


Abstract
Epigenetics are the heritable changes in gene expression or cellular phenotype caused by mechanisms other than changes in the underlying DNA sequence. After mitosis, it is thought that bookmarking transcription factors remain at promoters, regulating which genes become active and which remain silent. Herein, we demonstrate that poly(ADP-ribose)polymerase-1 (PARP-1) is a genome-wide epigenetic memory mark in mitotic chromatin, and we further show that the presence of PARP-1 is absolutely crucial for reactivation of transcription after mitosis. Based on these findings, a novel molecular model of epigenetic memory transmission through the cell cycle is proposed.
INTRODUCTION
Mitotic chromatin is transcriptionally inactive (1). The majority of transcription factors and ribonucleicacid (RNA) polymerases are excluded from mitotic chromatin, albeit a few are retained (2–7). Following the completion of cell division, regulatory proteins need to re-engage their genomic targets to restore appropriate gene transcription states (8–12). Epigenetic marking of mitotic chromatin is involved in precise post-mitotic restoration of proper transcriptional patterns to avoid disastrous regulatory consequences (13,14). Such marks include certain histone and DNA modifications that remain in mitotic chromatin (15–18). Deoxyribonucleic acid (DNA) methylation is known to suppress transcription upon completion of mitosis (19,20), whereas relation between specific histone modifications and gene expression states remains somewhat illusive. To reset different active states of genes after mitosis, transcription factors and their attendant co-regulators must find their appropriate sites in transcriptionally silent chromatin. Mechanisms responsible for re-engagement of transcription factors and their co-regulators in chromatin after cell division are in many respects similar to mechanisms responsible for chromatin programming during cell differentiation, when regulatory factors engage transcriptionally silent genes in progenitor cells and activate expression de novo. Histone modifications can help recruit transcription factors and co-regulators to target sites in chromatin (21–23). Early embryonic cells already possess distinct histone modification patterns at a subset of silent genes, apparently marking genes cinched to be activated (24). Nevertheless, these known modifications of histones and DNAs likely represent only a subset of epigenetic marks (25).
In a living cell, the activation of chromatin-associated PARP-1 leads to assembly of poly(ADP-ribose) (pADPr) via transfer of ADP-ribose to various substrates, including existing pADPr chain, PARP-1 itself and other acceptor proteins. Subsequent accumulation of pADPr neutralizes positively charged groups, mediating chromatin decondensation and stimulating transcription (26–30). pADPr chain averages 80 moieties, although length variation is substantial (31). PARP-1 is active as a dimer (32,33). A cluster of 10–28 Glu residues located near the center of the PARP-1 dimerization domain serves as the major acceptor of ADPr moieties. Auto-ADPr-modification of PARP-1 affects its dimerization domain, causing PARP-1 dimers to dissociate from each other and from active chromatin and lose activity. This negative feedback loop mediated by automodification limits the time during which PARP-1 remains active.
In steady state conditions, PARP-1 is unevenly distributed in the chromatin and often found near the promoter regions of specific genes (34,35). We hypothesized that PARP-1 is an epigenetic memory factor, capable of marking specific chromatin regions during mitosis to be reactivated at the start of interphase. We report the results of whole-genomic comparative analysis of PARP-1 distribution in untreated asynchronous human embryonic kidney (HEK293) cells and cells synchronized in the mitotic phase by nocodazole treatment. This experimental system allows accumulation of up to 99% of a total cell population in mitosis (36). These cells remain alive, and upon nocodazole removal immediately mitosis (Supplementary Figure S1A–D). Our experiments demonstrated that PARP-1 establishes stable epigenetic marks in metaphase chromatin at the transcription start sites of a large subset of genes and that the presence of these marks is necessary for transcriptional restart after mitosis.
MATERIALS AND METHODS
Cell lines and cell culture
Human Embryonic Kidney (HEK293) cells (catalog no. CRL-1573) were purchased from American Type Culture Collection (Boston, MA, USA) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with antibiotics (streptomycin, 100 mg/ml; penicillin, 100 IU/ml), and 10% (w/v) newborn calf serum (NCS) at 37ºC in a humidified atmosphere containing 5% CO2. Nocodazole, olabarib and Epidermal Growth Factor (EGF) were used to treat cells in specific experiments. Short hairpin (sh) RNA control and shRNA PARP-1-transfected cells were synchronized using nocodazole and released in the presence or absence of olaparib. PARP-1 protein in knockdown cells was assayed by western blot.
Immuno-electron-microscope and immunostaining
For EM immunocytochemistry, samples were prepared according to (37). The cells were fixed in 4% formaldehyde/0.2% glutaraldehyde in 0.1 M PHEM [60 mM PIPES, 25 mM HEPES, 2 mM MgCl2, 10 mM EGTA (pH 6.9)], cryoprotected in 2.3 M sucrose, mounted on aluminum pins and frozen in liquid nitrogen. Thin-frozen sections were then cut on a Leica EM UC6/FC6 cryomicrotome (Leica), collected on a drop of sucrose/methylcellulose mixture and placed on a formvar-carbon grid. The sections were labeled with primary antibody and the label was subsequently visualized by colloidal gold conjugated to protein A. Sections were stained/embedded in 2% methylcellulose/0.2% uranyl acetate and observed under a Tecnai 12 transmission electron microscope. Immunostaining was performed as described (38).
Expression vector and cell transfection
Cloning vector pBKSII (BKS) is commercially available from Agilent/Strategene, Inc. Short hairpin RNA (shRNA) expression vectors were generated as described previously (39). Short hairpins of 27–29 nucleotides for PARP-1 were expressed by the U6 promoter in the pENTR/DTopo vector (Invitrogen). PGEM1 plasmid containing the U6 promoter was used as the template for PCR with the Sp6 primer (CACCGATTTAGGTGACACTATAG) and PARP-1 primer (AAAAAAAG GACAAGAC GTACGCTAA GAACAAC CCCCAAG CTTCGGAG TTGTTCTTA GCGCACA TCTTGTCC CGGTGT TTCGTCCTT TCCACAA) (40). Cells were transfected by Effectene transfection reagent (Qiagen) according to manufacturer's instructions.
Chemicals and cell treatment
To synchronize cells, nocodazole (Sigma) was dissolved at 10 mg/ml in dimethyl sulfoxide (DMSO) and stored at −20°C. PARP-1 inhibitor, olaparib (AZD-2281, Selleckchem), was dissolved in DMSO (10 mM stock solution) and stored at −20°C. EGF (Sigma) was dissolved in phosphate buffered saline (PBS) and stored (10 μg/ml stock) at 4°C. Small aliquots of these stock solutions were used once.
Cell synchronization
The cells were initially plated at a density of 5 × 105 cells in a 10-cm dish. One day after seeding, the cells were washed twice with prewarmed PBS at 37°C and incubated with or without 100 ng/ml nocodazole-containing medium. Eighteen hours later, the media were removed and the cells were washed twice with prewarmed PBS. Mitotic arrested cells were then harvested and used either for western blotting or ChIP.
Olaparib and EGF treatment
Nocodazole-treated cells were released by changing fresh media after washing twice with PBS in the presence and absence of olaparib (10 μM) for 2 h, unless otherwise indicated, at 37°C in culture dishes, followed by harvesting the cells. For the EGF experiment, cells were treated with EGF (50 ng/ml) for 1 h (41). Cells were treated with nocodazole or olaparib or EGF in 60 mm dishes.
Flow cytometric analysis of cell cycle status
After nocodazole treatment, control or shRNAPARP-1 transfected cells were released at different time points (0, 1:30, 2:30, 4 and 5:30 h), then cells were harvested, washed twice with ice-cold PBS and fixed in 70% ethanol. Tubes containing the cell pellets were stored at −20°C. Afterward, the cells were centrifuged at 1000 g for 10 min and the supernatant was discarded. The pellets were resuspended in 30 μl of phosphate/citrate buffer (0.2 Na2HPO4 and 0.1 citric acid, pH 7.5) at room temperature for 30 min. Cells were then washed with 5 ml of PBS and incubated with 400 μl of propidium iodide (PI) solution (50 μg/ml PI, 1 mg/ml RNase A and 1 g glucose/l of PBS) for 30 min. The samples were analyzed on a Coulter Elite flow cytometer.
Antibodies
The following antibodies were used for the ChIP experiment: rabbit polyclonal anti-PARP (ab6079, Abcam), anti-H2AX (#39689, Active motif), anti-H2A.Z (#39943, Active motif), mouse anti-NFATC2 (ab2722, Abcam) and anti-H2A (Active motif, #39111). For western blot analysis, anti-PARP-1 (C2–10, Trevigen), anti-H3 (Millipore), anti-H3S10 (Millipore), anti-H3T3 (Millipore), mouse monoclonal anti-α tubulin (Sigma), anti-NFATC2 (Thermo, #PA5-19164), anti-RNAPolII (Covance, 8WG16) and anti-pADPr (Trevigen, 10H) were used. For electron microscopy (EM) immunocytochemistry, anti-H1 antibody (sc8030, Santa Cruz) and anti-PARP-1 (ab6079, Abcam) were used. Either goat anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (Sigma) were used.
Western blotting
Western blotting was performed on nocodazole-arrested released shRNAcontrol cells or shRNAPARP-1-transfected cells using the detection kit from Amersham/GE Healthcare (#RPN2106), according to the manufacturer's instructions. For semi-quantitative protein analysis, whole cell extracts (0.8 × 105 cells) were prepared by boiling cells for 10 min in sodium dodecyl sulfate (SDS) sample buffer [25 mM Tris (pH 6.8), 2% β-mercaptoethanol, 3% SDS, 0.1% bromophenol blue, 5% glycerol] at 1 × 107 cells/ml. Lysed cell extracts were resolved by SDS-PAGE (4–10% NuPAGE, Invitrogen) and transferred to nitro-cellulose membrane (BioRad) by i-Blot dry blotting system (Invitrogen). Detection was performed with ECL-Plus (Amersham) and autoradiography film (HyBlot CL).
RNA isolation and qPCR
Total RNA was isolated according to the manufacturer's instructions (Qiagen) from shRNAcontrol and shRNAPARP-1-transfected cells or olaparib-treated cells. Purified total RNA was treated by Deoxyribonuclease I (Qiagen). First strand of cDNA was synthesized from 5 μg of purified DNAse-treated total RNA, according to manufacturer's instructions (Invitrogen). Quantitative PCR was performed on a StepOnePlus q-PCR System (Applied Biosystems) using 2× SYBR Green master PCR mix (Applied Biosystems). All amplifications were performed in triplicate using 2.0 μl of complementary DNA (cDNA) per reaction. Triplicate mean values were calculated according to the ΔΔCt quantification method using GAPDH gene transcription as reference for normalization. Changes in expression were quantitated by the ΔΔ threshold cycle (ΔΔCt) method as described (42). Control reactions lacking reverse transcriptase yielded little to no signals.
RESULTS
PARP-1 remains bound to chromatin during mitosis
We first compared the PARP-1 protein distribution in interphase chromatin and metaphase-arrested chromosomes (Figure 1A), confirming the metaphase status of DNA by co-staining with phosphoH3/Ser10, a well-known marker of mitotic chromatin (43) (Supplementary Figure S1). PARP-1 remained bound to condensed chromatin during mitosis (Figure 1A). To assess the distribution PARP-1 in metaphase chromatin, we performed ultrastructural analysis of metaphase chromosomes using immuno-electron microscopy (EM) (37) and immunofluorescence. EM immunocytochemistry deploying anti-PARP-1 and the antibody to linker histone H1 (Figures 1B–D) revealed a number of well-defined domains in the condensed chromatin occupied by PARP-1. Confocal microscopy (Figure 1E) confirmed that PARP-1 and H1 localized to distinct non-overlapping blocks in mitotic chromatin (Figure 1E). Similar anti-correlation in PARP-1 and H1 distributions has been previously observed in interphase chromatin (28,35).
Figure 1.
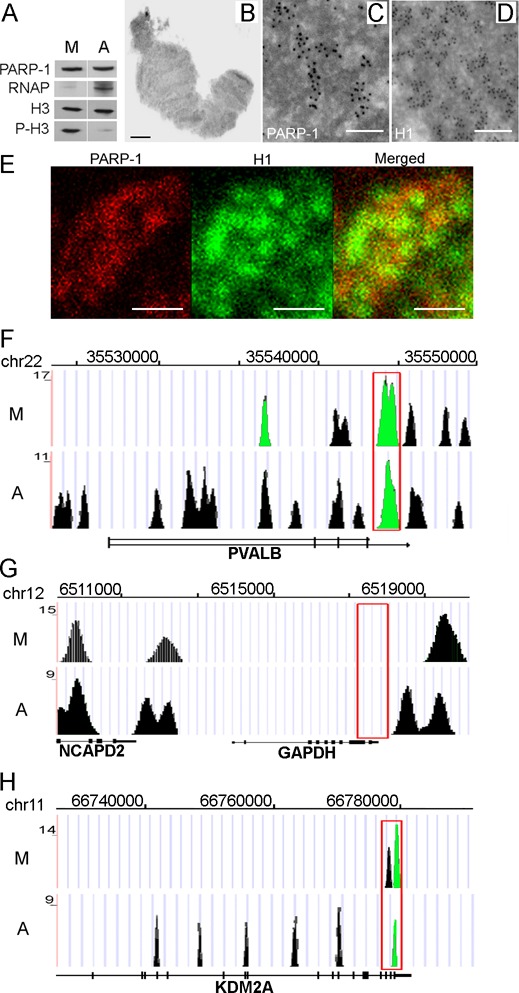
PARP-1 is associated with chromatin during mitosis. (A) Equal amounts of total chromatin proteins extracted from asynchronous cells and cells arrested in mitosis were analyzed after PAGE on western blot using anti-PARP-1, anti-RNAP (RNA polymerase II), anti-H3 and anti-phospho-Ser10-H3 (marker of mitotic chromatin) antibodies (B–D). Panel (B) shows typical single mitotic chromosome used for Immuno-EM analysis. Immuno-EM analysis of PARP-1 (C) and H1 histone (D) localization in mitotic chromatin. Scale bars correspond to 1 mm (B) and 100 nm (C and D). (E) Confocal microscopy analysis of PARP-1 (red) and H1 histone (green) localization in mitotic chromatin. Lack of overlap between red and green areas indicates the extent to which PARP-1 and H1 occupy different genomic domains. Scale bars correspond to 5 nm. (F–H) The identification of PARP-1 protein binding in mitotic (M) and interphase (asynchronous) (A) chromatin using ChIP-Seq assays. UCSC genome browser view is presented based on ChIP-Seq data. Red box represents the area of promoter sequences. Significant peaks are shown as green. (F) PARP-1 binding in the previously reported PARP-1-dependent gene PVALB. (G) Absence of PARP-1 binding along PARP-1-independent GAPDH locus. (H) PARP-1 binds along KDM2A gene sequences.
To determine exact genomic distribution of PARP-1 in metaphase chromosomes, we performed ChIP-Seq assays with anti-PARP-1 antibody (see ‘Materials and Methods’ section) in both asynchronous and mitosis-arrested cells (Supplementary Table S1). We used ChIP-Seq signal with Input and IgG as a background control and confirmed the specificity of ChIP-Seq protocol by determining PARP-1 occupancy at previously reported PARP-1-dependent loci, PVALB and GDF15 (35), versus constitutively expressed PARP-1-independent genes, GAPDH and Actin. PARP-1 binding was enriched at PARP-1-dependent genes (Figure 1F–H; Supplementary Figure S1E), but completely absent at PARP-1-independent gene sequences in both metaphase and interphase chromatin (Figure 1G; Supplementary Figure S1E; Supplementary Figure S2).
PARP-1 accumulates near transcriptional start sites in mitotic chromatin
Analysis of cell-phase specific PARP-1 occupancy identified three groups of PARP-1 binding sites: interphase-specific, mitotic-specific and shared (Figure 2), indicating that PARP-1 performs different functions during different segments of the cell cycle. We identified 12 823 interphase PARP-1 binding sites (Supplementary Table S2) and 25 388 mitotic sites (Supplementary Table S3) (Supplementary Figures S3 and S4). Analysis of PARP-1 occupancy relative to annotated genomic features in both mitotic and interphase chromatin revealed marked binding enrichment near transcriptional start sites (TSSs) (Figure 2A). Based on technical limitations of ChIP-seq resolution, any distance less than 200 bp was considered below method resolution and assumed to represent the same binding site. Only 135 binding sites that shared between interphase and mitotic chromatin were located closer than 200 bp from one another (Supplementary Table S4) (Figure 2B). The overwhelming majority of PARP-1 binding sites was different between mitotic and interphase chromatin, strongly suggesting that PARP-1 becomes redistributed during the transition from mitosis to interphase (Figure 2C).
Figure 2.
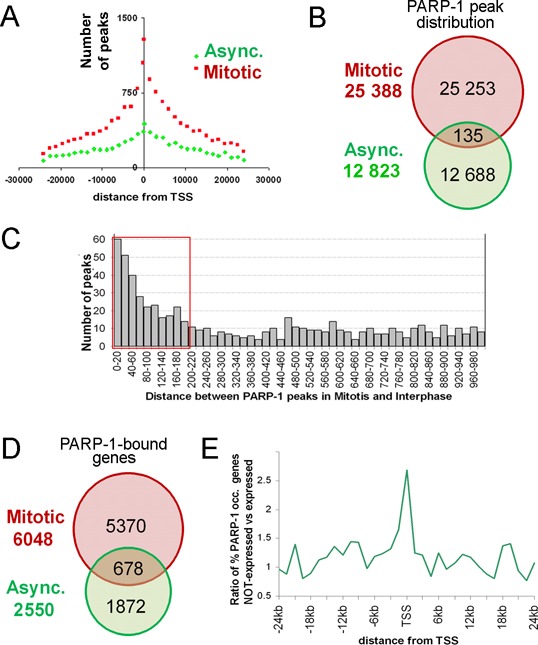
Identification of PARP-1 binding sites in mitotic and interphase chromatin in HEK293 cells. (A) PARP-1 binding sites predominantly group around transcriptional start sites (TSS) during mitosis (red) genome-wide and spread out more evenly in asynchronous cells (green). (B) PARP-1 binding in mitotic cells (red) and asynchronous cells (green). Shared sites in mitotic and asynchronous cells are shown in yellow. (C) Prevalent distance of PARP-1 binding site overlaps between mitosis and interphase. Red box highlights 200 bp (nucleosomal region) distances. (D) PARP-1 binding is associated with non-overlapping sets of genes in mitosis versus interphase: 6048 transcription units have at least one peak in the mitosis stage, 2550 transcription units have at least one peak in interphase, and 678 transcription units have peaks in mitosis and interphase (shared). (E) interphase-specific PARP-1 binding correlates with gene silencing. Using RNA-seq data, we tested whether the difference between the fraction of PARP-1-occupied genes among non-expressed genes and the fraction of PARP-1-occupied genes among expressed genes is statistically significant (48).
Next, we examined how PARP-1 occupancy corresponds to known genes by comparing its binding sites to annotated transcriptional units. While 6048 transcriptional units were identified as being occupied by PARP-1 in mitotic chromatin and 2550 were occupied by PARP-1 during interphase, only 678 transcriptional units were occupied during both segments of the cell cycle (Figure 2D; Supplementary Table S5). This overlap is significantly less than expected by chance (P = 10−43, hypergeometric test), thus indicating that PARP-1 is redistributed away from ‘mitotic’ TSSs to a new set of ‘interphase’ TSSs. These findings strongly suggest that PARP-1 marks specific TSSs during mitosis and that upon completion of mitosis become redistributed to a novel non-overlapping set of TSSs.
The functional significance of PARP-1 redistribution from mitosis to interphase is supported by earlier studies of the role of PARP-1 in transcription activation. Specifically, PARP-1 automodifies itself with pADPr during PARP-1-dependent transcriptional activation, leading to electrostatic repulsion (44–47) between the newly assembled pADPr and DNA, which results in dissociation of the automodified PARP-1 and chromatin (27,30). Consequently, continuing PARP-1 presence at TSS regions during interphase suggests that transcriptional activation has not occurred and that these loci will remain inactive throughout interphase. Indeed, significant number of genes that have interphase-specific PARP-1 binding site within ±1.5 kb (upstream or downstream) of TSSs were silent (P = 5 × 10−6 by a hypergeometrical test; Figure 2E) in HEK293 transcriptome (48) demonstrating a remarkable anti-correlation between PARP-1 binding in the interphase and gene expression (Figure 2E). Therefore, taken together, our results suggest that the difference in PARP-1 distribution between mitotic and interphase chromatin is caused by its automodification following gene activation after mitosis, while silent genes keep PARP-1 bound to promoters.
PARP-1 colocalizes with H2A.Z-bearing nucleosome in mitotic chromatin
Computation prediction of nucleosome binding affinities in the areas centered at PARP-1 peaks suggests 100% co-occurrence between PARP-1 binding and the presence of a nucleosome (Figure 3A). Unfortunately, modeling unambiguous predictions of nucleosome positioning near the TSS is problematic due to deviations from regular nucleosomal arrays caused by Pol II and general transcription machinery recruitment. Therefore, we determined experimentally the localizations of PARP-1 and TSS-associated nucleosomes using ChIP with PARP-1 and core histones antibodies. Earlier research on Drosophila demonstrated that mononucleosomes represent binding platforms for PARP-1 (49). The nucleosomes containing histone variant H2Av, a homologue of mammalian H2A.Z and H2AX, are preferred sites for PARP-1 binding in vivo (30,50). Consistent with these findings, we observed PARP-1/H2A.Z co-occupancy at TSSs of all tested mitotic PARP-1 binding sites in HEK293 cells (Figure 3B). The profile of PARP-1 binding corresponds perfectly to the distributions of H2A.Z and H2AX histone variants, exclusive of H2A histone localization (Figure 3C and Supplementary Figure S5). Taken together, these findings suggest that H2A.Z/X nucleosomes are colocalized with PARP-1 in promoter regions in mammalian genome.
Figure 3.
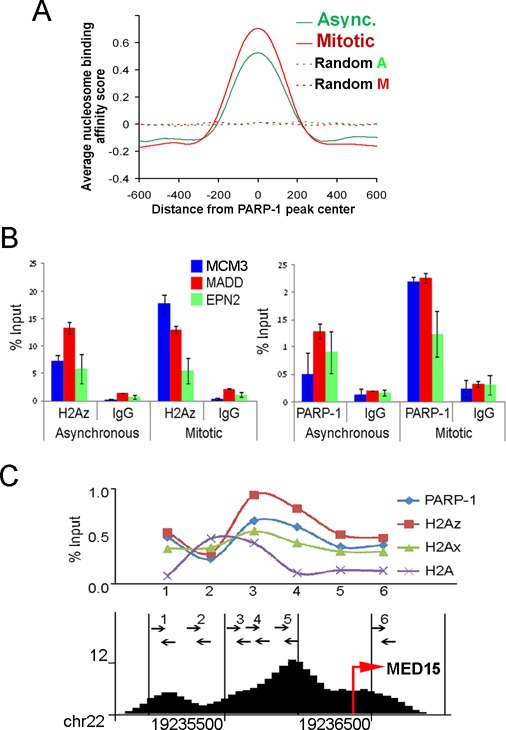
PARP-1 colocalizes with H2A.Z-bearing nucleosome. (A) PARP-1 binding in relation to nucleosome position. 1200 bp DNA sequences centered at PARP-1 peaks were tested to computationally predict nucleosome binding affinity (NBA). Average NBA scores for mitotic and interphase cells demonstrate that PARP-1 binding sites coincide with nucleosome position. (B) PARP-1 binding sites colocalize with H2A.Z histone. Three randomly chosen genes from the mitotic-specific gene list were subjected to ChIP analysis using anti-H2A.Z, anti-PARP-1 and anti-IgG antibody. PARP-1 and H2A.Z are simultaneously present at all tested sites. (C) PARP-1 binding site within MED15 locus colocalizes with H2A.Z/H2AX histones, but not with H2A. Arrows indicate positions of oligonucleotide primers, which were used in the ChIP assay, along the human genome sequence and ChIP-seq PARP-1 peaks (bottom).
PARP-1 binding sites co-occur with NFATC2 transcriptional factor binding sites genome-wide
To address which specific genes are marked by PARP-1 during mitosis, we performed consensus sequence comparison for the mitotic and interphase sites of PARP-1 occupancy. This analysis revealed GGAAAGG consensus sequences in proximity to approximately 65% of all sites of PARP-1 localization in mitosis and interphase (Figure 4A and Supplementary Figure S6). This sequence is similar to the known binding sites of transcriptional factor NFATC2 (51). NFATC2 serves as a transcriptional factor controlling inducible expression of interleukin gene family (52,53). Recent research has shown that transcription factors from NFAT family act as regulators of oncogenic transformation (54,55). NFATC2 remains bound to chromatin during interphase, as does PARP-1 (Figure 4B). We determined NFATC2 occupancy at a randomly selected group of PARP-1-positive peaks. Indeed, three tested PARP-1-positive loci OR51B2, ZSCAN4 and RRAS2 demonstrated significant occupancy by NFATC2, while the PARP-1 negative locus TMCC2 showed no NFATC2 binding in either mitosis or interphase (Figure 4C–D). Genome-wide co-occurrence of PARP-1 and NFATC2 binding sites suggests that genes marked by PARP-1 may share a common mechanism of regulation by NFATC2. This hypothesis is supported by earlier reports that NFATC2 directly interacts with PARP-1 and PARP-1 mediated poly(ADP-ribosyl)ation (56,57).
Figure 4.
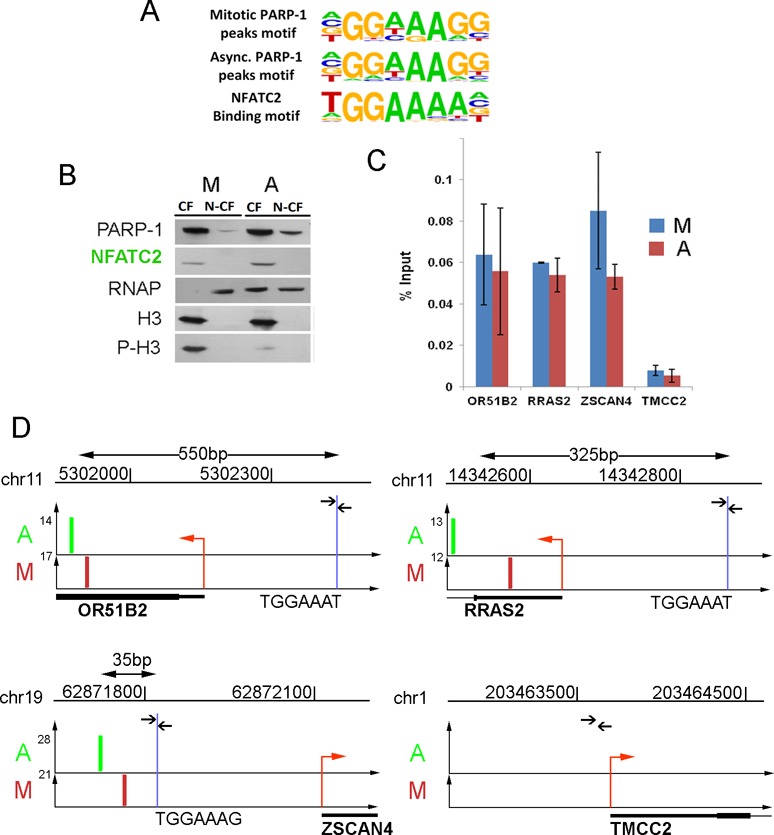
PARP-1 binding sites co-occur with NFATC2 binding consensus. (A) Consensus binding motif found in proximity to PARP-1 binding sites. The most significant motifs associated with PARP-1 binding sites in mitotic and asynchronized cells were compared to known NFATC2 binding motif. (B) NFATC2 remains associated with chromatin during mitosis. Equal amounts of total chromatin proteins extracted from asynchronous and mitosis-arrested cells were analyzed by PAGE and western blot using anti-NFATC2, anti-PARP-1, anti-RNAP (RNA polymerase II), anti-H3 and anti-phospho-Ser10-H3 (marker of mitotic chromatin) antibodies. CF, chromatin fraction; N-CF, non-chromatin fraction. (C) NFATC2 binds to TGGAAA sequence in three randomly picked PARP-1-positive loci (OR51B2, ZSCAN4, RRAS2) and did not bind to a PARP-1-negative locus (TMCC2). Blue bars indicate NFATC2 binding in mitosis, red—in interphase. (D) The locations of predicted NFATC2 binding (blue line) sites relative to PARP-1 binding sites in mitosis (M) and interphase (A) within OR51B2, ZSCAN4, RRAS2 and TMCC2 loci. Arrows indicate positions of oligonucleotide primers around NFATC2 consensus sequence, which were used in the ChIP assay. Position of PARP-1 binding signal is shown in green for interphase and in red for mitosis. The distance between the PARP-1 binding site and NFATC2 consensus sequence is shown above each figure. Red arrow indicates the location and direction of transcription within each locus.
Functional classification of genes occupied by PARP-1
We used Gene Signature Enrichment Analysis (GSEA) to identify 33 gene ontology (GO) categories that are enriched in at least one PARP-1 occupancy group (see Figure 2; occupied in mitosis, but no longer occupied in interphase, occupied in both mitosis and interphase, and occupied in interphase). We performed GSEA using genes containing PARP-1 binding sites and separately with genes that contained NFATC2 motifs around PARP-1 binding site (Figure 5). Clusters of genes that are occupied by PARP-1 specifically during mitosis (Figure 5, clusters 1, 2 and 3) contain several enriched GO categories that are related to cell adhesion, intercellular interaction, receptor-mediated signaling and protein secretion. Curiously, very few GO categories, including defense response, carbohydrate binding and protein kinase A binding, are overrepresented among genes that are occupied by PARP-1 during interphase. Consistent with functional systems regulated by NFATC2 these sites are enriched in GO categories related to the circulatory system (cluster 1c), defense response and carbohydrate-binding genes (cluster 4).
Figure 5.

The distribution of PARP-1 binding sites and NFATC2 consensus between functional groups of genes in human genome. GO enrichment among genes from different categories based on PARP-1 occupancy during mitosis and interphase. Using DAVID software, genes displaying different PARP-1 occupancy patterns during mitosis and interphase were tested separately for enrichment with respect to gene ontology terms including categories for biological processes (bp), molecular functions (mf) and cellular components (cc). The occupancy pattern is indicated on the bottom as follows: the mitotic group (M) has PARP-1 peak(s) in mitosis within 10 kb from TSS, mitotic gene in interphase (M gone in I) had PARP-1 peak(s) in mitosis within 1.5 kb, but lost them during interphase [this region shown to be important for silencing during interphase by PARP-1], mitotic and interphase (M and I) has peaks within 10 kb of TSS in both mitosis and interphase and interphase (I) has peaks in interphase only. An additional GO enrichment test was performed for genes that contained NFATC2 motif within 200 bp of the PARP-1 peak center. Intensity of red shading on the heatmap corresponds to the degree of significance enrichment.
Bookmarking promoters with PARP-1 during mitosis is required for transcription restart after mitosis completion
Our data demonstrate that PARP-1 binds promoters of a subset of genes during mitosis. Following mitosis, the amount of pADPr in chromatin increases dramatically (Supplementary Figure S7A). Therefore, it seems likely that PARP-1 bookmarks specific genes during mitosis and contributes their transcriptional reactivation by poly(ADP-ribosyl)ation of chromatin post-mitotically. To test whether PARP-1 is required for transcriptional restart after mitosis, we compared primary transcript levels of PARP-1 associated genes in PARP-1 knockdown and control cells after mitosis. We transfected HEK293 cells with shRNAcontrol and shRNAPARP-1 using Effectine transfection reagent (Qiagen) for 36 h. To synchronize cells in mitosis we treated cells with nocodazole (100 ng/ml) for 18 h. Synchronized cells were released by washing with PBS and culturing for 2 h. The protein levels of PARP-1 were evaluated by western blotting. shRNA transfection resulted in 65% PARP-1 depletion and reduced total pADPr by 88% (Figure 6A and B), but did not affect cell cycle progression (Supplementary Figure S7B–C). We found that a shorter treatment with shRNA failed to sufficiently deplete PARP-1 because of high stability of the PARP-1 protein. A prolonged treatment results in a greater PARP-1 depletion but affected cell viability due to secondary effects.
Figure 6.
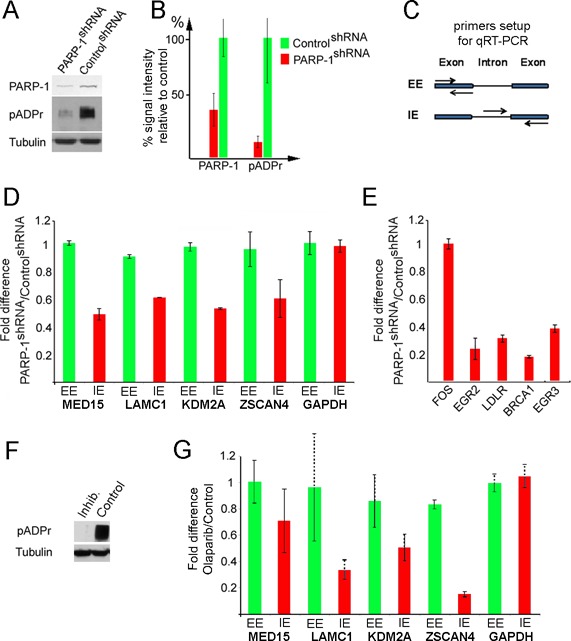
PARP-1 and its enzymatic activity are required to restart post-mitotic transcription. (A and B) Generating synchronous PARP-1 knockdown cells (Supplementary Figure S7B). PARP-1 level and enzymatic activity in PARP-1 shRNA and control shRNA-expressing cells. Each experiment was repeated three times. (A) Mouse monoclonal anti-PARP-1 and anti-pADPr (10H) antibodies were used to detect the presence of PARP-1 protein and poly(ADP) ribose moieties. Anti-tubulin antibody was used as a loading control. Quantification of PARP-1 and pADPr amounts based on three repeated experiments was performed using ImageQuant software (B). (C) Two types of primer pairs were employed to detect total mRNA (EE) and nascent RNA (IE). Arrows represent primers. (D and E) Comparative analyses of transcriptional re-start after mitosis in control and PARP-1 depleted cells for randomly chosen PARP-1-dependent genes (D) and EGF-PARP-dependent genes (E). Each experiment was repeated three times. The fold difference in q-RT-PCR signal between PARP-1 knockdowns and control is marked along the y-axis. Transcription of EGF-dependent genes was stimulated by EGF treatment as described elsewhere (41). (F) PARP-1 inhibition in human cells (Supplementary Figure S8). Comparative analysis of PARP-1 activity (pADPr) in cells treated with PARP-1 inhibitor, olaparib and in untreated control cells. Mouse monoclonal anti-pADPr (10H) was used to detect pADPr. Anti-tubulin antibody was used as a loading control. (G) Comparative analyses of transcriptional re-start after mitosis in cells treated with PARP-1 inhibitor, olaparib and in untreated control cells, for randomly chosen PARP-1-dependent genes. Each experiment was repeated three times. The fold difference in q-RT-PCR signal versus input between control and olaparib-treated samples is shown along the y-axis.
We compared post-mitotic levels of primary transcripts (Figure 6A and B) for two groups of genes selected from the pool of PARP-1 mitotic-specific sites: randomly selected genes (MED15, LAMC1, ZSCAN4 and KDM2A) (Supplementary Figure S7D) and EGF hormone-dependent genes (FOS, EGR2, LDLR, BRCA1 and EGR3) (41). To distinguish total mRNA from new (nascent) RNA, we designed two pairs of primers for each gene: exon to exon (EE) and intron to exon (IE) (Figure 6C). Newly transcribed RNA (IE) cannot be detected during mitosis. After mitosis, the amount of nascent RNA increases rapidly for PARP-1-occupied genes, but slowly for a PARP-1-independent gene (GAPDH) (Supplementary Figure S7E).
Following mitosis, the total amount of mRNA detected by qPCR using EE primers remained the same (Figure 6D), as was evidenced by the unchanged volume of cytoplasm. However, the production of new RNA, as detected by IE primers, was reduced by at least two-fold in PARP-1 knockdown cells relative to control cells (Figure 6D,E). A similar decline in the level of nascent transcripts was observed in cells pretreated with the specific PARP-1 inhibitor (Olaparib) instead of depleting PARP-1 using shRNA (Supplementary Figure S8 and 6F–G). Thus, PARP-1 binding to TSSs does facilitate transcriptional reactivation of PARP-1 associated genes after mitosis by a mechanism that relies on PARP-1-dependent poly(ADP-ribosyl)ation.
DISCUSSION
The condensed state of mitotic chromatin poses a challenge for transmission of epigenetic information across cell division, as most transcriptional factors and RNA polymerases dissociate from mitotic chromatin during this process (2,6). The few factors that remain attached must be sufficient to restore proper active chromatin patterns upon the completion of cell division. Here, we report a new PARP-1-dependent bookmarking mechanism of mitotic chromatin. Our findings show PARP-1 acts as an epigenetic marker for a large subset of genes. Our data strongly suggest that being stably bound to transcription start sites, PARP-1 can trigger quick and robust transcription restart of these genes following the completion of mitosis. We demonstrate that effector proteins, such as PARP-1 can serve as epigenetic marks that remain stably associated with certain promoters, along with posttranslational modification of histones or DNA.
Previously, activation of PARP-1 has been implicated in stimulating transcription by loosening chromatin during developmental processes and in response to environmental stress (26–28,50,58). Upon PARP-1 activation, poly(ADP-ribose), a nucleic acid analogue which is twice as negatively charged as DNA, is synthesized from NAD+ and either form long polymers or modifies acceptor proteins (59). The negatively charged poly(ADP-ribose) then participate in electrorepulsive shuttling of chromatin proteins, such as histones and ultimately lead to their dissociation from DNA, which results in chromatin loosening and increased accessibility of DNA for transcription factors and general transcription machinery (26,27,30,60,61).
In agreement with these findings, we observed that mitotic and interphase PARP-1 cistromes are essentially non-overlapping and that PARP-1 becomes redistributed in chromatin following completion of mitosis, leaving promoters of genes that are associated with PARP-1 driven activation. Intriguingly, the retention of PARP-1 near TSSs during interphase strongly correlates with gene silencing.
The localization and dynamics of PARP-1 in mitotic and interphase chromatin is consistent with PARP-1 acting as an epigenetic memory mark. Indeed, (i) PARP-1 remains stably bound to TSS of a specific subsets of genes, (ii) PARP-1 correlates with the renewed transcription of these genes upon mitosis exit and (iii) PARP-1 dissociates from these genes upon restart of transcription. As PARP-1 dissociates from many of its targets during interphase, the use of mitotic chromatin for genome-wide identification of PARP-1-bound promoters produced a more comprehensive list of PARP-1-dependent genes, as compared to screening interphase chromatin of asynchronous cells. In that respect PARP-1 acts as an active epigenetic mark that can regulate its own catalytic activity and the state and accessibility of surrounding chromatin (Figure 7), as opposed to passive marks such as histone modifications that require recruitment of additional modification-sensing effectors.
Figure 7.
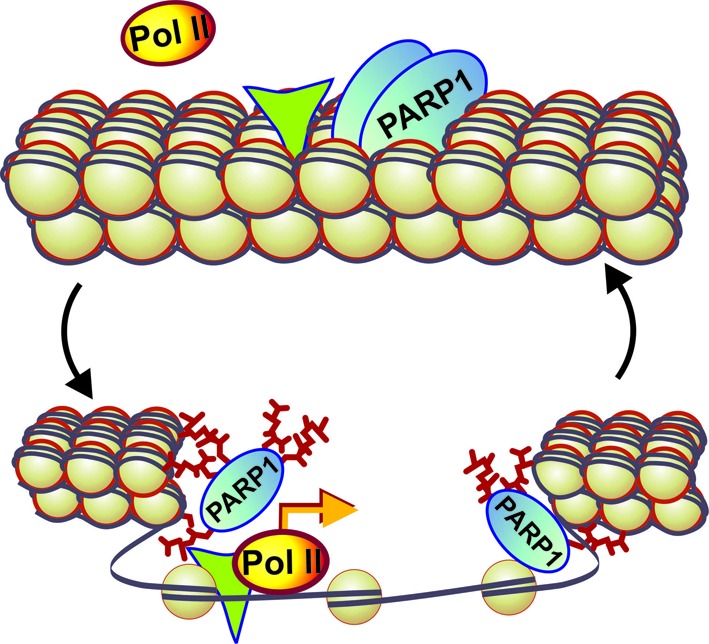
PARP-1 acts as an active epigenetic memory marker to regulate post-mitotic transcription. Based on our finding we propose the following model of PARP-1 action as an epigenetic mark: PARP-1 remains bound to chromatin during mitosis and facilitates transcription after mitosis by poly(ADP-ribosyl)ating chromatin, effectively opening it. Upon depletion or inhibition of PARP-1, chromatin remains condensed after mitosis, thereby blocking or delaying transcriptional restart. Green triangle indicates NFATC2 transcription factor binding in proximity to PARP-1 binding site.
Remarkably, PARP-1 is required for the expression of several genes that are responsible for products that are involved in interactions between cells and with extracellular matrix. As mitotic cells typically lose contact with other cells and the extracellular matrix, marking genes responsible for establishing cellular identity by an epigenetic factor, is necessary for rapid restoration of cell niche interactions, preventing uncontrolled cell migration and potentially tumorigenic de-differentiation. Upon the completion of mitosis, PARP-1 opens up chromatin around TSSs of these genes, thereby allowing access to transcription machinery and reactivating transcription. PARP-1 association with genes involved in metabolic and developmental pathways, cell adhesion, communication, proliferation and apoptosis suggests that PARP-1 may serve as a central developmental/pathogenic hub, controlling such basic organismal functions as cellular proliferation and adhesion, while deregulation of its production and localization can lead to pathogenesis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Italo Tempera for providing shRNA constructs and Dr P. Wickramsinghe from Wistar Bioinformatics Facility for data analysis, as well as Jim Oesterling for technical assistance and advice during the performance of ChIP-Seq experiments and data analysis. Electron microscope analysis was provided by the FCCC EM Core Facility. Dr Alana O'Reily, Dr Kate Pechenkina, Dr Colin Thomas, Dr Kenneth Zaret, Dr Christoph Seeger and Dr Jean-Pierre Issa provided comments on the manuscript.
FUNDING
National Institutes of Health [R01 GM077452, R01 DK082623 to A.V.T.]. Funding for open access charge: National Institutes of Health [R01 GM077452, R01 DK082623].
Conflict of interest statement. None declared.
REFERENCES
- 1.John S., Workman J.L. Bookmarking genes for activation in condensed mitotic chromosomes. Bioessays. 1998;20:275–279. doi: 10.1002/(SICI)1521-1878(199804)20:4<275::AID-BIES1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Martizez-Balbas M.A., Dev A., Rabindran S.K., Ozato K., Wu C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 3.Michelotti E.F., Sanford S., Levens D. Marking of active genes on mitotic chromosomes. Nature. 1997;388:895–899. doi: 10.1038/42282. [DOI] [PubMed] [Google Scholar]
- 4.Chen D., Hinkley C.S., Henry R.W., Huang S. TBP dynamics in living human cells: constitutive association of TBP with mitotic chromosomes. Mol. Biol. Cell. 2002;13:276–84. doi: 10.1091/mbc.01-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christova R., Oelgeschläger T. Association of human TFIID-promoter complexes with silenced mitotic chromatin in vivo. Nat. Cell Biol. 2002;4:79–82. doi: 10.1038/ncb733. [DOI] [PubMed] [Google Scholar]
- 6.Gottesfeld J.M., Forbes D.J. Mitotic repression of the transcriptional machinery. Trends Biochem. Sci. 1997;22:197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- 7.Delcuve G.P., He S., Davie J.R. Mitotic partitioning of transcription factors. J. Cell. Biochem. 2008;105:1–8. doi: 10.1002/jcb.21806. [DOI] [PubMed] [Google Scholar]
- 8.Dey A., Nishiyama A., Karpova T., McNally J., Ozato K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell. 2009;20:4899–4909. doi: 10.1091/mbc.E09-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaidi S.K., Young D.W., Montecino M.A., Lian J.B., van Wijnen A.J., Stein J.L., Stein G.S. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat. Rev. Genet. 2010;11:583–589. doi: 10.1038/nrg2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadauke S., Udugama M.I., Pawlicki J.M., Achtman J.C., Jain D.P., Cheng Y., Hardison R.C., Blobel G.A. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012;150:725–737. doi: 10.1016/j.cell.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caravaca J.M., Donahue G., Becker J.S., He X., Vinson C., Zaret K.S. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013;27:251–260. doi: 10.1101/gad.206458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Follmer N.E., Wani A.H., Francis N.J. A polycomb group protein is retained at specific sites on chromatin in mitosis. PLoS Genet. 2012;8:e1003135. doi: 10.1371/journal.pgen.1003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolinoy D.C., Jirtle R.L. Environmental epigenomics in human health and disease. Environ. Mol. Mutat. 2008;49:4–8. doi: 10.1002/em.20366. [DOI] [PubMed] [Google Scholar]
- 14.Skinner M.K. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics. 2011;6:838–842. doi: 10.4161/epi.6.7.16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kouskouti A., Talianidis I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 2005;24:347–357. doi: 10.1038/sj.emboj.7600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valls E., Sanchez-Molina S., Martinez-Balbas M.A. Role of histone modifications in marking and activating genes through mitosis. J. Biol. Chem. 2005;280:42592–42600. doi: 10.1074/jbc.M507407200. [DOI] [PubMed] [Google Scholar]
- 17.Zhao R., Nakamura T., Fu Y., Lazar Z., Spector D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011;13:1295–1304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaidi S.K., Young D.W., Montecino M., Van Wijnen A.J., Stein J.L., Lian J.B., Stein G.S. Bookmarking the genome: maintenance of epigenetic information. J. Biol. Chem. 2011;286:18355–18361. doi: 10.1074/jbc.R110.197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 20.Goll M.G., Bestor T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 21.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 22.Clayton A.L., Hazzalin C.A., Mahadevan L.C. Enhanced histone acetylation and transcription: a dynamic perspective. Mol. Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Bernstein B.E., Meissner A., Lander E.S. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 25.Sarge K.D., Park-Sarge O.K. Mitotic bookmarking of formerly active genes: keeping epigenetic memories from fading. Cell Cycle. 2009;8:818–823. doi: 10.4161/cc.8.6.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tulin A., Spradling A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science. 2003;299:560–562. doi: 10.1126/science.1078764. [DOI] [PubMed] [Google Scholar]
- 27.Petesch S.J., Lis J.T. Activator-induced spread of poly(ADP-ribose) polymerase promotes nucleosome loss at Hsp70. Mol. Cell. 2012;45:64–74. doi: 10.1016/j.molcel.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M.Y., Mauro S., Gevry N., Lis J.T., Kraus W.L. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Krishnakumar R., Kraus W.L. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell. 2010;39:736–749. doi: 10.1016/j.molcel.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas CJ., Kotova E., Andrake M., Adolf-Bryfogle J., Glaser R., Regnard C., Tulin A.V. Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via poly-ADP-ribosylation. Mol. Cell. 2014;53:831–842. doi: 10.1016/j.molcel.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rippmann J.F., Damm K., Schnapp A. Functional characterization of the poly(ADP-ribose) polymerase activity of tankyrase 1, a potential regulator of telomere length. J. Mol. Biol. 2002;323:217–224. doi: 10.1016/s0022-2836(02)00946-4. [DOI] [PubMed] [Google Scholar]
- 32.Kameshita I., Matsuda M., Nishikimi M., Ushiro H, Shizuta Y. Reconstitution and poly(ADP-ribosyl)ation of proteolytically fragmented poly(ADP-ribose) synthetase. J. Biol. Chem. 1986;261:3863–3868. [PubMed] [Google Scholar]
- 33.Kameshita I., Matsuda Z., Taniguchi T., Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 1984;259:4770–4776. [PubMed] [Google Scholar]
- 34.Tulin A., Stewart D., Spradling A.C. The Drosophila heterochromatic gene encoding poly(ADP-ribose) polymerase (PARP) is required to modulate chromatin structure during development. Genes Dev. 2002;16:2108–2119. doi: 10.1101/gad.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnakumar R., Gamble M.J., Frizzell K.M., Berrocal J.G., Kininis M., Kraus W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science. 2008;319:819–821. doi: 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- 36.Lodhi N., Tulin A.V. PARP-1 genomics: chromatin immunoprecipitation approach using anti-PARP-1 antibody (ChIP and ChIP-Seq) Methods Mol. Biol. 2011;780:191–208. doi: 10.1007/978-1-61779-270-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tokuyasu K.T. Immunochemistry on ultrathin frozen sections. Histochem. J. 1980;12:381–403. doi: 10.1007/BF01011956. [DOI] [PubMed] [Google Scholar]
- 38.Kotova E., Jarnik M., Tulin A.V. Uncoupling of the transactivation and transrepression functions of PARP1 protein. Proc. Natl. Acad. Sci. U.S.A. 2010;107:6406–6411. doi: 10.1073/pnas.0914152107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paddison P.J., Caudy A.A., Bernstein E., Hannon G.J., Conklin D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tempera I., Deng Z., Atanasiu C., Chen C.J., D'Erme M., Lieberman P.M. Regulation of epstein-Barr virus OriP replication by poly(ADP-ribose) polymerase 1. J. Virol. 2010;84:4988–4997. doi: 10.1128/JVI.02333-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byun J.S., Fufa T.D., Wakano C., Fernandez A., Haggerty C.M., Sung M., Gardner K. ELL facilitates RNA polymerase II pause site entry and release. Nat. Commun. 2012;3:633. doi: 10.1038/ncomms1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Hans F., Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene. 2001;20:3021–3027. doi: 10.1038/sj.onc.1204326. [DOI] [PubMed] [Google Scholar]
- 44.Kotova E., Jarnik M., Tulin A.V. Poly (ADP-ribose) Polymerase 1 is required for protein localization to Cajal body. PLoS Genet. 2009;5:e1000387. doi: 10.1371/journal.pgen.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caiafa P., Guastafierro T., Zampieri M. Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 2009;23:672–678. doi: 10.1096/fj.08-123265. [DOI] [PubMed] [Google Scholar]
- 46.Zhang T., Berrocal J.G., Yao J., DuMond M.E., Krishnakumar R., Ruhl D.D., Ryu K.W., Gamble M.J., Kraus W.L. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD +synthase. J. Biol. Chem. 2012;287:12405–12416. doi: 10.1074/jbc.M111.304469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sultan M., Schulz M.H., Richard H., Magen A., Klingenhoff A., Scherf M., Seifert M., Borodina T., Soldatov A., Parkhomchuk D., et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 49.Pinnola A., Naumova N., Shah M., Tulin A.V. Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. J. Biol. Chem. 2007;282:32511–32519. doi: 10.1074/jbc.M705989200. [DOI] [PubMed] [Google Scholar]
- 50.Kotova E., Lodhi N., Jarnik M., Pinnola A.D., Ji Y., Tulin A.V. Drosophila histone H2A variant (H2Av) controls poly(ADP-ribose) polymerase 1 (PARP1) activation in chromatin. Proc. Natl. Acad. Sci. U.S.A. 2011;108:6205–6210. doi: 10.1073/pnas.1019644108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Macián F., López-Rodríguez C., Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 52.Walters R.D., Drullinger L.F., Kugel J.F., Goodrich J.A. NFATc2 recruits cJun homodimers to an NFAT site to synergistically activate interleukin-2 transcription. Mol. Immunol. 2013;56:48–56. doi: 10.1016/j.molimm.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serfling E., Avots A., Klein-Hessling S., Rudolf R., Vaeth M., Berberich-Siebelt F. NFATc1/αA: the other face of NFAT factors in lymphocytes. C0. 2012;10:16. doi: 10.1186/1478-811X-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tie X., Han S., Meng L., Wang Y., Wu A. NFAT1 is highly expressed in, and regulates the invasion of, glioblastoma multiforme cells. PLoS One. 2013;8:e66008. doi: 10.1371/journal.pone.0066008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J.F, Zhao S.H., Wu S.S. Depleting NFAT1 expression inhibits the ability of invasion and migration of human lung cancer cells. Cancer Cell. Int. 2013;13:41. doi: 10.1186/1475-2867-13-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Opeyemi O.A., Soto-Nieves N., Nieves E., Yang T.T., Yang X., Yu R.Y., Suk H.Y., Macian F., Chow C.W. Regulation of transcription factor NFAT by ADP-ribosylation. Molecular and cellular biology. 2008;28:2860–2871. doi: 10.1128/MCB.01746-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valdor R., Schreiber V., Saenz L., Martínez T., Muñoz-Suano A., Dominguez-Villar M., Ramírez P., Parrilla P., Aguado E., García-Cózar F. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Molecular immunology. 2008;45:863–1871. doi: 10.1016/j.molimm.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 58.Petesch S.J., Lis J.T. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell. 2008;134:74–84. doi: 10.1016/j.cell.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 60.Poirier G.G., de Murcia G., Jongstra-Bilen J., Niedergang C., Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U.S.A. 1982;79:3423–3427. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Realini C.A., Althaus F.R. Histone shuttling by poly(ADP-ribosylation) J. Biol. Chem. 1992;267:18858–188865. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.