

Abstract
Existing reports on the frequencies of neurodegenerative diseases are typically based on clinical diagnoses. We sought to determine these frequencies in a prospectively-assessed, community-based autopsy series. Included subjects had normal cognitive and movement disorder assessments at study entry. Of the 119 cases meeting these criteria, 52% were female, median age of study entry was 83.5 years (range 67 to 99), and median duration from first visit until death was 4.3 years (range 0-10). At autopsy a clinico-neuropathological diagnosis was made in 30 cases (25%). Clinicopathological diagnoses included 20 (17%) with Alzheimer's disease (AD), 7 (6%) with vascular dementia, 4 (3%) with progressive supranuclear palsy, (1; 0.8%) with dementia with Lewy bodies, (1; 0.8%) with corticobasal degeneration and (1; 0.8%) with multiple system atrophy. Of those 87 subjects (73%) still clinically normal at death, 33 (38%) had extensive AD pathology (pre-clinical AD), 17 (20%) had incidental Lewy bodies and 4 (5%) had incidental pathology consistent with progressive supranuclear palsy. Diagnoses are not mutually exclusive. Although limited by a relatively small sample size, the neuropathological outcome of these initially normal elderly subjects represents a rough estimate of the incidence of these neurodegenerative conditions over a defined time period.
Keywords: Alzheimer's disease, progressive supranuclear palsy, vascular dementia, Parkinson's disease, dementia with Lewy bodies, pathology, epidemiology
Introduction
Estimating incidence and prevalence rates of neurodegenerative diseases can be problematic. There have been numerous studies with different populations (1-5). These reports are limited in that they typically have been focused only on Alzheimer's disease (AD) and few have autopsy confirmation on a substantial fraction of their population. There are several notable longitudinal clinicopathological studies, including the Vantaa 85+ cohort, Religious Orders Study, Rush Memory and Aging Project, CC75C study, CFAS, Adult Changes in Thought study, Cache County study, the Honolulu Asian Aging Study, and our own Arizona Study of Aging and Neurodegenerative Disorders (AZSAND) (6-16). These studies have generated considerable insight into risk and protective factors for AD but no study has been undertaken to evaluate the entire spectrum of defined clinicopathological outcomes of prospectively followed elderly persons who were normal at the time of study entry. The purpose of our study is to report final clinico-neuropathological diagnoses in prospectively followed cognitively normal elderly individuals lacking parkinsonism or other clinicopathologically defined movement disorders at the time of their initial research assessments.
Materials and Methods
Case selection
All subjects were enrolled in an ongoing longitudinal clinicopathological study, the AZSAND. To achieve clinical assessments as well as autopsy, subjects are enrolled into the Banner Sun Health Research Institute Brain and Body Donation Program (BBDP: www.brainandbodydonationprogram.org) (12). Our subjects are mainly drawn from the retirement communities of northwestern greater Phoenix especially Sun City, Sun City West and Sun City Grand. The majority of subjects are recruited directly from the community through public speaking events, media reports and monthly public tours of the Institute. All enrolled subjects or their legal representatives sign an Institutional Review Board-approved informed consent form before the time of death allowing both clinical assessments during life and several options for brain and/or bodily organ donation after death. The BBDP database was queried for subjects who had come to autopsy and had a complete neuropathological examination between January 1997 and December 2012. Only subjects that were normal at the time of study entry were included. Study entry was defined as the first epoch with both cognitive and movement examinations performed less than 1 year apart. Normal was defined as subjects who at the time of their first examinations were cognitively normal (lacking diagnoses of dementia or mild cognitive impairment) and lacked diagnoses of any defined neurodegenerative disease, parkinsonism, Huntington's disease, or motor neuron disease, as well as normal pressure hydrocephalus.
Clinical assessments
Annual physical, neurological, cognitive, and movement examinations have been previously described (12, 17). In brief, all subjects had a standardized neuropsychological test battery that included global testing as well as testing of specific cognitive domains. Furthermore, patients were examined for physical illness that could give rise to cognitive or neurological symptoms. They also were examined for evidence of parkinsonism and other movement disorders. Following each evaluation subjects were given a movement disorders diagnosis which included: 1) Clinically probable Parkinson's disease (PD): defined as 2 of the 3 cardinal signs (rest tremor, bradykinesia, rigidity), no symptomatic cause, a response to dopaminergic medications (based on patient report or clinical findings) and continued response if still being treated, or if lack of current response then an explanation for why treatment was no longer working (i.e. inadequate dose due to side effects, etc.); 2) Clinically possible PD: defined as 2 of the 3 cardinal signs, no symptomatic cause, symptoms or signs were present for 5 years or less, dopaminergic treatment had not been tried or an adequate trial had not clearly occurred; 3) Progressive supranuclear palsy (PSP): defined as meeting NINDS-PSP clinical criteria (18) for diagnosis with key features including age over 40, early postural instability, square wave jerks and supranuclear gaze palsy; 4) parkinsonism not otherwise specified (NOS): defined as subjects with parkinsonism but who either did not respond to an adequate dose of dopaminergic medication or had disease duration greater than 5 years and have not been treated or given an adequate trial of dopaminergic treatment, or appeared to have another etiology for PD signs including a neurodegenerative condition such as AD, corticobasal degeneration (CBD), multiple system atrophy (MSA), dementia with parkinsonian features, or secondary parkinsonism such as vascular or drug-induced.
Alzheimer's disease was clinically diagnosed using DSM-IV and NINCDS-ADRDA criteria (19). Vascular dementia (VaD) was clinically diagnosed using NINDS-AIREN criteria (20) and dementia with Lewy bodies (DLB) was clinically diagnosed according to the DLB Consortium criteria (21). Mild cognitive impairment (MCI) was diagnosed using guidelines set forth by Petersen et al. and Winblad et al. (22, 23). Specifically, the clinical diagnosis of MCI required subjective cognitive complaint(s) not associated with significant functional decline as well a deficit of approximately ≥ 1.5 standard deviations (SD) below the expected age-corrected mean score in one or more of five designated cognitive domains (frontal/executive, memory, visuospatial, attention, and language). Dysfunction in a cognitive domain had to be shown by a consistent pattern of impaired performance on neuropsychological measures that loaded on that cognitive domain (i.e., more than 1 abnormal test within a domain). These criteria were used in conjunction with clinical judgment at a consensus conference with a behavioral neurologist and neuropsychologist. For subjects not seen within 18 months of death, a standardized telephone interview was conducted with a knowledgeable contact, typically next of kin, to evaluate for both cognitive and movement disorders.
Tissue processing and neuropathological diagnosis
All subjects underwent autopsy and a standardized neuropathological assessment, resulting in the assignment of clinicopathological diagnoses according to published recommendations (12, 24). For clinicopathological diagnoses, subjects received a diagnosis of PD if they had two or more of the three cardinal clinical signs as well as Lewy bodies and pigmented neuron loss in the substantia nigra (25). Subjects received a clinicopathological diagnosis of AD if they had a clinical history of dementia and were classified as “intermediate” or “high” according to the NIA-Reagan criteria (26). Dementia with Lewy bodies (DLB) was distinguished from Parkinson's disease (PD) with dementia (27) according to consensus criteria published by the DLB Consortium; the diagnosis of DLB was assigned if dementia was diagnosed prior to or within one year of the onset of parkinsonism and if the distribution and density of Lewy body-type pathology met “intermediate” or “high” criteria. A clinicopathological diagnosis of VaD was determined using criteria by Roman and colleagues (20). Other diagnoses followed previously published guidelines (24, 28-31). Tissue processing methods have been previously described (12, 32). Briefly, the cerebrum was cut in the coronal plane at the time of brain removal into 1 cm thick slices and then divided into left and right halves. The slices from the right half were frozen between slabs of dry ice while the slices from the left half were fixed by immersion in neutral-buffered 4% formaldehyde for 48 hours at 4 degrees C. Formaldehyde-fixed paraffin-embedded sections were stained with hematoxylin and eosin, while large-format, 40-80 μm-thick formaldehyde-fixed sections were stained for plaques, tangles and other features using Gallyas, Thioflavin-S and Campbell-Switzer methods (33). Thioflavin-S is one of two methods recommended and validated for neuritic plaque density grading by the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) (34). The Braak neurofibrillary tangle (NFT) staging protocol was originally described using the Gallyas stain (35) on similarly-thick sections. Subjects with Lewy body-related histopathology were classified according to the Unified Staging System for Lewy Body Disorders (36) after immunohistochemical detection of phosphorylated α-synuclein on 5 μm paraffin sections (37). All subjects were genotyped for apolipoprotein E (ApoE) using a modification of a standard method (38, 39).
Statistical analysis
All analyses were conducted with SAS statistical software (Version 9.2 SAS Institute Inc., Cary, NC) and Sigma Plot 12.0 (Systat Software, Inc., San Jose, CA, USA). Group comparisons were done using the Mann-Whitney U-test.
Results
Of 1440 autopsied BBDP subjects queried, 119 (8%) subjects fit our inclusion and exclusion criteria. Of these 119 subjects, 52% were female, mean age at study entry was 83.5 years (range 67 to 99) and median duration from first visit until death was 4.3 years (range 0-10). Of the 119 subjects, 21% had at least one ApoE–ε4 allele. Demographic details and frequencies of final clinicopathological diagnosis are located in Table 1. A full description of each of the 119 cases including gender, age at death, Braak NFT stage, CERAD neuritic plaque densities, Lewy pathology stage, and full diagnosis is listed in Supplemental Table 1.
Table 1.
Details of clinicopathological diagnoses of subjects who lacked dementia as well as parkinsonism at initial visits. Table is divided by subjects who were clinicopathologically normal, those who progressed to a defined neurodegenerative or cerebrovascular disease, and other abnormalities not meeting a clinicopathological diagnosis; the type of pathologic diagnosis is listed below each main group. Diagnostic groups within each main category listed above are not mutually exclusive. Age is listed as mean ± standard deviation.
| N | % out of 119 | females, N (%) | age of death | |
|---|---|---|---|---|
| Clinicopathological Normal at Autopsy | 87 | 73.1% | 46 (53%) | 87 ± 6.6 |
| Pre-Clinical Alzheimer's Disease | 33 | 27.7% | 21 (63%) | 90 ± 4.9 |
| Incidental Lewy Bodies | 17 | 14.3% | 8 (47%) | 88 ± 5.1 |
| Incidental Progressive Supranuclear Palsy | 4 | 3.4% | 3 (75%) | 91 ± 9.4 |
|
| ||||
| Neurodegenerative Disease at Autopsy | 30 | 25.2% | 17 (57%) | 89 ± 7.5 |
| Alzheimer's Disease | 20 | 16.8% | 12 (60%) | 89 ± 7.3 |
| Vascular Dementia | 7 | 5.9% | 6 (86%) | 95 ± 3.7 |
| Progressive Supranuclear Palsy | 4 | 3.4% | 2 (50%) | 95 ± 10.1 |
| Neurofibrillary Tangle Predominant Dementia | 3 | 2.5% | 1 (33%) | 89 ± 5.7 |
| Parkinson's Disease | 3 | 2.5% | 2 (67%) | 90 ± 10.7 |
| Dementia with Lewy Bodies | 1 | 0.8% | 1 (100%) | 90 |
| Corticobasal Degeneration | 1 | 0.8% | 1 (100%) | 75 |
| Multiple System Atrophy | 1 | 0.8% | 0 (0%) | 75 |
|
| ||||
| Other abnormalities | 2 | 1.7% | 0 (0%) | 86± 9.2 |
Eighty-seven (73%) of the initially normal subjects remained without a defined clinicopathological diagnosis upon death (termed normal). Within these normal subjects, 17/87 (20.0%) had incidental Lewy body disease (iLBD) and 4/87 (4.7%) had PSP pathology; for comparative purposes, clinical and pathological features of the iPSP and PSP cases are given in Table 2 and Figure 1. Thirty-three of eighty-seven subjects (38%) had pre-clinical AD, meaning they contained a density and distribution of plaques and tangles sufficient to meet pathology criteria for “intermediate” probability according to the NIA-Reagan criteria but did not have a clinical history of dementia. Of the pre-clinical AD cases, 16 had CERAD neuritic plaque scores of moderate, and 17 were CERAD frequent while for neurofibrillary tangles, 13 were Braak NFT stage III and 20 were Braak NFT stage IV (Supplemental Table 1). The group means for both Braak NFT stage and CERAD neuritic plaque density were significantly lower in the pre-clinical AD subjects as compared to those with clinicopathologically manifest AD.
Table 2.
Progressive supranuclear palsy features in each incidental PSP and PSP case. Case # corresponds to those in Supplementary Table 1. NFT= neurofibrillary tangles, Tft= tufted astrocytes, Thrn = thorned astrocytes, CB= Coiled bodies.
| incidental PSP | PSP | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Case # | 9 | 30 | 55 | 71 | 97 | 109 | 110 | 111 |
|
| ||||||||
| Clinical Features | n/a | n/a | n/a | Parkinsonism NOS | Dementia | Early Falls, Parkinsonism NOS | Typical PSP | Abnormal Gait and Posture |
| Pathologic Features | ||||||||
| Frontal Cortex | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | Tft | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT | NFT, Tft, Thrn, CB |
| Temporal Cortex | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT | - | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | - | NFT, Tft, Thrn, CB |
| Parietal Cortex | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | - | Tft | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | Tft, Thrn, CB |
| Putamen | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | - | - | NFT, Tft, CB | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT, Thrn |
| Subthalamic Nucleus | NFT | NFT, Tft, Thrn, CB | - | - | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT, Thrn, CB | NFT, Tft, Thrn, CB |
| Globus Palllidus | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | - | - | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT | NFT, Thrn, CB |
| Thalamus | - | NFT, Tft, Thrn, CB | - | - | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT, Thrn, CB | NFT, Tft, CB |
| Substantia Nigra | NFT | NFT, Tft, Thrn, CB | NFT | - | NFT, Tft, Thrn, CB | NFT, Tft, Thrn, CB | NFT, Thrn, CB | NFT |
| Substantia Nigra Pigment Loss | Moderate | Mild | Mild | Mild | Mild | Severe | Mild | Severe |
| Pons | NFT | NFT, Tft, Thrn, CB | NFT | - | NFT, Tft, Thrn, CB | NFT, Thrn, CB | - | |
| Dentate Nucleus | NFT, Tft | NFT, Tft, Thrn, CB | NFT | - | NFT, Thrn | NFT, Tft, CB | - | NFT, Tft, CB |
Figure 1.
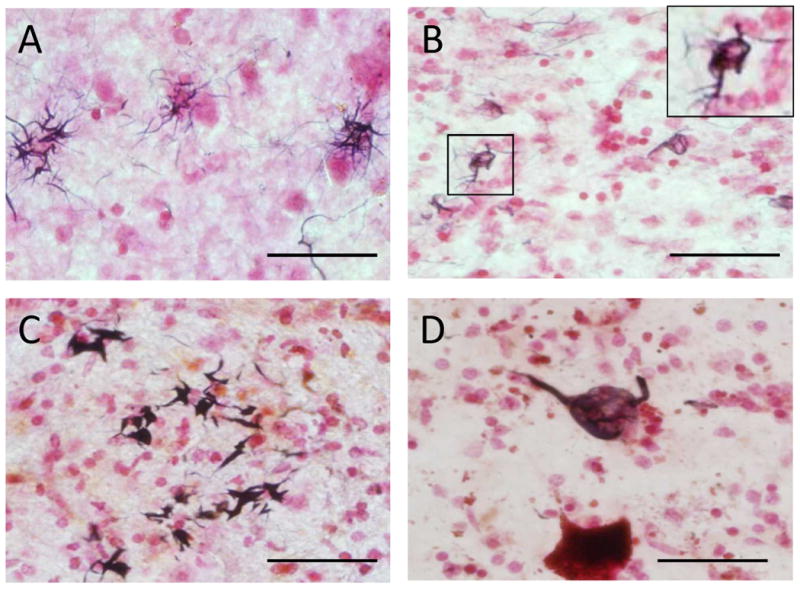
Tau histology in progressive supranuclear palsy (PSP) cases. (A) Gallyas stain of tufted astrocytes in the putamen; (B) Gallyas stain of coiled bodies in pontine base, enlargement of boxed image in upper right corner; (C) Gallyas stain of thorn-shaped astrocytes in the cerebellar dentate nucleus; (D) AT8 immunohistochemistry stain of a globose tangle in the substantia nigra. Scale bar = 50 μm.
There were some subjects who developed a clinical diagnosis but lacked a corresponding pathologic diagnosis. Nine subjects had a clinical diagnosis of parkinsonism (NOS) as they did not meet specific pathologic criteria for any diagnosis. Of these, 5 had one or more cerebral infarctions (see Supplemental Table 1); case 8 had one lacunar sized infarct in the corona radiata cerebral white matter and several cortical micro-infarcts in the frontal, parietal and occipital lobes; case 72 had one lacunar infarct in the putamen; case 85 had one old micro-infarct in the putamen; case 88 had an acute capsular hemorrhage of the basal ganglia and a single old micro-infarct of the thalamus, and case 119 contained multiple lacunar sized infarcts, two of which were in the putamen and one in the substantia nigra. This last-named case could possibly be considered to be “vascular parkinsonism”. Two cases had Lewy bodies restricted to the amygdala and olfactory bulb and none to mild substantia nigra depigmentation (in the Supplemental Table 1: case 56 and 94) and 2 had non-specific glial tauopathies (case 25 had argyrophilic grains and thorned shaped astrocytes in the mesial temporal lobe, and case 56 contained focal cortical tufted astrocytes).
Thirty subjects (25%) progressed to at least one type of clinicopathologically defined neurodegenerative or cerebrovascular disease at the time of autopsy. The most common diagnosis was dementia due to AD (20/119; 16.8%), followed by VaD (7; 5.9%), PSP (4; 3.4%), PD (3; 2.5%), tangle-predominant dementia (3; 2.5%), DLB (1; 0.8%), CBD (1; 0.8%) and MSA (1; 0.8%). These groups were not mutually exclusive; there was considerable overlap amongst diagnostic groups (see Supplementary Table 1 for complete diagnostic categorization of all cases). Of those with AD, 50% had Lewy bodies (9 had Lewy bodies insufficient for a pathological diagnosis of DLB and 1 had DLB) while 6 subjects had VaD, 1 had PSP and 1 had CBD. There was one case with concurrent PD and PSP. There were no cases, within the entire series of 119, with a diagnosis of hippocampal sclerosis. There were 2 cases whose final clinical and autopsy findings did not allow their classification into either the normal or clinicopathological disease category. These included: one case with glioblastoma multiforme and one case with vascular cognitive impairment (cognitive impairment and multiple infarcts but not meeting criteria for VaD).
Discussion
This study describes the clinicopathological outcomes of elderly BBDP volunteers who at the time of study entry did not have evidence for a defined clinicopathological neurodegenerative disorder. The overall conversion rate to a neurodegenerative disease or cerebrovascular disease was 25%. The majority of subjects (73%) remained clinicopathologically normal until death; many of these cases, however, did display hallmark pathologies of some neurodegenerative or cerebrovascular diseases (Table 1).
Of those who remained normal, nearly 40% had enough plaques and tangles to be pathologically defined as AD, although they did not have dementia clinically, for these cases we use the term pre-clinical AD as some have done (40). Other studies have described similar findings of individuals lacking cognitive changes and having varying degrees of AD pathology (40-44). We found 20% of subjects who remained clinically normal had incidental Lewy bodies; previous studies have described similar frequencies of iLBD (17, 36, 45-47). Our finding of some cases with incidental PSP has previously been reported in more depth (48); we know of only one other report of iPSP, an 84 year old Japanese man who died of heart failure with no other neurological abnormalities (49).
With respect to dementia diagnoses, 17% of subjects who were normal at enrollment developed AD. A recent article published by the Alzheimer's Association estimates that 32% of those over age 85 have clinically diagnosed AD (50). The Vantaa 85+ study demonstrated a 33% prevalence of pathologically detected AD while clinically defined AD had a prevalence of 16% (51). For VaD, population prevalence has been reported to range between 0-8%, and 4-16% within dementia (8, 52, 53). Our frequency of 6% for VaD is within this reported range. In our series there were 3 (2.5%) cases that meet commonly-accepted criteria for tangle-predominant dementia. Other studies have examined this in elderly individuals and concluded these cases comprise approximately 5% of aged individuals (54-56). Other tauopathies, such as globular glial tauopathies, were not present (57-60). We had only one case with DLB in our study so it is difficult to compare to other findings in the literature. Despite this, our 0.8% is within the reported DLB population prevalence range of 0-5% (1, 8).
For movement disorders, we report a surprisingly high rate of PSP as compared to clinically based studies lacking autopsy confirmation (5, 61-64). We found 3.4% of normal elderly subjects developed PSP. Nath and colleagues reported clinically diagnosed PSP to have a national prevalence of 1 per 100,000 in the United Kingdom (61). Bower and colleagues reported in Olmstead County the annual incidence rate of clinically-defined PSP was 14.7 per 100,000 (5). Progressive supranuclear palsy is likely to be radically underestimated in clinical studies as clinical identification of PSP depends largely on the presence of the characteristic vertical gaze palsy but this can be absent in the majority of PSP cases confirmed at autopsy (65). In contrast to the clinical ascertainment of PSP, a recent autopsy study by Kovacs and colleagues agrees with our much higher rate, finding neuropathologically confirmed PSP to be present in 2% of their community-drawn autopsy population (66). Their study further noted that these individuals generally did not present with typical clinical features of PSP, such as the vertical gaze palsy (66). We have outlined the clinical and pathological presentations for our PSP cases and iPSP cases in Table 2 and Figure 1. Of 4 individuals with a diagnosis of clinicopathological PSP, only one had the classic presentation of Steele-Richardson syndrome with vertical gaze palsy; two had parkinsonism NOS and another presented with cognitive impairment that progressed to dementia without motor signs. One of those with parkinsonism NOS had early falls but normal eye movements and one had gait difficulty and postural instability (Table 2). All 4 of the PSP cases had prominent Gallyas-positive tufted astrocytes (Figure 1A) coiled bodies (Figure 1B,) thorned astrocytes (Figure 1C), and neurofibrillary tangles (Figure 1D), meeting both classic neuropathologic criteria of PSP (through the presence of neurofibrillary tangles in several of the characteristic brain regions) as well as modern criteria emphasizing the presence of tufted astrocytes and other Gallyas or tau-positive glia (24, 31, 67). As described in our previous report (48), the clinically normal cases with PSP-like pathology (iPSP) generally had lower densities of the glial pathologies and a reduced topographical brain distribution (see Table 2). Although two of these cases lacked the full classical neuropathological criteria for PSP, we still feel that, because of the prominence and frequency of tufted astrocytes, they are most appropriately classified as iPSP rather than with nonspecific tauopathies such as globular glial tauopathy (57-60), the glial tauopathy described to be associated with argyrophilic grains (68), the thorn-shaped astrocytes described in normal aging (69), the glial tauopathy described in chronic traumatic encephalopathy (70) or those nonspecific multisystem tauopathies associated with clinical dementia and/or parkinsonism (57, 71). We have described 3 of the iPSP cases listed in Table 2 in our previous study; Case 9 in Table 2 corresponds to Case 1 in the previous study, Case 30 to Case 2, and Case 55 to Case 3 (48).
As for other movement disorders found in our series, the population prevalence of clinically diagnosed PD has been reported to range from 0-3%, and like AD, increases with age (3, 62, 72). The most recent Olmstead county study examining the full spectrum of parkinsonism showed incidence rates for PD to be 14.2/100,000 person-years, PSP to be 0.9, MSA to be 0.8, and CBD to be 0.2; however, only 19% of subjects had autopsy confirmation (73). Previous studies on MSA in the UK population and Olmstead county had estimated annual incidence rates of 3 and 4.4 subjects per 100,000, respectively (5, 63). CBD prevalence has been estimated at 4.9-7.3 subjects per 100,000, with an annual incidence rate of 0.02 subjects per 100,000 (74-76). With only 1 subject each of CBD and MSA in our series, and both being generally rare neurological diseases with typical onset decades before our average age of study entry, it is difficult to compare our findings with others, however, as with PSP, these conditions do not always have the classical clinical syndromes and are likely to be underdiagnosed without autopsy.
In our series there were 9 cases with parkinsonism NOS. These parkinsonism NOS cases had a variety of pathologic abnormalities such as infarctions, non- substantia nigra Lewy bodies, and glial tauopathies. For the cases with infarctions, some of these might be termed vascular parkinsonism but because a great majority of older subjects contain similar lesions and do not have parkinsonism, we classified these cases as parkinsonism NOS. One case had Lewy bodies in the amygdala and olfactory bulb but none in the substantia nigra; also there was none to mild substantia nigra pigment loss, so would not meet standard diagnostic criteria for PD. One case with parkinsonism NOS had focally frequent cortical tufted astrocytes but did not have the full nuclear distribution required for the classical definition of PSP (77) and so this case is problematic. It is possible that tufted astrocytes in the cortex may have been responsible for the parkinsonism, but on the other hand the basal ganglia did not have glial or other aggregated tau deposits so again this is not clear.
The complexity and comorbidities of pathologic diagnoses in aging subjects have been commented on previously (57, 78, 79). Since AD is the most common neurodegenerative disease in the elderly it is not surprising that it had the greatest overlap with other diseases. These variations in the pathological phenotypes (i.e. concomitant diagnoses) many help explain the observed heterogeneity in clinical presentations, often making it difficult to make an accurate clinical diagnosis (80). Concomitant diagnoses within diseases may constitute distinct subtypes of the disease, which may alter the stage of the disease at death, and/or the pathological spread of either disease through the brain. With our study, given the low numbers of any particular overlapping groups we could not analyze these relationships. This concept, however, is taken into account in the current consensus guidelines for DLB diagnoses, with the likelihood of a typical DLB clinical presentation depending on the severity of the often-concomitant AD pathologies (21).
A limitation of this study is that the sample selection was based on elderly, predominantly community-dwelling volunteers residing mainly in Maricopa County, Arizona. The population is overwhelmingly composed of well-educated, Caucasian, middle and upper income individuals originating most commonly from the Midwestern and Northeastern United States. However, unlike in most brain banks, our subjects are mainly recruited through community outreach, not from doctors' offices or through hospital autopsies done for other reasons. Also to be considered is the possibility of volunteer bias, since individuals who volunteer may have different characteristics than those who do not. Additionally, it is recognized that normal subjects volunteering for dementia studies might be enriched for incipient dementia. If this were true for our presently-studied group, one might expect a higher apolipoprotein E-ε4 carriage rate than what has been reported in population-based studies. While our ε4 prevalence of 21% at study entry falls near the middle of the reported normal population range of 10-30% (81-83), some meta-analyses have reported a decreased prevalence of the ε4 allele with age, i.e. to about 18% by age 85 (84) or even to 12% or lower (85). If these lower age-related prevalence figures are reliable, then it is possible that our prevalence for the ε4 allele, and thus our progression rate to AD-type dementia may be higher than expected for the entire US population. Another limitation derives from our recruitment from retirement communities with an average age at death of 83.7 yrs. This effectively screens out subjects with early onset conditions such as frontotemporal lobar degenerations and those who have developed any of these diseases at a younger age. For these reasons, caution should be exercised in extrapolating these findings to the general population. Caution is also advisable due to geographical variation in the prevalence of neurodegenerative diseases (86).
The strength of this study is that all subjects received serial research-quality clinical examinations as well as neuropathological examinations and thus this report constitutes the first comprehensive analysis of clinicopathological outcomes in prospectively followed, initially-normal elderly individuals. Not only does this study provide rough estimates of the incidence rates for the major defined neurodegenerative diseases, but also provides estimates for the proportion of elderly individuals who have sufficient or near-sufficient pathology to meet diagnostic criteria for a major neurodegenerative disorder but lack clinical signs and symptoms. The findings also emphasize the frequent overlapping neurodegenerative diagnoses that exist in elderly subjects. This information may help explain the frequent ambiguities in biomarker studies aimed at predicting these pathologies and further highlights diagnostic difficulties, both clinically and pathologically, encountered in the assessment of elderly individuals.
Supplementary Material
Acknowledgments
We thank Dr. Hirohiko Akiyama for generously donating the antibody used to detect phosphorylated alpha-synuclein. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium) and the Michael J. Fox Foundation for Parkinson's Research.
References
- 1.Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34:561–6. doi: 10.1093/ageing/afi190. [DOI] [PubMed] [Google Scholar]
- 2.Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 3.de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–35. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 4.Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bower JH, Maraganore DM, McDonnell SK, et al. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997;49:1284–8. doi: 10.1212/wnl.49.5.1284. [DOI] [PubMed] [Google Scholar]
- 6.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 7.Bennett DA, Schneider JA, Buchman AS, et al. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology. 2005;25:163–75. doi: 10.1159/000087446. [DOI] [PubMed] [Google Scholar]
- 8.Brayne C, Richardson K, Matthews FE, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis. 2009;18:645–58. doi: 10.3233/JAD-2009-1182. [DOI] [PubMed] [Google Scholar]
- 9.Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70:774–80. doi: 10.1002/ana.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White L, Small BJ, Petrovitch H, et al. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–7. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 11.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–46. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 12.Beach TG, Sue LI, Walker DG, et al. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue banking. 2008;9:229–45. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tschanz JT, Welsh-Bohmer KA, Skoog I, et al. Dementia diagnoses from clinical and neuropsychological data compared: the Cache County study. Neurology. 2000;54:1290–6. doi: 10.1212/wnl.54.6.1290. [DOI] [PubMed] [Google Scholar]
- 14.Zandi PP, Anthony JC, Hayden KM, et al. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59:880–6. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
- 15.Polvikoski T, Sulkava R, Rastas S, et al. Incidence of dementia in very elderly individuals: a clinical, neuropathological and molecular genetic study. Neuroepidemiology. 2006;26:76–82. doi: 10.1159/000090252. [DOI] [PubMed] [Google Scholar]
- 16.Zaccai J, Ince P, Brayne C. Population-based neuropathological studies of dementia: design, methods and areas of investigation--a systematic review. BMC Neurol. 2006;6:2. doi: 10.1186/1471-2377-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler CH, Connor DJ, Hentz JG, Sabbagh MN, Caviness JN, Shill HA, Noble B, Beach TG. Incidental Lewy body disease: clinical comparison to a control cohort. Mov Disord. 2010;25:642–6. doi: 10.1002/mds.22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): Report of the NINDS-SPSP International Workshop. Neurology. 1994;47:1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- 19.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 20.Roman GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–60. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 21.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies - Third report of the DLB consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 22.Hughes AJ, Daniel SE, Blankson S, et al. A clinicopathologic study of 100 cases of Parkinson's disease. Arch Neurol. 1993;50:140–8. doi: 10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- 23.Jellinger KA, Attems J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006;112:253–60. doi: 10.1007/s00401-006-0088-2. [DOI] [PubMed] [Google Scholar]
- 24.Dickson DW. Required techniques and useful molecular markers in the neuropathologic diagnosis of neurodegenerative diseases. Acta Neuropathol. 2005;109:14–24. doi: 10.1007/s00401-004-0950-z. [DOI] [PubMed] [Google Scholar]
- 25.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet neurol. 2009;8:1150–7. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 26.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 27.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22:1689–707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 28.Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol. 1964;10:333–59. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 29.Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol. 1968;18:20–33. doi: 10.1001/archneur.1968.00470310034003. [DOI] [PubMed] [Google Scholar]
- 30.Litvan I. The clinical and pathologic hallmarks of progressive supranuclear palsy. Curr Opin Neurol. 1997;10:346–50. doi: 10.1097/00019052-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 31.Dickson DW, Rademakers R, Hutton ML. Progressive supranuclear palsy: pathology and genetics. Brain Pathol. 2007;17:74–82. doi: 10.1111/j.1750-3639.2007.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beach TG, Sue LI, Walker DG, et al. Striatal Amyloid Plaque Density Predicts Braak Neurofibrillary Stage and Clinicopathological Alzheimer's Disease: Implications for Amyloid Imaging. J Alzheimers Dis. 2012;28:869–76. doi: 10.3233/JAD-2011-111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–6. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 34.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 35.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 36.Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta neuropathologica. 2009;117:613–34. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beach TG, White CL, Hamilton RL, et al. Evaluation of alpha-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol. 2008;116:277–88. doi: 10.1007/s00401-008-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–8. [PubMed] [Google Scholar]
- 39.Beach TG, Sue L, Scott S, et al. Hippocampal sclerosis dementia with tauopathy. Brain Pathol. 2003;13:263–78. doi: 10.1111/j.1750-3639.2003.tb00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jicha GA, Abner EL, Schmitt FA, et al. Preclinical AD Workgroup staging: pathological correlates and potential challenges. Neurobiol Aging. 2012;33:622 e1–e16. doi: 10.1016/j.neurobiolaging.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Troncoso JC, Martin LJ, Dal Forno G, et al. Neuropathology in controls and demented subjects from the Baltimore Longitudinal Study of Aging. Neurobiol Aging. 1996;17:365–71. doi: 10.1016/0197-4580(96)00028-0. [DOI] [PubMed] [Google Scholar]
- 42.Tomlinson BE, Blessed G, Roth M. Observations on the brains of non-demented old people. J Neurol Sci. 1968;7:331–56. doi: 10.1016/0022-510x(68)90154-8. [DOI] [PubMed] [Google Scholar]
- 43.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42:1681–8. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 44.Crystal H, Dickson D, Fuld P, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology. 1988;38:1682–7. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 45.Frigerio R, Fujishiro H, Ahn TB, et al. Incidental Lewy body disease: do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol Aging. 2011;32:857–63. doi: 10.1016/j.neurobiolaging.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klos KJ, Ahlskog JE, Josephs KA, et al. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology. 2006;66:1100–2. doi: 10.1212/01.wnl.0000204179.88955.fa. [DOI] [PubMed] [Google Scholar]
- 47.Gibb WR. Idiopathic Parkinson's disease and the Lewy body disorders. Neuropathol Appl Neurobiol. 1986;12:223–34. doi: 10.1111/j.1365-2990.1986.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 48.Evidente VG, Adler CH, Sabbagh MN, et al. Neuropathological findings of PSP in the elderly without clinical PSP: possible incidental PSP? Parkinsonism Relat Disord. 2011;17:365–71. doi: 10.1016/j.parkreldis.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oshima K, Tsuchiya K, Iritani S, et al. [An autopsy case of “progressive supranuclear palsy” without psychiatric or neurological signs] Brain Nerve. 2004;56:157–61. [PubMed] [Google Scholar]
- 50.2013 Alzheimer's disease facts and figures. Alzheimers Dement. 2013;9:208–45. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Polvikoski T, Sulkava R, Myllykangas L, et al. Prevalence of Alzheimer's disease in very elderly people: a prospective neuropathological study. Neurology. 2001;56:1690–6. doi: 10.1212/wnl.56.12.1690. [DOI] [PubMed] [Google Scholar]
- 52.Ott A, Breteler MM, van Harskamp F, et al. Prevalence of Alzheimer's disease and vascular dementia: association with education. The Rotterdam study. BMJ. 1995;310:970–3. doi: 10.1136/bmj.310.6985.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corrada MM, Berlau DJ, Kawas CH. A population-based clinicopathological study in the oldest-old: the 90+ study. Curr Alzheimer Res. 2012;9:709–17. doi: 10.2174/156720512801322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson PT, Abner EL, Schmitt FA, et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:774–84. doi: 10.1097/NEN.0b013e3181aacbe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janocko NJ, Brodersen KA, Soto-Ortolaza AI, et al. Neuropathologically defined subtypes of Alzheimer's disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol. 2012;124:681–92. doi: 10.1007/s00401-012-1044-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santa-Maria I, Haggiagi A, Liu X, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol. 2012;124:693–704. doi: 10.1007/s00401-012-1017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kovacs GG, Molnar K, Laszlo L, et al. A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol. 2011;122:205–22. doi: 10.1007/s00401-011-0819-x. [DOI] [PubMed] [Google Scholar]
- 58.Kovacs GG, Majtenyi K, Spina S, et al. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008;67:963–75. doi: 10.1097/NEN.0b013e318187a80f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miki Y, Mori F, Hori E, et al. Hippocampal sclerosis with four-repeat tau-positive round inclusions in the dentate gyrus: a new type of four-repeat tauopathy. Acta Neuropathol. 2009;117:713–8. doi: 10.1007/s00401-009-0531-2. [DOI] [PubMed] [Google Scholar]
- 60.Ahmed Z, Bigio EH, Budka H, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. 2013;126:537–44. doi: 10.1007/s00401-013-1171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nath U, Ben-Shlomo Y, Thomson RG, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. 2001;124:1438–49. doi: 10.1093/brain/124.7.1438. [DOI] [PubMed] [Google Scholar]
- 62.Chio A, Magnani C, Schiffer D. Prevalence of Parkinson's disease in Northwestern Italy: comparison of tracer methodology and clinical ascertainment of cases. Mov Disord. 1998;13:400–5. doi: 10.1002/mds.870130305. [DOI] [PubMed] [Google Scholar]
- 63.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354:1771–5. doi: 10.1016/s0140-6736(99)04137-9. [DOI] [PubMed] [Google Scholar]
- 64.Golbe LI, Davis PH, Schoenberg BS, et al. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988;38:1031–4. doi: 10.1212/wnl.38.7.1031. [DOI] [PubMed] [Google Scholar]
- 65.Birdi S, Rajput AH, Fenton M, et al. Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Mov Disord. 2002;17:1255–64. doi: 10.1002/mds.10211. [DOI] [PubMed] [Google Scholar]
- 66.Kovacs GG, Milenkovic I, Wohrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol. 2013;126:365–84. doi: 10.1007/s00401-013-1157-y. [DOI] [PubMed] [Google Scholar]
- 67.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–9. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 68.Togo T, Sahara N, Yen SH, et al. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol. 2002;61:547–56. doi: 10.1093/jnen/61.6.547. [DOI] [PubMed] [Google Scholar]
- 69.Schultz C, Ghebremedhin E, Del Tredici K, et al. High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol Aging. 2004;25:397–405. doi: 10.1016/S0197-4580(03)00113-1. [DOI] [PubMed] [Google Scholar]
- 70.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–35. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bigio EH, Lipton AM, Yen SH, et al. Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol. 2001;60:328–41. doi: 10.1093/jnen/60.4.328. [DOI] [PubMed] [Google Scholar]
- 72.de Rijk MC, Breteler MM, Graveland GA, et al. Prevalence of Parkinson's disease in the elderly: the Rotterdam Study. Neurology. 1995;45:2143–6. doi: 10.1212/wnl.45.12.2143. [DOI] [PubMed] [Google Scholar]
- 73.Savica R, Grossardt BR, Bower JH, et al. Incidence and Pathology of Synucleinopathies and Tauopathies Related to Parkinsonism. JAMA neurology. 2013:1–7. doi: 10.1001/jamaneurol.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Togasaki DM, Tanner CM. Epidemiologic aspects. Adv Neurol. 2000;82:53–9. [PubMed] [Google Scholar]
- 75.Winter Y, Bezdolnyy Y, Katunina E, et al. Incidence of Parkinson's disease and atypical parkinsonism: Russian population-based study. Mov Disord. 2010;25:349–56. doi: 10.1002/mds.22966. [DOI] [PubMed] [Google Scholar]
- 76.Kouri N, Whitwell JL, Josephs KA, et al. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol. 2011;7:263–72. doi: 10.1038/nrneurol.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Litvan I, Agid Y, Calne D, Campbell G, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- 78.Matthews FE, Brayne C, Lowe J, McKeith I, Wharton SB, Ince P. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. 2009;6:e1000180. doi: 10.1371/journal.pmed.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schneider JA, Arvanitakis Z, Bang W, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 80.Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology. 2005;25:111–24. doi: 10.1111/j.1440-1789.2005.00605.x. [DOI] [PubMed] [Google Scholar]
- 81.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 82.Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–78. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 83.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKay GJ, Silvestri G, Chakravarthy U, Dasari S, et al. Variations in apolipoprotein E frequency with age in a pooled analysis of a large group of older people. Am J Epidemiol. 2011;173:1357–64. doi: 10.1093/aje/kwr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewis SJ, Brunner EJ. Methodological problems in genetic association studies of longevity--the apolipoprotein E gene as an example. Int J Epidemiol. 2004;33:962–70. doi: 10.1093/ije/dyh214. [DOI] [PubMed] [Google Scholar]
- 86.Russ TC, Batty GD, Hearnshaw GF, et al. Geographical variation in dementia: systematic review with meta-analysis. Int J Epidemiol. 2012;41:1012–32. doi: 10.1093/ije/dys103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.