

Abstract
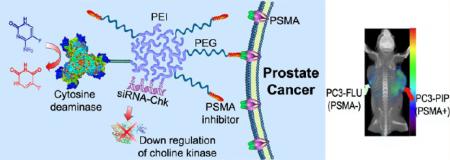
Theranostic imaging, where diagnosis is combined with therapy, is particularly suitable for a disease that is as complex as cancer, especially now that genomic and proteomic profiling can provide an extensive “fingerprint” of each tumor. With such information, theranostic agents can be designed to personalize treatment and minimize damage to normal tissue. Here we have developed a nanoplex platform for theranostic imaging of prostate cancer (PCa). In these proof-of-principle studies, a therapeutic nanoplex containing multimodal imaging reporters was targeted to prostate-specific membrane antigen (PSMA), which is expressed on the cell surface of castrate-resistant PCa. The nanoplex was designed to deliver small interfering RNA (siRNA) along with a prodrug enzyme to PSMA-expressing tumors. Each component of the nanoplex was carefully selected to evaluate its diagnostic aspect of PSMA imaging and its therapeutic aspects of siRNA-mediated down-regulation of a target gene and the conversion of a prodrug to cytotoxic drug, using noninvasive multimodality imaging. Studies performed using two variants of human PC3-PCa cells and tumors, one with high PSMA expression level and another with negligible expression levels, demonstrated PSMA-specific uptake. In addition, down-regulation of the selected siRNA target, choline kinase (Chk), and the conversion of the nontoxic prodrug 5-fluorocytosine (5-FC) to cytotoxic 5-fluorouracil (5-FU) were also demonstrated with noninvasive imaging. The nanoplex was well-tolerated and did not induce liver or kidney toxicity or a significant immune response. The nanoplex platform described can be easily modified and applied to different cancers, receptors, and pathways to achieve theranostic imaging, as a single agent or in combination with other treatment modalities.
Keywords: prostate cancer, theranostic imaging, siRNA therapy, prodrug enzyme therapy, PSMA
The convergence of chemistry with molecular biology and imaging is providing some of the most exciting advances in nanomedicine. In cancer medicine, the ideal treatment would eliminate cancer cells without damaging normal tissue. Most conventional chemotherapies exhibit toxicity not only to the cancer cells but also to several types of normal tissues.1,2 Small interfering RNA (siRNA)-mediated silencing of specific target mRNAs3,4 has significant potential to down-regulate cancer-specific pathways in cancer treatment.5,6 Several novel strategies have been developed including the use of dendritic nanocarriers,7,8 iron oxide and gold nanoparticles,9,10 and polymers11,12 to deliver siRNA in vivo. A recent study has described the use of self-assembled nucleic acid nanoparticles that provide monodisperse nanoparticles with a well-defined size for the effective delivery of siRNA in vivo.13 Many of these strategies have incorporated imaging reporters into the siRNA delivery nanoplatforms to visualize siRNA delivery.14,15 By attaching a cancer cell-specific targeting peptide or ligand, these nanoplatforms have been transformed into theranostic agents that combine detection with treatment and can be designed to target multiple cancer-specific pathways or networks by incorporating several siRNAs within a single delivery vehicle.13 Similarly, prodrug enzyme therapy, where a drug-activating enzyme delivered to the tumor converts a nontoxic prodrug to a cytotoxic drug,16 is being actively investigated to minimize normal tissue damage.17,18 A combination of both strategies can be exploited to enhance cancer-selective therapy. Noninvasive real-time imaging, a major component in the applications of theranostic agents, provides multiple advantages toward achieving the goal of effective cancer treatments without systemic toxicity. Since tumor vasculature is typically heterogeneous,19,20 the ability to image the delivery of therapeutics (e.g., siRNA and prodrug-activating enzyme) within the tumor ascertains effective delivery. Noninvasive detection of target mRNA down-regulation, through direct changes of the product or through surrogate markers, provides a means of determining the delivery of an effective dose of siRNA to the tumor. Visualization of the prodrug-activating enzyme in the tumor and its clearance from normal tissue can be leveraged to time prodrug administration, thereby minimizing normal tissue damage. Moreover, detecting the conversion of the prodrug to the active drug within the tumor would verify that the prodrug enzyme was functional within the tumor microenvironment. The use of clinically relevant imaging modalities would considerably accelerate the implementation of such treatment concepts.
Here, in proof-of-principle studies, we describe the design, development, and testing of a platform for theranostic imaging of prostate cancer (PCa). Our prototype theranostic nanoplex was synthesized as three covalently linked core components: (i) the pro-drug-activating enzyme bacterial cytosine deaminase (bCD), (ii) the multimodal imaging reporter carrier poly-L-lysine (PLL) labeled with a near-infrared (NIR) fluorescent probe Cy5.5, and (iii) the vector for siRNA delivery and for labeling with [111In]DOTA for single photon emission computed tomography (SPECT): polyethylenimine-polyethylene glycol (PEI-PEG) co-grafted polymer. The three compartments were covalently conjugated, and the siRNA was associated with the PEI-PEG co-grafted polymer through electrostatic interactions. For targeting to the prostate-specific membrane antigen (PSMA), a low molecular weight, urea-based PSMA-targeting moiety21,22 (2-(3-[1-carboxy-5-[7-(2,5-dioxopyrrolidin-1-yloxycarbonyl)heptanoylamino]pentyl]-ureido)pentanedioic acid (MW 572.56) was used for conjugating maleimide-PEG-NH2 (MW ~3400) to PEI. PSMA is a type II integral membrane protein that has abundant expression on the surface of PCa, particularly in androgen-independent, advanced, and metastatic disease.23,24
The radiolabel provided clinical translatability, while the optical reporters were valuable for in vivo as well as microscopic evaluation of nanoplex distribution in cells, cellular organelles, and in ex vivo tissue samples. To avoid the likely loss of enzymatic activity that would occur if direct multiconjugations of bCD were undertaken, imaging reporters were instead conjugated to the PLL moiety using two different cross-linkers that allowed for facile specific bridging between bCD and the siRNA delivery vector (Figures 1 and 2). Cy5.5 was used as the fluorescent moiety in PLL. Due to its emission in the NIR region (680 900 nm), Cy5.5 is advantageous for in vivo optical imaging because tissue autofluorescence and the absorption by intrinsic chromophores are relatively low in this spectral region. In the PEI-PEG/siRNA compartment, rhodamine was conjugated to the PEI polymer to track the intracellular and in vivo distribution of the siRNA vector and siRNA with microscopy.
Figure 1.

Synthetic procedure of generating nanoplex 1.
Figure 2.

Schematic representation of the structure of nanoplex 1 designed with the PSMA-targeting moiety, and the structure of nanoplex 2 that does not contain the PSMA-targeting moiety.
We selected the prodrug enzyme bCD because it converts a nontoxic prodrug 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU)25 that can be detected by 19F magnetic resonance spectroscopy (MRS).26 We have previously established that choline kinase (Chk), the enzyme that converts choline to phosphocholine (PC), is significantly up-regulated in aggressive breast cancer cells and plays an important role in tumor growth, invasion, and metastasis.27,28 We also have strong evidence that Chk down-regulation can enhance the effect of 5-FU.29 Building upon our insights in targeting choline metabolism,30,31 and because changes in choline metabolism can be easily detected clinically with MR spectroscopic imaging (MRSI) or with [11C]choline positron emission tomography (PET) studies, here we focused on using siRNA to down-regulate Chk in the choline pathway. The data collectively demonstrate the feasibility of using the platform to combine detection and treatment.
RESULTS
Nanoplex Synthesis
The bCD was produced as previously described.32,33 A brief synthesis route of the nanoplex is outlined in Figure 1. Initially, N-hydroxysuccinimide (NHS) ester of the low molecular weight urea-based PSMA inhibitor (PI) (2-(3-[1-carboxy-5-[7-(2,5-dioxopyrrolidin-1-yloxycarbonyl)heptanoylamino]pentyl]-ureido)pentanedioic acid, MW 572.56), that is, a functionalized targeting moiety, was generated. PI-NHS was conjugated with maleimide-PEG-NH2 (3.4 kDa)(Nanocs. Inc., New York, NY) to form PI-PEG-maleimide. N-Succinimidyl-S-acetylthiopropionate (SATP) (Pierce, Rockford, IL) was conjugated to PEI (Sigma, Milwaukee, WI) (25 kDa) at a 10:1 molar ratio of SATP/PEI, and then the SATP moiety was reduced to form a free sulfydryl group. Reaction between this sulfydryl and PI-PEG-maleimide generated PI-PEG-PEI (compound 3). Compound 3 was labeled with NHS-rhodamine (Sigma, Milwaukee, WI) and NHS-DOTA using previously described in-house synthesis34 to form compound 4. Compound 4 was reacted with 111InCl3 in sodium acetate buffer (pH ~4.6 5.5) to form 111In-labeled PEI (compound 5). That radioactive compound was conjugated with succinimidyl 4-formylbenzoate (SFB) (Pierce, Rockford, IL) in HEPES buffer at pH 8.4 to form 6. PLL (poly-L-lysine) (Sigma, Milwaukee, WI) (~20 kDa) was labeled with Cy5.5-NHS (GE Healthcare, Piscataway, NJ), SATP, and succinimidyl 6-hydrazinonicotin-amide acetone hydrazine (SANH) (Pierce, Rockford, IL) to produce 7. Conjugation of 6 and 7 at pH 7.4 produced the PEI-PLL copolymer, which was reduced to form 8 that contained a free sulfydryl group. Treatment of bCD with N-[ε-maleimidocaproyloxy)succinimide ester (EMCS) (Pierce, Rockford, IL) produced 9. Equimolar amounts of 9 and 8 were cross-linked through the reaction of maleimide and sulfydryl to provide the bCD-PLLPEI, 10. Finally, binding of siRNA with 10 gave the PSMA-targeting bCD-PLL-PEI/siRNA nanoplex termed nanoplex 1. We also synthesized nanoplex 2, which was identical to nanoplex 1, but without the PSMA-targeting moiety as a nontargeted control reagent. The final structures of nanoplex 1 and 2 are shown in Figure 2.
During synthesis, the amounts of PEI and PLL were measured through the absorption coefficients of rhodamine (attached on PEI), Cy5.5 (attached on PLL), and bCD at 279 nm as previously described.34 The final molar ratio of PEI/PLL/bCD was 1:1.1:1.1. Size-exclusion chromatography was used to determine the molecular weight of 375 kDa of the nanoplex. The longitudinal size and zeta-potential of the nanoplex were 65 nm (Figure 3A) and 1.6 mV (Figure 3B), respectively, as measured by dynamic light scattering (DLS). The average polydispersity index (PDI) of the nanoplex measured by DLS was found to be 0.19 ± 0.03. Although the nanoplex had a narrow size distribution, the PDI indicates that the nanoparticles did not undergo self-organization to form near-monodisperse supraparticles with a PDI of 0.1. The size distribution was confirmed by transmission electron microscopy (TEM), as shown in Figure 3C.
Figure 3.

(A) Representative size distribution of nanoplex 1. (B) Average zeta-potential of bCD, nanoplex 1 without siRNA, and nanoplex 1 with siRNA by dynamic light scattering (DLS). Values represent mean ± SEM of three measurements. (C) Transmission electron microscopy (TEM) image of nanoplex 1.
Cytosine and 5-FC were used as substrates to evaluate the activity of the prodrug enzyme. The kinetic constants were determined by monitoring changes in the absorbance of cytosine versus 5-FC at saturating substrate concentrations, as reported previously by us.34 Nanoplex 1 was found to have Km values similar to those found for native bCD for both substrates. These results indicated that conjugation of bCD to PEI-PLL did not hamper the function of bCD. Electrophoretic gel mobility shift assay indicated that nanoplex 1 retained strong binding with siRNA at an N/P ratio of 50.
Cellular Characterization of Nanoplex 1. To evaluate the specificity of nanoplex 1 to PSMA, laser fluorescent confocal microscope imaging was applied to investigate the uptake of nanoplexes 1 and 2 in PC3-Flu (PSMA negative) and PC3-PIP (PSMA positive) cells (Figure 4). After 2 h of incubation, the uptake of 5 nM nanoplex 1 in PC3-PIP was high whereas the uptake of 5 nM of nanoplex 2 in PC3-PIP was much lower. When excess PMPA (2-phosphonomethylpentanedioic acid)35,36 was added to block PSMA, the uptake of nanoplex 1 in PC3-PIP cells decreased to levels similar to the uptake of nanoplex 2. When nanoplex 1 or 2 was added to PC3-Flu cells that have low PSMA expression, the uptake was low. When the concentration was reduced to 0.5 nM, fluorescence was only observed when PC3-PIP cells were treated with nanoplex 1, which has PSMA-specific binding.
Figure 4.

Confocal fluorescence microscopic images of PC3-PIP and PC3-Flu cells incubated for 2 h with 0.5 nM or 5 nM nanoplex. Images are displayed for each concentration tested. Row 1: PC3-PIP cells treated with nanoplex 1. Row 2: PC3-PIP cells treated with nanoplex 1 after blocking with PMPA. Row 3: PC3-PIP cells treated with nanoplex 2. Row 4: PC3-Flu cells treated with nanoplex 1. Row 5: PC3-Flu cells treated with nanoplex 2. Scale bar: 50 μm.
The MTT assay demonstrated that nanoplex 1 had almost no effect on cell viability at concentrations lower than 2 μM. The transient transfection delivery efficiency of siRNA with nanoplex 1 into PC3-PIP cells was evaluated with immunoblotting. As shown in Figure 5A, down-regulation of Chk by the nanoplex 1 –siRNA complex was dependent on the concentration of siRNA used. After 24 h incubation, a concentration of 100 nM siRNA-Chk (lane 3) showed the largest down-regulation of Chk protein, to nearly undetectable levels, relative to 50 or 20 nM (lanes 4 and 5, respectively). With the latter two treatments, Chk protein levels remained similar to those seen without siRNA or with scrambled siRNA treatments (lanes 1 and 2, respectively).
Figure 5.

(A) Representative immunoblot showing that in PC3-PIP cells the down-regulation of Chk following 24 h of incubation with nanoplex 1 was dependent on the concentration of siRNA-Chk incorporated into the nanoplex (N/P ratio is 50). GAPDH protein levels were used for protein loading assessment. Lane 1: PC3-PIP cells treated with nanoplex 1 without siRNA-Chk. Lane 2: PC3-PIP cells treated with nanoplex 1 with 100 nM scrambled siRNA. Lane 3: PC3-PIP cells treated with nanoplex 1 with 100 nM siRNA-Chk. Lane 4: PC3-PIP cells treated with nanoplex 1 with 50 nM siRNA-Chk. Lane 5: PC3-PIP cells treated with nanoplex 1 with 20 nM siRNA-Chk. (B) Therapeutic efficacy of siRNA and prodrug in PC3-PIP cells. PC3-PIP cells were treated with nanoplex 1 without siRNA-Chk (control), nanoplex 1 with siRNA-Chk (siRNA-Chk), nanoplex 1 without siRNA-Chk but with 5-FC (5-FC), and nanoplex 1 with siRNA-Chk and 5-FC for 24, 48, and 72 h (siRNA-Chk+5-FC). (1, Treatment for 24 h; 2, treatment for 48 h; 3, treatment for 72 h; nanoplex concentration = 350 nM, N/P = 50, siRNA-Chk concentration = 80 nM, 5-FC concentration = 3 mM. Values represent mean ± SEM of three or more assays for each treatment; *, P < 0.05; **, P < 0.01; ***, P < 0.001.)
The therapeutic efficacy of siRNA, prodrug, and combination therapy in PC3-PIP cells is presented in Figure 5B. With nanoplex 1 (350 nM) alone, cell viability remained above 95% throughout the 72 h incubation period. With siRNA-Chk bound to nanoplex 1 (350 nM nanoplex, 80 nM siRNA, N/P = 50), cell viability decreased to about 65% after 24 h and to less than 60% after 48 and 72 h of incubation. When cells were incubated with 350 nM of nanoplex 1 with 3 mM 5-FC but without siRNA for 1–3 days, cell viability was reduced to about 40% after 24 h, 25–30% after 48 h, and nearly 20% at 72 h. Finally, when cells were incubated with 350 nM nanoplex 1 complexed with 80 nM siRNA plus 3 mM 5-FC, the decrease in cell viability after 24 h was 30%, which was significantly lower than either therapy used alone at this time point. This significant difference was also observed at 48 h, but by the 72 h time point, there was no significant difference in viability between treatment with 5-FC alone and the combined treatment. This was most likely due to the longer exposure of cells to 5-FU resulting in a convergence of cell viability values for cells treated with 5-FC alone and cells treated with siRNA and 5-FC.
Higher Specific Uptake of the Targeted Nanoplex in PSMA Overexpressing Tumors
Immunoblot analysis of PC3-PIP and PC3-Flu cell extracts confirmed the differential expression of PSMA, as shown in the representative immunoblot in Figure 6A. SPECT/CT images obtained from mice bearing PC3-PIP and PC3-Flu tumors revealed a significantly higher uptake of the targeted nanoplex in PSMA-overexpressing PC3-PIP tumors compared to PC3-Flu tumors (Figure 6B,C). To confirm the specificity of this uptake, blocking experiments were performed by injecting antibody directed against PSMA 5 h before injecting nanoplex 1. Optical imaging analysis, performed on tissue slices without or with PSMA blocking in mice bearing PC3-PIP and PC3-Flu tumors, demonstrated increased uptake in PC3-PIP tumors compared to PC3-Flu, which was reduced with blocking, confirming the in vivo results obtained by SPECT imaging (Figure 6D).
Figure 6.

(A) Representative immunoblot showing PSMA protein expression in PC3-PIP and PC3-Flu cells. GAPDH was used as a loading control. (B) Representative SPECT images of a SCID mouse bearing PC3-PIP and PC3-Flu tumors. Mice were injected i.v. with 776 μCi of 111In-labeled PSMA-targeted nanoplex 1 (150 mg/kg in 0.2 mL of PBS). SPECT images were acquired in 64 projections at 30 s/projection. Following tomography, CT images were acquired in 512 projections to allow co-registration. Decay-corrected transaxial SPECT imaging slice (slice thickness 5 mm) of a representative mouse showed clear accumulation of radioactivity in PC3-PIP tumor at 48 h. (C) ROI analysis of tumors and muscle showed significant accumulation of activity in PC3-PIP tumors at 48 h post-injection. Values represent mean ± SEM (n = 4, *P < 0.05 with PC3-Flu tumor uptake as the comparative reference). (D) Nanoplex accumulation in PC3-PIP and PC3-Flu tumors without and with blocking. Representative tumors excised at 48 h after nanoplex injections are shown. Images were acquired on the Caliper Spectrum scanner to detect Cy5.5 signal. For the blocking studies, 100 μg of anti-PSMA antibody was injected i.v. in PC3-PIP and PC3-Flu tumor bearing mice. Five hours after injection of antibody, 75 mg/kg of nanoplex 1 was injected i.v. in the same mouse.
In Vivo Assessment of Chk Inhibition and bCD Activity
To assess the efficacy of siRNA-Chk to down-regulate Chk, we acquired in vivo 1H MRSI of PC3-PIP tumors 48 h after administration of nanoplex 1. As shown in Figure 7A–D, we observed a significant decrease of the total choline (tCho) signal that consists of free choline, PC, and glycerophosphocholine. Prior to injection, tCho was detected throughout large portions of each tumor. However, tCho decreased significantly within 48 h post-injection in the tumor and was largely localized to a thin rim at the tumor periphery. On average, tCho levels decreased to about 30% of pretreatment values at 48 h post-injection. Moreover, by performing 19F MRS, we observed that the prodrug enzyme bCD was still active at 24 and 48 h post-injection, as it continued to convert the prodrug 5-FC to 5-FU over this time (Figure 7E).
Figure 7.

(A) In vivo tCho density maps from 2D CSI datasets acquired from arepresentative PC3-PIPtumor (~400 mm3) before and 48 h after i.v. injection of the PSMA-targeted nanoplex 1 (150 mg/kg). Parameters used were TE = 120 ms, TR= 1000 ms, 4 scans per phase encode step. CSI spectra were acquired at 9.4 T with an in-plane spatial resolution of 1 mm × 1 mm from a 4 mm thick slice. (B) Corresponding in vivo tCho maps from the same 2D CSI data sets. (C) Representative one voxel spectra from 2D CSI represented in A and B. (D) tCho concentration calculated in arbitrary units before and at 48 h after injection of nanoplex 1. Values represent median ± SEM (n = 3, *P < 0.05). (E) In vivo 19FMR spectra acquired from a PC3-PIP tumor (~400 mm3) at 24 and 48 h after i.v. injection of the PSMA-targeted nanoplex (150 mg/kg) carrying bCD and siRNA-Chk. Spectra were acquired after a combined i.v. and i.p. injection of 5-FC (450 mg/kg) on a Bruker Biospec 9.4 T spectrometer using a 1 cm solenoid coil tunable to 1H and 19F frequency. Following shimming on the water proton signal, serial nonselective 19F MR spectra were acquired starting 20 min after the 5-FC injection and continued every 30 min for 110 min with a repetition time of 0.8 s, a number of scans of 2000, and a spectral width of 10 kHz.
Assessment of Toxicity and Immunogenicity
Alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine, and blood urea nitrogen measurements were performed to assess the hepatic and renal toxicity of the nanoplex (Figure 8A,B). No significant differences were observed in these four parameters between the treated group injected with nanoplex 1 and the control group injected with PBS. We also studied the immunogenicity of nanoplex 1 by measuring the white blood cell (WBC) count after three repeated injections in imnunocompetent mice (Figure 8C). Total WBC counts did not increase after the injections. Instead, we observed a decrease of WBCs. This was mainly because lymphocyte numbers decreased although the values remained within the normal range (0.9–9.3 million/mL). Red blood cells and platelets were not affected by nanoplex injections (Figure 8D). We compared the effects of nanoplex injections to those induced by similar injections of Feridex. At the dose used, Feridex was found to significantly decrease blood cell counts and increase hematocrit compared to nanoplex 1 (Figure 8C,D).
Figure 8.

(A) Alanine aminotransferase (ALT) and aspar-tate aminotransferase (AST) measurements at 48 h post-injection of 150 mg/kg nanoplex 1 per mouse (n = 4). (B) Creatinine and blood urea nitrogen measurements at 48 h post-injection of 150 mg/kg nanoplex 1 per mouse (n = 4). (C,D) Results from the immunogenicity studies (150 mg/kg of nanoplex 1 injected every 3 days for a total of three injections). Values represent mean ± SEM (*P < 0.05; **P < 0.01; ***P < 0.001, n = 4); M/mL denotes million/mL.
DISCUSSION
Here in proof-of-principle studies, we have shown the feasibility of using a nanoplex platform to achieve theranostic imaging of PCa. Our chosen PCa diagnostic target, PSMA, is being actively investigated for diagnostic imaging and as a therapeutic target.37,38 PSMA expression is correlated with androgen insensitivity and may play a role in tumor invasiveness.39 Recently, measurement of PSMA expression through molecular imaging in vivo enabled monitoring of anti-androgen therapy.40 Here we used a small molecule based on the glutamate-urea-X (X is an R-amino acid derivative) motif to achieve PSMA-specific retention of nanoplex 1 through electrostatic interaction with the extracellular active site of PSMA. The three carboxylic acid groups of the PSMA-targeting moiety are necessary for binding with PSMA, with the urea providing interaction with Zn2+ at the active site.41 A 3.4 kDa PEG chain is required to separate the targeting moiety from the nanoplex since the targeting moiety must reach deep within PSMA for productive binding.
Although the nanoplex can, at relatively high concentrations, enter cells through endocytosis, our cellular and in vivo binding specificity studies demonstrated that the PSMA-targeting moiety enhanced the uptake of the nanoplex in PSMA-expressing cells and tumors. We observed increased retention of nanoplex 1 in PSMA-over-expressing tumors in vivo. Further evidence of specificity was provided by the blocking studies in which the differential retention was eliminated once PSMA was blocked with prioradministrationofanti-PSMA antibodies.
The SPECT-CT imaging data detected an increased accumulation of nanoplex 1 in PC3-PIP tumors at 48 h after injection. The higher permeability of the tumor vasculature provided a natural selection process for allowing the nanoplex to leak out extensively in tumors but not in normal tissue.42 However, because of the specificity of the PSMA-targeting moiety, the accumulation of nanoplex 1 was much higher in PC3-PIP than in PC3-Flu tumors. Recent studies have reported the presence of PSMA on human tumor neovasculature43 although this has not been shown in mouse vasculature that supports the growth of human tumor xeno-grafts in mice. PSMA expression on tumor neovasculature therefore did not contribute to the differences observed here.
It is apparent from the SPECT images that there was significant accumulation of the nanoplex in the liver. In addition, mouse (but to a lesser extent human) kidneys express PSMA.44 Any tissue where the nanoplex localizes will result in the formation of high concentrations of 5-FU. However down-regulation of Chk, the siRNA target selected, does not affect nonmalignant cells.29 In addition, the liver contains high levels of dihydropyrimidine dehydrogenase (DPD), which catabolizes 5-FU to dihydrofluorouracil (DHFU).45 This may, however, be a problem for other prodrug enzyme systems where the end product is not 5-FU. The vasculature itself maybe damaged if the prodrug is delivered while the concentration of the nanoplex in the blood is high, but based on the imaging data, maximum retention in the tumor occurs when nanoplex concentrations in the blood are negligible. The major advantage of having noninvasive imaging reporters on the nanoplex is that it is possible to visualize the nanoplex and inject the prodrug when the ratio of prodrug enzyme concentration in tumor to normal tissue is highest. The ability to target the nanoplex to PSMA resulted in significantly more retention in PSMA-expressing tumors than just the enhanced permeability and retention effect due to leaky tumor vasculature. Of course once 5-FU is formed locally in the tumor it is possible that not all of it will be metabolized by cancer cells, and a small fraction will reenter the bloodstream. Normal tissue damage arising from this limitation is, however, likely to be relatively insignificant compared to direct systemic administration of therapeutic doses of 5-FU.
We have previously shown34 that a single dose of the nontargeted siRNA and bCD containing nanoplex together with a single dose of 5-FC resulted in a 6-fold slower tumor doubling time, compared to a 3-fold slower tumor doubling time with just bCD alone, in a breast cancer xenograft model. Our purpose here was to demon-stratethe abilityto selectively target thenanoplex to PSMA that is expressed by aggressive PCa cells. With specific targeting we anticipate that tumor retention and the 5-FU formed will be even higher than without targeting.
We found that branched PEI can be used as an efficient siRNA delivery vector because of its buffering effect,46 which resulted in endosomal release of endocytosed siRNA into cytoplasm.34 There were approximately 10 PEG chains on the surface of one PEI molecule. The molar excess of PEG served as a bridge between the PSMA-targeting moiety and the nanoplex and also sterically shielded the relatively large net positive charge on the surface of the PEI, which might otherwise hinder the functioning of the PEI and nanoplex. Shielding of the positive charge reduces the toxicity of PEI, in part, through decreased interactions of the PEI with blood and cellular components.47,48 Conjugation of the PEI in the nanoplex with hydrophilic PEG increased the IC50 to about 30 times that of the nanoplex without PEG modification. The prodrug enzyme bCD displayed high stability, and importantly, it was possible to detect enzyme activity noninvasively with 19F MRS.34 Our studies demonstrated that bCD maintained high activity even after conjugation with PEI. We chose PLL as the linker between the PEI and bCD to minimize interactions between PEI and bCD and thus maintain bCD activity. The bCD enzyme attached to the nanoplex was active up to 48 h post-injection and was able to convert the nontoxic prodrug 5-FC into the toxic 5-FU efficiently throughout the time course of these experiments. The siRNA directed against Chk induced a decrease in the tCho signal due to a decrease of PC, which was visible in vivo with 1H MRSI. Proton and 19F MRS techniques are noninvasive and can be easily translated to the clinic. Here we showed that it was possible to noninvasively assess the conversion of 5-FC into 5-FU in the tumor, along with the efficacy of Chk down-regulation by acquiring 19F spectra and tCho maps with 1H MRSI, respectively.
The immunogenicity studies performed in immuno-competent mice did not detect any major effects of repeated injections on mouse immune cell counts. All values stayed within the normal range. However, more extensive toxicity and immunogenicity studies will be required prior to translation to the clinic. In addition, the application of humanizing protein technology to bCD may further reduce potential immunogenic effects.
To achieve systemic therapy safely, it is important to have control over where the toxic species are delivered preferentially within the tumor, leaving normal tissues unharmed. That control can be achieved through targeting a nanoplex and being able to measure the delivery of that nanoplex through imaging. There is also a compelling need to find effective treatments for metastatic disease, as it typically becomes refractory to treatment. The targeted nanoplex that we have developed, and which carries multimodality imaging reporters together with siRNA and a prodrug enzyme, will be useful for theranostic imaging of metastatic PCa. It can also be extended into a platform technology toward many cancer subtypes and therapeutic targets. Down-regulation of specific pathways using siRNA further provides unique opportunities to target cancer cells selectively while sparing normal tissue. The nanoplex platform described here has the ability to deliver multiple siRNA. The strategy developed here can be extended, in the future, to down-regulate multi-drug-resistant pathways or repair enzymes with the goal of increasing the efficacy, safety, and efficiency of chemoor radiation therapies.
MATERIALS AND METHODS
siRNA
The siRNA-Chk duplex directed against human Chk mRNA (sense: 5′-CAUGCUGUUCCAGUGCUCCUU-3′ and anti-sense: 5′-GGAGCACUGGAACAGCAUGUU-3′) and the scrambled siRNA were purchased from Dharmacon (Lafayette, CO) and designed using their ON-TARGET plus program.
Determination of Size Distribution and Zeta-Potential of Nanoplex 1
The hydrodynamic radius and size distribution of nanoplex 1 were determined by DLS. Nanoplex 1 was diluted in PBS at pH 7.4 buffer at a concentration of 1 mg/mL and filtered through a 0.45 μm filter before measurement. The average zeta-potential of bCD, nanoplex 1 without siRNA, and nanoplex 1 with siRNA in 0.1 M NaCl solution was estimated from 20 acquisitions in a clear zeta cell cuvette with a concentration of approximately 2.5 mg/mL.
Transmission Electron Microscopy
Nanoplex 1 was visualized by TEM using 300 mesh carbon-coated copper grids at 80 kV and 120 000× magnification. Nanoplex 1 was prepared at a concentration of 0.01 μg/μL. Approximately 5 μL of the nanoplex solution was loaded on the Cu grid for 2 min followed by blotting of the excess liquid and stained for another 2 min with 1% phosphotungstic acid followed by blotting. The grid was air-dried for another 5 min and visualized under a transmission electron microscope (Hitachi 7600, Hitachi High Technologies America, Inc., Pleasanton, CA). The particle size was measured in digital images by NIH program ImageJ (http://rsbweb.nih.gov/ij/).
Cell Culture
Human PCa PC3 cells transfected to overexpress PSMA (PC3-PIP) or transfected with the plasmid alone (PC3-Flu) were obtained from Dr. Warren Heston (Cleveland Clinic, Cleveland, OH). Cells were maintained in RPMI 1640 (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum in a humidified incubator at 37 °C/5% CO2.
In Vitro Cell Culture Studies
The cytotoxicity of the nanoplex was evaluated by an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Sigma, Milwaukee, WI). PC3-PIP cells (2 × 103 cells/well) in 96-well plates were incubated for 24 h in RPMI 1640 prior to treatment. To evaluate the therapeutic efficacy induced by down-regulation of Chk, the cells were treated with nanoplex 1 (N/P = 50, 80 nM siRNA/350 nM nanoplex 1). To test the therapeutic efficacy of the prodrug strategy, the cells were treated with nanoplex 1 (350 nM) without siRNA with the addition of 5-FC (3 mM). To evaluate the combined therapeutic efficacy of siRNA and prodrug strategy, cells were treated with nanoplex 1 (N/P = 50, 80 nM siRNA/ 350 nM nanoplex 1) with the addition of 5-FC (3 mM).
Confocal Laser Scanning Fluorescence Microscopy
The cells were treated with either nanoplex 1 or 2 at different concentrations for 2 h, then washed three times with PBS buffer at pH 7.4. An ethanol/acetic acid/formaldehyde (85/5/10) solution was used to fix the treated cells, following which cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 5 min. Fluorescence microscopic images of PC3-PIP and PC3-Flu cells were generated on a Zeiss LSM 510 META confocal laser scanning microscope (Carl Zeiss, Inc., Oberkochen, Germany). Rhodamine and DAPI fluorescence images were obtained using λex = 543 nm and λem = 560 nm and λex = 405 and λem = 420–480 nm filter sets, respectively.
Immunoblot Analysis of PC3-PIP Cells
PC3-PIP cells were treated with different concentrations of the nanoplex for 24 h, following which cells were collected after washing three times with ice-cold PBS buffer. Proteins were extracted using RIPA buffer with protease inhibitor cocktail (1/500, Sigma, St. Louis, MO), dithiothreitol (1/1000, 1 M stock), phenylmethylsulfonyl fluoride (1/200, 0.2 M stock), sodium orthovanadate (1/500, 0.5 M stock), and sodium fluoride (1/500, 0.5 M stock). About 100 μg of protein was resolved on 10% SDS-PAGE, transferred onto nitrocellulose membranes, and probed with a mouse monoclonal antibody against PSMA (Abcam, Cambridge, MA) or with a custom-made polyclonal antibody against Chk (Proteintech Group, Inc., Chicago, IL) as previously described.49,50 Appropriate horseradish peroxidase (HRP)-conjugated secondary antibody donkey anti-mouse antibody was used at 1/2000 dilution. A mouse monoclonal antibody against GAPDH (Sigma, St. Louis, MO) at 1/1000 was used as loading control. Immunoblots were developed using the Super-Signal West Pico chemiluminescent substrate kit (Pierce Biotechnology, Inc., Rockford, IL). Intratumoral Chk levels in cells before and after treatment were visually evaluated by immunoblot analyses.
Mouse Model and Tumor Implantation
All in vivo studies were done in compliance with guidelines established by the Institutional Animal Care and Use Committee of The Johns Hopkins University. PC3-PIP and PC3-Flu human PCa cells (2 × 106 cells/mouse) were inoculated subcutaneously in severe combined immunodeficient (SCID) male mice. Tumors were palpable within one week after implantation and reached a volume of approximately 300 to 400 mm3 within three weeks, at which time they were used for experiments. Immunogenicity and toxicity studies were performed in immunocompetent Balb/C mice.
SPECT/CT Imaging
SPECT imaging of SCID mice bearing PC3-PIP and PC3-Flu tumors was performed with [111In]DOTA-radiolabeled nanoplex 1 (770 ± 208 μCi, 150 mg/kg dose injected intravenously in 0.2 mL of PBS, n = 4). A dedicated small-animal SPECT/CT system (Gamma Medica X-SPECT, Northridge, CA) was used for image acquisition. SPECT/CT images were obtained at 48 h post-injection with an energy window of 170–250 keV. Tomographic data were acquired in 64 projections over 360° at 40 s/projection. Following SPECT, CT images were acquired with 512 projections. Data were reconstructed using an ordered subsets-expectation maximization (OS-EM) algorithm and analyzed using AMIDE software (SourceForge; http://sourceforge.net/projects/amide/). To calculate the amount of accumulated radioactivity, images were normalized to the injected dose and regions of interest were drawn over the whole tumor.
In Vivo MRS
PC3-PIP tumor bearing mice were anesthetized with a mixture of ketamine (25 mg/kg) and acepromazine (2.5 mg/kg) injected i.p. before all MR studies. Anesthetized mice were imaged on a 9.4 T Bruker Biospec spectrometer (Bruker Biospin Co., Billerica, MA) using a solenoid coil placed around the tumors. Body temperature of the animals in the magnet was maintained by a thermostat-regulated heating pad.
In Vivo 1H MRS
MRSI was performed using a two-dimensional (2D) chemical shift imaging (CSI) sequence. A reference image from a 4 mm thick central slice of the tumor was acquired using a spin–echo sequence. Water-suppressed MRSI was performed on the same 4 mm thick central slice, with an in-plane resolution of 1 mm × 1 mm per pixel using a 2D CSI sequence with VAPOR water suppression and the following parameters: echo time (TE) of 120 ms, repetition time (TR) of 1000 ms, field of view of 1.6 cm × 1.6 cm, phase encode steps of 16 (16 × 16 matrix), number of scans (NS) 4, block size 512, and sweep width of 7000 Hz. Water MR spectroscopic images were also acquired without water suppression on the same slice, with TE = 20 ms and NS = 1, and with all other parameters remaining the same. Spectroscopic images of the tCho signal at 3.2 ppm and the water signal at 4.7 ppm were generated from the MRSI data sets using an in-house IDL program. These images were imported in the freeware NIH program ImageJ (http://rsbweb.nih.gov/ij/) for analyses.
In Vivo 19F MRS
All 19F MRS experiments were done using a solenoid coil tunable to 1H or 19F frequency. Typically, after injection of 5-FC (450 mg/kg), anesthetized mice (n = 3) were placed on a plastic cradle to allow positioning of the tumor in the RF coil. Following shimming on the water proton signal, serial 19F nuclear MR spectra were acquired from the tumor every 30 min for 110 min using a one-pulse sequence (flip angle, 60°; repetition time, 0.8 s; number of average, 2000; spectral width, 10 kHz). 19F MR spectra were processed with an in-house XsOs nuclear magnetic resonance software developed by Dr. D. Shungu (Cornell University, New York, NY). The chemical shift of the 5-FU resonance was set to 0 ppm.
Blocking Experiments and Ex Vivo Optical Imaging Studies
For the binding specificity (blocking) studies, 100 μg of anti-PSMA mouse monoclonal antibody (Clone GCP-05, Abcam, Cambridge, MA) was injected i.v. in a volume of 0.05 mL pf PBS in PC3-PIP and PC3-Flu tumor bearing mice. Five hours after injection of antibody, 1.5 mg of nanoplex 1 (75 mg/kg) was injected i.v. in the same mice. Mice were sacrificed 48 h after nanoplex injection. Tumors, muscle, and kidney were excised, and optical images were obtained on the IVIS Caliper Spectrum optical scanner (Caliper Life Sciences, Hopkinton, MA). A Cy5.5 excitation (615–665 nm) and emission (695–770 nm) filter set was used to acquire the Cy5.5 fluorescence data. Cy5.5 fluorescence images were acquired using a λex = 615–665 nm and λem = 695–770 nm filter set, 1 s exposure time, and the fluorescence intensity was scaled as units of ps −1 cm −2 sr −1.
Blood Analysis
All blood analyses were performed by the Johns Hopkins Phenotyping and Pathology Core. ALT, AST, creatinine, and blood urea nitrogen measurements were done on mouse serum 48 h post-injection of 150 mg/kg nanoplex 1. For the immunogenicity studies, blood cell counts were performed on heparinized blood samples from immunocompetent Balb/C mice that were injected i.v. with 150 mg/kg nanoplex 1 every 3 days for a total of three injection. An additional comparison was made with immunocompetent mice injected with Feridex (Advanced Magnetics Inc., Cambridge, MA) injected at a dose of 10 mg/kg of Feridex, which is in the range of typical concentrations used in preclinical studies,51 with the same injection schedule as for the nanoplex.
Acknowledgment
This work was supported by NIH P50 CA103175, R01 CA138515, and R01 CA134675. We thank F. Wildes, Y. Mironchik, J. Fox, G. Green, and G. Cromwell forvaluable technical support. We gratefully acknowledge the support of Dr. J. S. Lewin.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information Available: Detailed synthesis, characterization, enzymatic kinetics, cytotoxicity, stability studies. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.Hassett MJ, O'Malley AJ, Pakes JR, Newhouse JP, Earle CC. Frequency and Cost of Chemotherapy-Related Serious Adverse Effects in a Population Sample of Women with Breast Cancer. J. Natl. Cancer Inst. 2006;98:1108–1117. doi: 10.1093/jnci/djj305. [DOI] [PubMed] [Google Scholar]
- 2.Ladewski LA, Belknap SM, Nebeker JR, Sartor O, Lyons EA, Kuzel TC, Tallman MS, Raisch DW, Auerbach AR, Schumock GT, et al. Dissemination of Information on Potentially Fatal Adverse Drug Reactions for Cancer Drugs from 2000 to 2002: First Results from the Research on Adverse Drug Events and Reports Project. J. Clin. Oncol. 2003;21:3859–3866. doi: 10.1200/JCO.2003.04.537. [DOI] [PubMed] [Google Scholar]
- 3.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 4.Meister G, Tuschl T. Mechanisms of Gene Silencing by Double-Stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 5.Devi GR. siRNA-Based Approaches in Cancer Therapy. Cancer Gene Ther. 2006;13:819–829. doi: 10.1038/sj.cgt.7700931. [DOI] [PubMed] [Google Scholar]
- 6.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in Humans from Systemically Administered siRNA via Targeted Nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ofek P, Fischer W, Calderon M, Haag R, Satchi-Fainaro R. In Vivo Delivery of Small Interfering RNA to Tumors and Their Vasculature by Novel Dendritic Nanocarriers. FASEB J. 2010;24:3122–3134. doi: 10.1096/fj.09-149641. [DOI] [PubMed] [Google Scholar]
- 8.Merkel OM, Mintzer MA, Librizzi D, Samsonova O, Dicke T, Sproat B, Garn H, Barth PJ, Simanek EE, Kissel T. Triazine Dendrimers as Nonviral Vectors for In Vitro and In Vivo RNAi: The Effects of Peripheral Groups and Core Structure on Biological Activity. Mol. Pharmaceutics. 2010;7:969–983. doi: 10.1021/mp100101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu G, Xie J, Zhang F, Wang Z, Luo K, Zhu L, Quan Q, Niu G, Lee S, Ai H, et al. N-Alkyl-PEI-Functionalized Iron Oxide Nanoclusters for Efficient siRNA Delivery. Small. 2011;7:2742–2749. doi: 10.1002/smll.201100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng D, Giljohann DA, Chen DL, Massich MD, Wang XQ, Iordanov H, Mirkin CA, Paller AS. Topical Delivery of siRNA-Based Spherical Nucleic Acid Nanoparticle Conjugates for Gene Regulation. Proc. Natl. Acad. Sci. U.S.A. 2012;109:11975–11980. doi: 10.1073/pnas.1118425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gary DJ, Lee H, Sharma R, Lee J-S, Kim Y, Cui ZY, Jia D, Bowman VD, Chipman PR, Wan L, et al. Influence of Nano-Carrier Architecture on In Vitro siRNA Delivery Performance and In Vivo Biodistribution: Polyplexes vs Micelleplexes. ACS Nano. 2011;5:3493–3505. doi: 10.1021/nn102540y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng M, Librizzi D, Klc A, Liu Y, Renz H, Merkel OM, Kissel T. Enhancing In Vivo Circulation and siRNA Delivery with Biodegradable Polyethylenimine-Graft-Polycaprolac-tone-Block-Poly(ethylene glycol) Copolymers. Biomaterials. 2012;33:6551–6558. doi: 10.1016/j.biomaterials.2012.05.055. [DOI] [PubMed] [Google Scholar]
- 13.Lee H, Lytton-Jean AKR, Chen Y, Love KT, Park AI, Karagiannis ED, Sehgal A, Querbes W, Zurenko CS, Jayaraman M, et al. Molecularly Self-Assembled Nucleic Acid Nanoparticles for Targeted In Vivo siRNA Delivery. Nat. Nanotechnol. 2012;7:389–393. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taratula O, Garbuzenko O, Savla R, Wang YA, He H, Minko T. Multifunctional Nanomedicine Platform for Cancer Specific Delivery of siRNA by Superparamagnetic Iron Oxide Nanoparticles-Dendrimer Complexes. Curr. Drug Delivery. 2011;8:59–69. doi: 10.2174/156720111793663642. [DOI] [PubMed] [Google Scholar]
- 15.Kenny GD, Kamaly N, Kalber TL, Brody LP, Sahuri M, Shamsaei E, Miller AD, Bell JD. Novel Multifunctional Nanoparticle Mediates siRNA Tumour Delivery, Visualisation and Therapeutic Tumour Reduction in Vivo. J. Controlled Release. 2011;149:111–116. doi: 10.1016/j.jconrel.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 16.Xu G, McLeod HL. Strategies for Enzyme/Prodrug Cancer Therapy. Clin. Cancer. Res. 2001;7:3314–3324. [PubMed] [Google Scholar]
- 17.Russell PJ, Khatri A. Novel Gene-Directed Enzyme Prodrug Therapies Against Prostate Cancer. Expert Opin. Invest. Drugs. 2006;15:947–961. doi: 10.1517/13543784.15.8.947. [DOI] [PubMed] [Google Scholar]
- 18.Singh Y, Palombo M, Sinko PJ. Recent Trends in Targeted Anticancer Prodrug and Conjugate Design. Curr. Med. Chem. 2008;15:1802–1826. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain RK. Transport of Molecules in the Tumor Interstitium: A Review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 20.Fukumura D, Jain RK. Tumor Microvasculature and Microenvironment: Targets for Anti-Angiogenesis and Normalization. Microvasc. Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozikowski AP, Nan F, Conti P, Zhang J, Ramadan E, Bzdega T, Wroblewska B, Neale JH, Pshenichkin S, Wroblewski JT. Design of Remarkably Simple, Yet Potent Urea-Based Inhibitors of Glutamate Carboxypeptidase II (NAALADase). J. Med. Chem. 2001;44:298–301. doi: 10.1021/jm000406m. [DOI] [PubMed] [Google Scholar]
- 22.Banerjee SR, Pullambhatla M, Byun Y, Nimmagadda S, Foss CA, Green G, Fox JJ, Lupold SE, Mease RC, Pomper MG. Sequential SPECT and Optical Imaging of Experimental Models of Prostate Cancer with a Dual Modality Inhibitor of the Prostate-Specific Membrane Antigen. Angew. Chem., Int. Ed. 2011;50:9167–9170. doi: 10.1002/anie.201102872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulke N, Varlamova OA, Donovan GP, Ma D, Gardner JP, Morrissey DM, Arrigale RR, Zhan C, Chodera AJ, Surowitz KG, et al. The Homodimer of Prostate-Specific Membrane Antigen Is a Functional Target for Cancer Therapy. Proc. Natl. Acad. Sci. U.S.A. 2003;100:12590–125905. doi: 10.1073/pnas.1735443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Bennett M, Thorpe PE. Anti-Tumor Effects and Lack of Side Effects in Mice of an Immunotoxin Directed Against Human and Mouse Prostate-Specific Membrane Antigen. Prostate. 2004;61:1–11. doi: 10.1002/pros.20074. [DOI] [PubMed] [Google Scholar]
- 25.Malet-Martino M, Jolimaitre P, Martino R. The Prodrugs of 5-Fluorouracil. Curr. Med. Chem. Anticancer Agents. 2002;2:267–310. doi: 10.2174/1568011023354146. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Penet MF, Winnard P, Jr., Artemov D, Bhujwalla ZM. Image-Guided Enzyme/Prodrug Cancer Therapy. Clin. Cancer Res. 2008;14:515–522. doi: 10.1158/1078-0432.CCR-07-1837. [DOI] [PubMed] [Google Scholar]
- 27.Glunde K, Ackerstaff E, Mori N, Jacobs MA, Bhujwalla ZM. Choline Phospholipid Metabolism in Cancer: Consequences for Molecular Pharmaceutical Interventions. Mol. Pharmaceutics. 2006;3:496–506. doi: 10.1021/mp060067e. [DOI] [PubMed] [Google Scholar]
- 28.Glunde K, Jie C, Bhujwalla ZM. Molecular Causes of the Aberrant Choline Phospholipid Metabolism in Breast Cancer. Cancer Res. 2004;64:4270–4276. doi: 10.1158/0008-5472.CAN-03-3829. [DOI] [PubMed] [Google Scholar]
- 29.Mori N, Glunde K, Takagi T, Raman V, Bhujwalla ZM. Choline Kinase Down-Regulation Increases the Effect 5-Fluorouracil in Breast Cancer Cells. Cancer Res. 2007;67:11284–11290. doi: 10.1158/0008-5472.CAN-07-2728. [DOI] [PubMed] [Google Scholar]
- 30.Janardhan S, Srivani P, Sastry GN. Choline Kinase: An Important Target for Cancer. Curr. Med. Chem. 2006;13:1169–1186. doi: 10.2174/092986706776360923. [DOI] [PubMed] [Google Scholar]
- 31.Glunde K, Jacobs MA, Bhujwalla ZM. Choline Metabolism in Cancer: Implications for Diagnosis and Therapy. Expert Rev. Mol. Diagn. 2006;6:821–829. doi: 10.1586/14737159.6.6.821. [DOI] [PubMed] [Google Scholar]
- 32.Li C, Wildes F, Winnard P, Jr., Artemov D, Penet MF, Bhujwalla ZM. Conjugation of Poly-L-Lysine to Bacterial Cytosine Deaminase Improves the Efficacy of Enzyme/Prodrug Cancer Therapy. J. Med. Chem. 2008;51:3572–3582. doi: 10.1021/jm800288h. [DOI] [PubMed] [Google Scholar]
- 33.Li C, Winnard PT, Jr., Takagi T, Artemov D, Bhujwalla ZM. Multimodal Image-Guided Enzyme/Prodrug Cancer Therapy. J. Am. Chem. Soc. 2006;128:15072–15073. doi: 10.1021/ja066199i. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Penet MF, Wildes F, Takagi T, Chen Z, Winnard PT, Artemov D, Bhujwalla ZM. Nanoplex Delivery of siRNA and Prodrug Enzyme for Multimodality Image-Guided Molecular Pathway Targeted Cancer Therapy. ACS Nano. 2010;4:6707–6716. doi: 10.1021/nn102187v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pomper MG, Musachio JL, Zhang J, Scheffel U, Zhou Y, Hilton J, Maini A, Dannals RF, Wong DF, Kozikowski AP. 11C-MCG: Synthesis, Uptake Selectivity, and Primate PET of a Probe for Glutamate Carboxypeptidase II (NAALADase). Mol. Imaging. 2002;1:96–101. doi: 10.1162/15353500200202109. [DOI] [PubMed] [Google Scholar]
- 36.Jackson PF, Cole DC, Slusher BS, Stetz SL, Ross LE, Donzanti BA, Trainor DA. Design, Synthesis, and Biological Activity of a Potent Inhibitor of the Neuropeptidase N-Acetylated R-Linked Acidic Dipeptidase. J. Med. Chem. 1996;39:619–622. doi: 10.1021/jm950801q. [DOI] [PubMed] [Google Scholar]
- 37.Marchal C, Redondo M, Padilla M, Caballero J, Rodrigo I, Garcia J, Quian J, Boswick DG. Expression of Prostate Specific Membrane Antigen (PSMA) in Prostatic Adeno-carcinoma and Prostatic Intraepithelial Neoplasia. Histol. Histopathol. 2004;19:715–718. doi: 10.14670/HH-19.715. [DOI] [PubMed] [Google Scholar]
- 38.Perner S, Hofer MD, Kim R, Shah RB, Li H, Moller P, Hautmann RE, Gschwend JE, Kuefer R, Rubin MA. Prostate-Specific Membrane Antigen Expression as a Predictor of Prostate Cancer Progression. Hum. Pathol. 2007;38:696–701. doi: 10.1016/j.humpath.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 39.Yao V, Parwani A, Maier C, Heston WD, Bacich DJ. Moderate Expression of Prostate-Specific Membrane Antigen, a Tissue Differentiation Antigen and Folate Hydro-lase, Facilitates Prostate Carcinogenesis. Cancer Res. 2008;68:9070–9077. doi: 10.1158/0008-5472.CAN-08-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans MJ, Smith-Jones PM, Wongvipat J, Navarro V, Kim S, Bander NH, Larson SM, Sawyers CL. Noninvasive Measurement of Androgen Receptor Signaling with a Positron-Emitting Radiopharmaceutical That Targets Prostate-Specific Membrane Antigen. Proc. Natl. Acad. Sci. U.S.A. 2011;108:9578–9582. doi: 10.1073/pnas.1106383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hillier SM, Maresca KP, Femia FJ, Marquis JC, Foss CA, Nguyen N, Zimmerman CN, Barrett JA, Eckelman WC, Pomper MG, et al. Preclinical Evaluation of Novel Glutamate-Urea-Lysine Analogues That Target Prostate-Specific Membrane Antigen as Molecular Imaging Pharmaceuticals for Prostate Cancer. Cancer Res. 2009;69:6932–6940. doi: 10.1158/0008-5472.CAN-09-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greish K. Enhanced Permeability and Retention of Macro-molecular Drugs in Solid Tumors: A Royal Gate for Targeted Anticancer Nanomedicines. J. Drug Target. 2007;15:457–464. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]
- 43.Wernicke AG, Edgar MA, Lavi E, Liu H, Salerno P, Bander NH, Gutin PH. Prostate-Specific Membrane Antigen as a Potential Novel Vascular Target for Treatment of Glioblastoma Multiforme. Arch. Pathol. Lab. Med. 2011;135:1486–1489. doi: 10.5858/arpa.2010-0740-OA. [DOI] [PubMed] [Google Scholar]
- 44.Kularatne SA, Wang K, Santhapuram HK, Low PS. Prostate-Specific Membrane Antigen Targeted Imaging and Therapy of Prostate Cancer Using a PSMA Inhibitor as a Homing Ligand. Mol. Pharmaceutics. 2009;6:780–789. doi: 10.1021/mp900069d. [DOI] [PubMed] [Google Scholar]
- 45.Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 46.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A Versatile Vector for Gene and Oligonucleotide Transfer into Cells in Culture and in Vivo: Polyethylenimine. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petersen H, Fechner PM, Martin AL, Kunath K, Stolnik S, Roberts CJ, Fischer D, Davies MC, Kissel T. Polyethylenimine-Graft-Poly(ethylene glycol) Copolymers: In-fluence of Copolymer Block Structure on DNA Complexation and Biological Activities as Gene Delivery System. Bioconjugate Chem. 2002;13:845–854. doi: 10.1021/bc025529v. [DOI] [PubMed] [Google Scholar]
- 48.Chollet P, Favrot MC, Hurbin A, Coll JL. Side-Effects of a Systemic Injection of Linear Polyethylenimine-DNA Complexes. J. Gene. Med. 2002;4:84–91. doi: 10.1002/jgm.237. [DOI] [PubMed] [Google Scholar]
- 49.Glunde K, Raman V, Mori N, Bhujwalla ZM. RNA Interference-Mediated Choline Kinase Suppression in Breast Cancer Cells Induces Differentiation and Reduces Proliferation. Cancer Res. 2005;65:11034–11043. doi: 10.1158/0008-5472.CAN-05-1807. [DOI] [PubMed] [Google Scholar]
- 50.Krishnamachary B, Glunde K, Wildes F, Mori N, Takagi T, Raman V, Bhujwalla ZM. Noninvasive Detection of Lentiviral-Mediated Choline Kinase Targeting in a Human Breast Cancer Xenograft. Cancer Res. 2009;69:3464–3471. doi: 10.1158/0008-5472.CAN-08-4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu EX, Tang H, Jensen JH. Applications of Ultrasmall Superparamagnetic Iron Oxide Contrast Agents in the MR Study of Animal Models. NMR Biomed. 2004;17:478–483. doi: 10.1002/nbm.923. [DOI] [PubMed] [Google Scholar]