., Gao Q., Powell B.E., Li Z., Xu M., et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev. Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baylin S.B., Herman J.G., Graff J.R., Vertino P.M., Issa J.P. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- 51.Issa J.P. CpG-island methylation in aging and cancer. Curr. Top. Microbiol. Immunol. 2000;249:101–118. doi: 10.1007/978-3-642-59696-4_7. [DOI] [PubMed] [Google Scholar]
- 52.Issa J.P., Ahuja N., Toyota M., Bronner M.P., Brentnall T.A. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577. [PubMed] [Google Scholar]
- 53.Deaton A.M., Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herman J.G., Baylin S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 55.Hansen K.D., Timp W., Bravo H.C., Sabunciyan S., Langmead B., McDonald O.G., Wen B., Wu H., Liu Y., Diep D., et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ono R., Taki T., Taketani T., Taniwaki M., Kobayashi H., Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002;62:4075–4080. [PubMed] [Google Scholar]
- 57.Kudo Y., Tateishi K., Yamamoto K., Yamamoto S., Asaoka Y., Ijichi H., Nagae G., Yoshida H., Aburatani H., Koike K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103:670–676. doi: 10.1111/j.1349-7006.2012.02213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seshagiri S., Stawiski E.W., Durinck S., Modrusan Z., Storm E.E., Conboy C.B., Chaudhuri S., Guan Y., Janakiraman V., Jaiswal B.S., et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu C.H., Peng K.L., Kang M.L., Chen Y.R., Yang Y.C., Tsai C.H., Chu C.S., Jeng Y.M., Chen Y.T., Lin F.M., et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568–579. doi: 10.1016/j.celrep.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 60.Chen Q., Chen Y., Bian C., Fujiki R., Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deplus R., Delatte B., Schwinn M.K., Defrance M., Mendez J., Murphy N., Dawson M.A., Volkmar M., Putmans P., Calonne E., et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vella P., Scelfo A., Jammula S., Chiacchiera F., Williams K., Cuomo A., Roberto A., Christensen J., Bonaldi T., Helin K., et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell. 2013;49:645–656. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 63.Yamazaki J., Taby R., Vasanthakumar A., Macrae T., Ostler K.R., Shen L., Kantarjian H.M., Estecio M.R., Jelinek J., Godley L.A., et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics. 2012;7:201–207. doi: 10.4161/epi.7.2.19015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.