

Abstract

Barbital is a hypnotic agent that has been intensely studied for many decades. The aim of this work was to establish a clear and comprehensible picture of its polymorphic system. Four of the six known solid forms of barbital (denoted I0, III, IV, and V) were characterized by various analytical techniques, and the thermodynamic relationships between the polymorph phases were established. The obtained data permitted the construction of the first semischematic energy/temperature diagram for the barbital system. The modifications I0, III, and V are enantiotropically related to one another. Polymorph IV is enantiotropically related to V and monotropically related to the other two forms. The transition points for the pairs I0/III, I0/V, and III/IV lie below 20 °C, and the transition point for IV/V is above 20 °C. At room temperature, the order of thermodynamic stability is I0 > III > V > IV. The metastable modification III is present in commercial samples and has a high kinetic stability. The solid-state NMR spectra provide information on aspects of crystallography (viz., the asymmetric units and the nature of hydrogen bonding). The known correlation between specific N–H···O=C hydrogen bonding motifs of barbiturates and certain IR characteristics was used to predict the H-bonded pattern of polymorph IV.
Keywords: barbital, thermal analysis, crystal polymorphism, IR spectroscopy, Raman spectroscopy, solid-state NMR spectroscopy, X-ray powder diffraction, thermodynamics, phase transition
Introduction
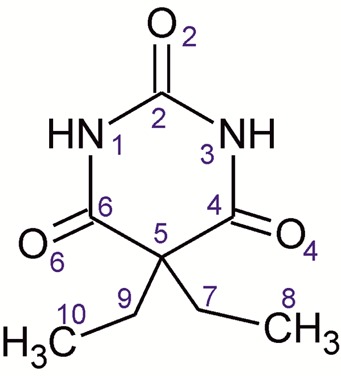
Barbital (Btl; 5,5-diethyl-2,4,6(1H,3H,5H)-pyrimidinetrione or 5,5-diethylbarbituric acid; Scheme 1)1 was the first commercially available barbiturate, which was applied as a sedative and hypnotic drug during the first half of the 20th century. This compound was marketed as tablets under the brand name Veronal (Bayer) and was also used in combination with other drug compounds. Because of its disadvantages2 (tolerance development, addiction potential, and lowering of blood pressure, heart rate, and respiration) and its high potential for abuse and fatalities, it has been replaced by alternative medications, in particular benzodiazepines. Btl is only rarely applied in human drug therapy today but is still listed in pharmacopoeias (e.g., in the European and in the Japanese Pharmacopoeias). It is used as a buffer substance and as a model or reference compound in various research disciplines in medicine, pharmacy, and chemistry. The global annual manufacture of barbital of approximately 100 tons accounts for 4% of the total production of barbiturates (2009), and within this group it is exceeded only by phenobarbital (77%), butalbital, and pentobarbital.3
Scheme 1. Molecular Structure of Btl and Atom Numbering Scheme.
Previous reports on the crystal polymorphism of Btl are listed in Table 1, including the methods employed and the forms examined. In this study, a nomenclature system will be applied where Roman numerals indicate the order of decreasing melting points (Kofler notation). The thermodynamically most stable polymorph at 25 °C is additionally indicated by a superscript zero, i.e., forms I0 to VI. The correlation between this and alternative nomenclature systems used by other authors is given in Table S1 (Supporting Information).
Table 1. Studies on Solid Forms of Btl.
| author(s) | form(s)a | method(s)b | ref |
|---|---|---|---|
| Fischer and Kofler (1932) | I0, III, IV | optical crystallography; HSM, sublimation studies | (4) |
| Lindpaintner (1939) | I0, III, IV, V | HSM, sublimation studies, melt film preparation | (5) |
| Brandstätter (1942) | I0, III, IV | HSM, isomorphism and polymorphism, | (16) |
| Kofler (1947, 1954) | I0, III, IV, V | crystallization (solvents, melt, sublimation), enantiotropic relationship of IV/V in binary system studies | (17) |
| Huang (1951) | I0, III, IV, V | PXRD of impure forms; PXRD of a contact preparation | (6a) |
| Huang (1953) | I0, III, IV, V | identification of solid solutions of aprobarbital (allypropymal) III and Btl-III (“diemal”) | (6b) |
| Cleverley and Williams (1959) | I0, III, Vc | PXRD; IR | (7) |
| Brandstätter-Kuhnert and Aepkers (1962, 1963) | I0, II, III, IV, V, VI | HSM on binary barbiturate systems | (8) |
| Nogami (1969) | I0, III, V | dissolution studies | (12a) |
| Craven et al. (1969, 1971) | I0, III, V | SXRD | (11) |
| Kuhnert-Brandstätter et al. (1971) | I0, III, IV, V | IR | (10) |
| Willis et al. (1972) | III | Raman spectroscopy | (18) |
| Kaliszsan et al. (1975) | I0, III, IVc, Vc | IR; poor quality spectra, interpretation very difficult | (19) |
| Sekiguchi et al. (1975) | I0, III, V | dissolution tests | (12b) |
| Grabowska et al. (1976) | I0, III, IVc, Vc | crystallization studies, thermal analysis, PXRD, interpretation on the basis of the data provided very difficult | (13) |
| McMullan et al. (1978) | III | SXRD from neutron data | (20) |
| Hollenbach et al. (1979–1982) | I0, III | formulation properties (compaction, dissolution etc.) | (12c−12h) |
| Burger and Ramberger (1979) | I0, II, III, IV, V, VI | thermochemical data, thermodynamic relationships between forms | (14) |
| Caillet and Claverie (1980) | I0, III, V | discussion of the structural differences | (21) |
| Craven et al. (1982) | III | charge density study | (22) |
| Chauvet et al. (1986) | I0, III | DTA, TGA | (23) |
| Fournival et al. (1987) | I0, III, V | DTA, TGA | (24) |
With reference to the nomenclature system used in this study.
Abbreviations: HSM, hot-stage microscopy studies; DTA, differential thermal analysis; TGA, thermogravimetric analysis; PXRD, powder X-ray powder diffraction; IR, infrared spectroscopy.
Assignment is doubtful.
Polymorphs I0, III, and IV were first reported by Fischer and Kofler in 1932,4 who reported their production, morphologies, thermal behavior, and melting points. In 1939, Lindpaintner found a new metastable form (V) by sublimation.5 The PXRD patterns of the polymorphs I0, III, IV, and V were reported by Huang.6 Cleverley and Williams recorded the infrared spectra of three Btl polymorphs.7 Thermoanalytical studies of binary mixtures of Btl with other 5,5-disubstituted barbiturates led Brandstätter-Kuhnert and Aepkers to the discovery of two other forms (II and VI),8 which crystallized only in the presence of an isomorphic additive (similar characteristics are known from five metastable forms of phenobarbital9). Modification II (mp 183.5 °C) forms an isomorphic series with dormin II (5-allyl-5-ethyl barbituric acid) and form II of aprobarbital (numal, 5-allyl-5-isopropylbarbituric acid). Polymorph VI (159 °C) is isomorphous with modifications I of thiothyr (5,5-diethyl-2-thiobarbituric acid) and I of inactin (thiobutabarbital, 5-ethyl-5-(1-methyl-propyl)-2-thiobarbituric acid). Having discovered these two forms and having determined their melting points, these authors decided to update the nomenclature of the known Btl forms following the Kofler notation and renamed some previously described phases.8
In 1971, Brandstätter-Kuhnert and Bachleitner-Hofmann10 reported the IR spectra of forms I0, III, IV, and V. The crystal structures of I0, III, and V were determined by Craven et al.11 Other studies by various groups12 dealing with the dissolution behavior, compaction properties, processing, etc. are not very consistent with regard to the information given on the stability and behavior of individual forms. Moreover, in many cases it is difficult for the reader to clearly identify what form was actually present, and the application of different nomenclature systems complicates this situation even more. The existence of Btl solvates is not explicitly mentioned in any of the reports, but it appears that a dimethylformamide solvate was present during the study by Grabowska and Kaliszan.13 A full set of thermochemical data for the forms I0, III, IV, and V was given by Burger and Ramberger.14 The stability of the polymorphs and their thermodynamic relationships were evaluated on this basis, but details on how the data have been obtained were not reported.
Our interest in reinvestigating this old and classic compound stems from teaching courses on hot-stage microscopy in which Btl is a model compound for the demonstration of solid–solid phase transitions and the concomitant sublimation of different polymorphs. This investigation is part of a wider study of barbiturates as a model class for structure–property relationships, packing features, and isomorphism.9,15 The main aim of the study was to establish a clear picture of the thermodynamic relationship between the Btl forms and to largely eliminate obscurities and inconsistencies from previous reports. All previously described polymorphs of Btl were reproduced, and new solvates were discovered in a selective solvent screening program. In this contribution, we provide new analytical data, including Raman and solid-state NMR and PXRD data for the polymorphs I0, III, IV, and V, which can be produced without isomorphic seeding. Relationships between these forms and their thermodynamic stability are interpreted with the help of a semischematic energy/temperature diagram based on experimental thermochemical data. The metastable forms II and VI, which can be produced by isomorphic seeding, and the new solvates will be described elsewhere.
Experimental Section
Materials
Barbital was purchased from Merck (Darmstadt, D, Art. 500535; purity 99.0%). All solvents used for crystallization experiments and solvent mediated transformation studies were of analytical grade.
Instruments and Procedures
Thermomicroscopy
Experiments were performed with an Olympus BH2 polarization microscope (Olympus Optical, J) equipped with a Kofler hot stage (Reichert, Vienna, A) and digital camera (Olympus ColorView IIIu).
Melt Film Preparations and Sublimation Experiments
A Kofler hot bench (Reichert, Vienna, Austria) was employed for the preparation of quench cooled melt films. After fusing a few milligrams of Btl between a glass slide and coverslip, the preparations were quickly cooled on the surface of a metal block. Sublimation experiments were performed between two glass slides separated by a spacer ring of 5–30 mm height. A series of preparations were placed side by side onto the surface of the hot bench (temperature range from 60 to 150 °C), and the deposited sublimates on the top glass slide were further investigated with optical- or IR microscopy.
Crystallization from Solvents
A selection of 20 solvents was used for crystallization experiments, which were performed under different conditions, mainly solvent evaporation at about 20 °C, as well as a limited number of fast and slow cooling crystallization experiments from hot saturated solutions (see Table S8 in the Supporting Information). After filtration, the crystals were air-dried and analyzed with PXRD and/or IR spectroscopy and/or DSC. The unstable solvates were tested (PXRD) without prior drying.
Lyophilization
A Lyolab B LSL secfroid lyophilizer (Aclens-Lausanne, CH) equipped with a mechanical fine vacuum pump 2400 A (Alcatel, Annecy, F) was used for freeze-drying experimets.
Solvent Mediated Transformation (SMT)
In a glass cylinder, the solid was suspended in water. The suspension was kept at 20 °C (thermostatted water bath) and stirred (magnetic stirrer, 900 rpm). A few milliliters of the suspension was removed at regular intervals, and the microcrystalline solid residue obtained after filtration was analyzed with PXRD and IR or Raman spectroscopy.
Differential Scanning Calorimetry (DSC)
Experiments were performed with a DSC 7 instrument (Perkin-Elmer, Norwalk, Ct., USA) using the Pyris 2.0 software. Between 1 and 3 mg of sample (weighed with a UM3 ultramicrobalance, Mettler, Greifensee, Switzerland) was placed in a 25 μL Al pan (purge gas: dry nitrogen; 20 mL min–1), which was then sealed (Btl has a high propensity for sublimation). The instrument was calibrated for temperature with pure benzophenone (mp 48.0 °C) and caffeine (mp 236.2 °C). The energy calibration was performed with pure indium (mp 156.6 °C; heat of fusion 28.45 J g–1). Errors quoted for temperature and enthalpy data are based on multiple (4 to 20) measurements and correspond with 95% confidence intervals.
Fourier Transform–Infrared (FT-IR) Spectroscopy
Crystal samples were prepared on ZnSe discs. In the case of the unstable form IV, a melt film was prepared between two ZnSe disks. FTIR spectra were recorded using a Bruker IFS 25 spectrometer and a Bruker IR microscope I (Bruker, Karlsruhe, D) with 15× Cassegrain objectives (spectral range 4000 to 600 cm–1, resolution 4 cm–1, and 100 scans).
Fourier Transform–Raman (FT-Raman) Spectroscopy
Spectra were recorded with a Bruker RFS 100 Raman spectrometer (Bruker, Ettlingen, D) equipped with a Nd:YAG Laser (1064 nm) as excitation source and a liquid-nitrogen-cooled high-sensitivity Ge detector (laser power 200 mW, 64 scans per spectrum, and resolution of 4 cm–1). Samples were placed on Al sample holders, except for that of the unstable form IV which was placed in a capillary tube.
Powder X-ray Diffraction (PXRD)
Diffraction patterns were recorded with a Siemens D-5000 diffractometer (Siemens, Karlsruhe, D; theta/theta goniometer, CuKα radiation, 1.5406 Å) with a 0.15° soller slit collimator, parallel beam optic (Goebel mirror; Bruker, Karlsruhe, D), and scintillation counter (tube voltage/current, 40 kV/35 mA; scan rate, 0.005° 2θ/s; 2θ range, 2° to 40°).
Solid-State NMR Studies
Experiments on forms I0, III, and V were performed at the University of Durham. Carbon-13 and nitrogen-15 cross-polarization magic-angle spinning (CPMAS) spectra were obtained at 75.40 and 30.39 MHz, respectively, using a Varian Unity Inova spectrometer based on a 7.05 T Oxford Instruments superconducting magnet. The probe accepts 7.5 mm (outside diameter) zirconia rotors, which were fitted with Kel-F end-caps. A flip-back procedure was implemented. Proton decoupling at a power equivalent to a frequency of ca. 60 kHz was employed during acquisition. The MAS rates were 5 kHz. Contact times were 10 ms. The numbers of transients were generally between 200 and 800 for 13C and between 10 000 and 40 000 for 15N. Recycle delays were 2 or 5 s. The 13C spectra were referenced via the high-frequency signal for a replacement sample of adamantane (δC = 38.4 ppm) and are reported relative to the resonance of neat TMS. The 15N spectra were analogously referenced to the resonance of neat nitromethane via the nitrate signal of ammonium nitrate (δN = −5.1 ppm).
Results and Discussion
Preparation of the Crystal Forms
The production and transformation pathways based on thermal methods are shown in Figure 1, and essential data of the polymorphs are summarized in Table 2. A summary of the crystallization methods from solvents and typical morphologies encountered is given in Figures S1 and S2 (Supporting Information).
Figure 1.

Phase transformation pathways of Bbtl polymorphs.
Table 2. Physicochemical, Spectroscopic, and Crystallographic Data for Polymorphs of Btla.
| property\modification | I0 | III | IV | V |
|---|---|---|---|---|
| Tfus [°C] | ||||
| HSM (refs (6 and 28)) | 190 | 183 | 181 | 176 |
| HSM (ref (5)) | 190 | 183 | 181 | 178 |
| DSC (onset)b | 189.9 ± 0.2 | 183.3 ± 0.3 | 181.2 ± 0.7 | 175.6 ± 0.2 |
| ΔfusH [kJ mol–1]b | 25.4 ± 0.1 | 26.8c | 24.8c | 27.3c |
| from ref (14) | 25.5 ± 0.5 | 27.05c | 24.95c | 27.25c |
| transformation | → I0 | → III | → III | |
| Ttrs (exp.) [°C]b | 140–160 | 100–120 | 110–130 | |
| ΔtrsH [kJ mol–1]b | +1.4 ± 0.1 | –2.0 ± 0.1 | +0.5 ± 0.2 | |
| from ref 14 | +1.55 ± 0.13 | –2.1 ± 0.2 | +0.25 ± 0.05 | |
| order of thermodynamic stability at 25 °C | 1 | 2 | 4 | 3 |
| density [g cm–3] at 22 °C | ||||
| dx (ref (13)) | 1.285 | 1.237 | – | 1.297 |
| dexp (ref (26)) | 1.284 | 1.242 | – | 1.303 |
| selected IR bands [cm–1] | ||||
| νN–H | 3241, 3111 | 3208, 3167, 3080 | 3213, 3094 | 3256 |
| νC=O | 1749, 1711, 1700 | 1765, 1719, 1680 | 1740, 1719, 1691 | 1770, 1723, 1677 |
| νN–H out-of-plane | 848 | 873 | 813 | 835 |
| selected Raman bands [cm–1] | ||||
| νC–H | 2989, 2961, 2941, 2881, 2745 | 2985, 2955, 2943, 2925, 2869, 2739 | 2988, 2966, 2941, 2884, 2761 | 2973, 2942, 2881, 2755 |
| νC=O | 1735, 1714, 1697 | 1763, 1734, 1702 | 1731, 1687 | 2756,1737, 1665 |
| δCH3 | 1462 | 1457, 1473 | 1459 | 1464 |
| δCH2 | 1448 | 1449 | 1445 | 1445 |
| δO=C–N | 630 | 626 | 623 | 627 |
| crystallographic data | ||||
| crystal system | trigonal | monoclinic | monoclinic | |
| space group | R3̅ | C2/c | P21 | |
| Z/Z′ | 18/1 | 4/0.5 | 8/4 | |
| N–H···O=C type (ref (15e)) | C-4 | C-2 | C-1d | L-1 |
| CSD refcode | DETBAA01 (ref (11a)) | DETBAA02 (ref (11a)) | DETBAA03 (ref (11b)) |
Abbreviations: Tfus = melting point; Ttrs = transition temperature; ΔfusH = enthalpy of fusion; ΔtrsH = transition enthalpy; dx = experimental density from single crystal data; dexp = experimental density from He pycnometry; Z = number of molecules in the unit cell; Z′ = number of molecules in asymmetric unit. Quoted errors correspond to a 95% confidence interval.
This work.
Calculated as (ΔfusH + ΔtrsH).
Inferred from characteristics of the IR spectrum.
Form I0
This polymorph was prepared by the following methods: (a) annealing the commercial form III at 160 °C, (b) sublimation at 180 °C, (c) slow-cooling of the melt, and (d) solvent mediating the transformation of either form III or V (or a mixture of these forms) in water at 25 °C. The transformations V→I° and III→I° were complete after 15 h and 3 days, respectively. Largely phase pure form I0 was additionally obtained by drying the acetonitrile and dimethylformamide solvates. Hexagonal needles or prisms of this form were obtained from diisopropylether, dioxane, n-hexane, n-heptane, and tetrahydrofurane by slow cooling the hot supersaturated solution. From other solvents such as water, acetone, or mixtures of these two solvents, form I0 also crystallized concomitantly with form III and form V. Moreover, Sekiguchi et al.12b obtained form I0 by freeze-drying a dioxane solution containing 10% barbital. Nogami et al.12a produced this modification by evaporating an alcohol solution of Btl to dryness and by applying a pressure of 300 to 500 MPa on crystals of polymorph III for 30 min.
Form III
Crystals of this polymorph can be recognized by their distinct prismatic morphology. Form III was most commonly encountered in solvent crystallization products but mostly along with form V and occasionally with form I0. Largely phase pure samples of form III were only obtained from dichloromethane, chloroform, and dimethylsulfoxide, through solvates that readily desolvate to form III on harvesting. Other solvents that yielded a higher fraction of form III were ethanol, diethylether, ethylacetate, and nitromethane. Crystals of III formed also in sublimation experiments above 100 °C.
Form IV
This polymorph can be produced on a microscopic scale by quench cooling the melt. In melt film preparations, it exists as spherulites displaying an internal broad beam structure and characteristic fissures. Fine needles of IV are obtained by sublimation (Figure 2). This modification is unstable at room temperature and transforms over time into more stable forms (primarily III). This process is accelerated by mechanical stress and exposure to air.
Figure 2.
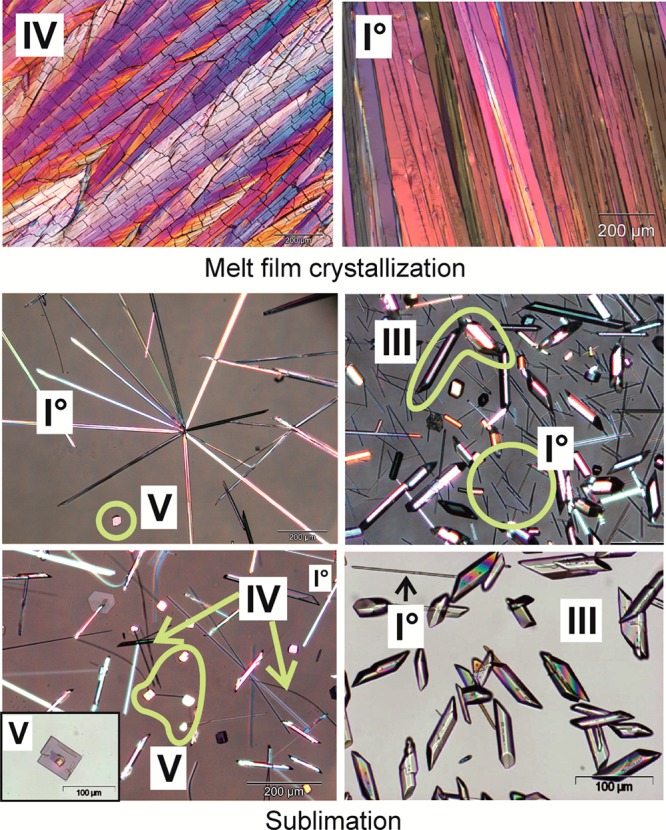
Polarized light micrographs of Btl crystals grown from the melt and by sublimation between 100 and 120 °C (I0, acicular; IV, fine curved needles; V, platelets; III, prismatic). A larger image can be found in the Supporting Information (S7).
Form V
The crystals of this metastable form are easily recognized by their rectangular, platy morphology. Phase-pure samples of form V were produced by lyophilization of an aqueous solution. In crystallization products obtained by rapid cooling of a hot saturated Btl solution in 1-propanol or acetone/water or by solvent evaporation of 1-butanol, methanol, pyridine, and ethyl acetate solutions, form V was identified as the dominant polymorph besides form III. Form V was also observed in sublimates, typically those produced below 100 °C.
Thermomicroscopy
Form I0 (mp 190 °C)
There was no solid–solid phase transition of the acicular crystals below their melting point. Sublimation and the subsequent growth of new needles (I0) on the coverslip were observed at 160 °C and melting of the sample at 190 °C.
Form III (mp 183 °C)
Sublimation started above 100 °C and was followed by the growth of prismatic sublimates (III) on the coverslip. The solid–solid transformation III→I0 ensued at approximately 150 °C in the main sample and 160 °C in the sublimates. Crystal edges became somewhat rounded during this process, but otherwise the external crystal habit remained unchanged.
Form IV (mp 181 °C)
A supercooled melt film of Btl crystallizes to either polymorph IV (after quench cooling) or polymorph I0 (after slow cooling). These two modifications are easily distinguishable from one another by their different appearance in polarized light (Figure 2) and different transformation behavior on heating. Both crystallize as aggregates with an internal broad beam structure, but only the crystals of form IV contained characteristic cross-cracks. Polymorph I0 does not transform on heating, but with IV as the initial phase, there are two solid–solid transformations (Figure 3). The transition IV→III (100–120 °C) started first at cracks and resulted in randomly oriented small crystals, which produce an irregular multicolor pattern under polarized light. The transformation III→I0 occurred at 140–150 °C and produced larger sections of uniform low-order interference colors (brown, gray, and black) in the sample.
Figure 3.

Polarized light microphotographs of melt film preparations illustrating the transformations of Btl polymorphs IV→III→I0.
Form V (mp 176 °C)
The platelets of this polymorph underwent a solid–solid transformation V→III on heating between 120 and 130 °C. This process was indicated by a change in interference colors, but the external habit of the crystals was preserved. At 150–160 °C, transformation III→I0 occurred, which was indicated by a change to brighter interference colors. An alternative preparation method for modification V from a melt film of polymorph IV was described by Kofler et al.25 After removal of the coverslip from the melt film preparation, some droplets of 96% ethanol were added, yielding pyramidal grains of form III and platelets of V. This experiment was reproduced by us and demonstrates clearly that at room temperature polymorph IV is less stable than V.
Differential Scanning Calorimetry
The DSC curves for forms I0, III, IV, and V are shown in Figure 4. The melting endotherm and enthalpy of fusion (Table 2) of polymorph I0 were determined directly at heating rates of 10 K min–1. It was not possible to record the pure fusion peaks and enthalpies of the forms III, IV, and V directly because each of them undergoes a solid–solid transformation to a more stable polymorph below its melting temperature. However, we determined the enthalpies of the polymorphic transitions III→I0, IV→III, and V→III (Table 2), and on this basis, it was possible to estimate heat of fusion values for all forms.
Figure 4.

DSC curves of Btl polymorphs (heating rate 10 K min–1). The insert shows an enlarged section of the heating curve of form V with two endothermic transitions.
The transition III→I0 at around 146 °C is endothermic, indicating that the two polymorphs are enantiotropically related to one another. The exothermic transition IV→III occurs at approximately 110 °C and is followed by the endothermic III→I0 transition at about 145 °C. Even though phase-pure polymorph IV can be readily prepared as a melt film on a glass slide, it was very difficult to do the same with a sample placed in a DSC aluminum pan by means of quench cooling. The aluminum surface appears to favor the nucleation of the more stable phases so that mixtures of forms I0, III, and IV (rarely V) were usually obtained and Figure S3 (Supporting Information) contains a neat example for a heating curve of a sample produced by quench cooling the melt in a DSC pan, showing the melting endotherm of all four forms. Therefore, more than 60 experiments were conducted to find the ideal conditions for producing the phase-pure form IV, in which we varied (a) the sample weight (1 to 10 mg), (b) the surface temperature (20, 4, and −20 °C) of the metal block to achieve different cooling rates of the melt, (c) the relaxation time of the quench cooled and recrystallized melt samples (0, 5, 10, 30, and 60 min), (d) the melting time at 200 °C (between 5 and 60 s), and (e) the DSC heating rate (5 to 50 K min–1). The main criterion for the amount of form IV was the enthalpy value obtained by integrating the exothermic transition peak (form IV to III) around 110 °C. The highest values for ΔtrsHIV→III were around −2.0 kJ mol–1, and lower transition enthalpies (−0.1 to −1.8 kJ mol–1) indicated the presence of form III and/or form I. Samples with the highest amount of form IV could finally be produced by melting a 1 mg sample within an aluminum pan (by heating on a Kofler hot bench for 20 s at 200 °C) and subsequent quench cooling on a metal block (−20 °C). The sample was then allowed to relax for 5 min before the DSC experiment was started. The endothermic transformation V→III occurred typically between 110 and 130 °C and was followed by the transition III→I0 between 145 and 160 °C (Figure 4). The presence of form III after the first transition was confirmed with IR microscopy.
The values measured by us for the heat of phase transitions agree very well with those reported by Burger and Ramberger14 (Table 2). This is somewhat surprising because convenient computer-assisted data evaluation methods were not available to these authors, whose work relied on an analog instrument, plotter, and planimeter.
Thermodynamic and Kinetic Stability
The thermodynamic relationships between III, IV, and V were derived from the melting points of the forms, the enthalpy of fusion of form I0, and the experimental transition enthalpies of polymorphs III, IV, and V (Table 2) by application of the Burger-Ramberger rules,26 and a semischematic energy/temperature (ET) diagram was constructed (Figure 5) based on these data. The heat of transition rule was crucial for the interpretation of this system because only the melting enthalpy of the highest melting form I0 can be measured accurately. DSC experiments showed two endothermic transitions on heating (III→I0 and V→III), which indicated that III/I0 and V/III are two pairs of enantiotropically related polymorphs. Therefore, the enthalpy curve of polymorph III runs below that of I0, and the curve of polymorph V below that of III. The transition IV→III is exothermic, which means that the two forms concerned are monotropically related to one another. The enthalpy of the transition IV→III (ΔtrsHIV→III = −2.0 kJ mol–1) is higher than that of III→I0 (ΔtrsHIII→I = 1.4 kJ mol–1). Accordingly, the enthalpy curve of polymorph IV runs above that of I0. It follows that the order of relative enthalpies is V < III < I0 < IV (Figure S4, Supporting Information). This is also the order of the Gibbs free energies of these polymorphs at 0 K (G = H) if we consider the third law of thermodynamics (S = 0 at T = 0 K) and the Gibbs energy function (G = H – TS). By connecting the corresponding points (enthalpies at 0 K) in the ET diagram with the melting points of the individual forms (indicated by open circles on the free energy curve of the melt), four intersections (transition points) of G curves are obtained. These intersections indicate an enantiotropic relationship for each of the pairs V/III, V/I0, III/I0 and V/IV. By contrast, the two G curves for each of the pairs IV/I0 and IV/III do not intersect, meaning that the corresponding relationships are monotropic. According to the DSC experiments, the phase transitions associated with the enantiotropic pairs III/I0 and V/III are close to 145 and 115 °C, respectively, which is in agreement with their order in the ET-diagram. However, solid–solid transitions are kinetically controlled and may therefore occur far above a true (thermodynamic) transition point.
Figure 5.
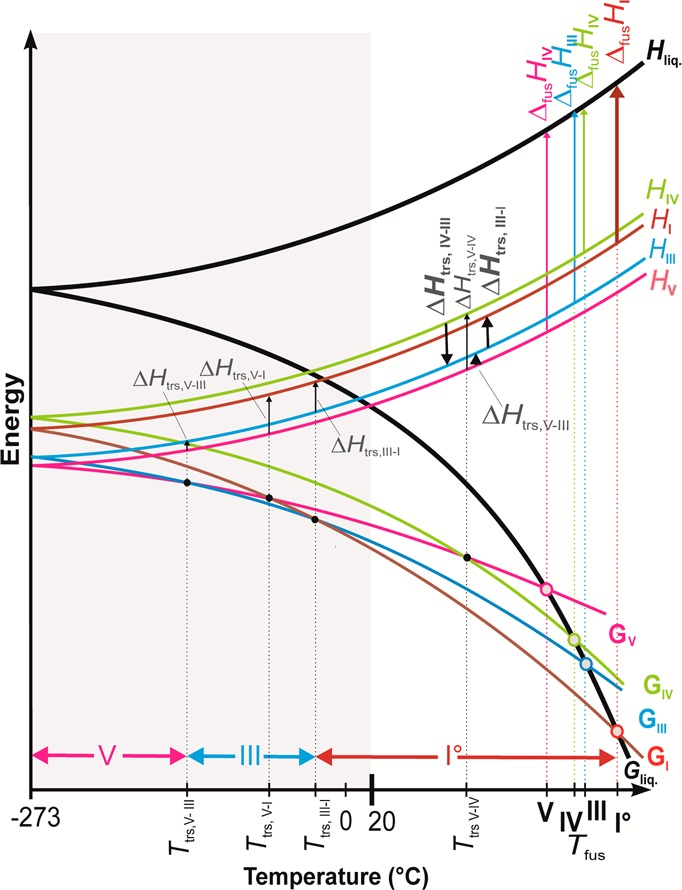
Energy/temperature diagram for the Btl forms I0, III, IV, and V (Tfus, melting point; G, Gibbs free energy; H, enthalpy; ΔfusH, enthalpy of fusion; Ttrs, transition point; ΔtrsH, transition enthalpy; liq, liquid phase). The bold vertical arrows indicate experimental enthalpies, and the horizontal two-headed arrows indicate ranges of thermodynamic stability of forms V, III, and I0.
Solution mediated transformation experiments were performed to overcome the kinetic barrier of these transformations and to verify whether the point of the III→I0 transition lies above or below 20 °C. These experiments showed that the transitions V→I0 and III→I0 both occur at approximately 20 °C. These results strongly indicate that I0 is the thermodynamically stable form between ambient temperature and its melting point, and they are also in agreement with the findings of Burger and Ramberger.14 However, a rather different order of stability at room temperature, V > III > I0, was postulated by Sekiguchi et al.12b on the basis of solubility studies. These authors determined 93 °C as the transition point of the pair I0/III. By contrast, our solution mediated transformation experiments showed that form I0 is indeed stable at 20 °C, and therefore, the transition point must be below this temperature. It should be noted that the findings obtained by Sekiguchi et al.12b are based on solubility data with “little statistical significance.”12b Polymorph III is thermodynamically unstable at 20 °C, but it has a high kinetic stability. This was shown by the PXRD analysis of a 20 year old sample (original label “mod III”), which still consisted of phase-pure form III. It is not unusual that a metastable polymorph is the predominant crystallization product from organic solvents because such processes often yield the kinetic rather than the thermodynamic form. The kinetic stability of modification V is clearly lower than that of form III. Accordingly, we found that a 20 year old sample (original label “mod V”) has undergone a complete transition V→III. The formation of modification III instead of the most stable polymorph I0 is a distinctive demonstration of Ostwald’s rule of stages and shows again that the activation energy barrier between III and I0 is high.
Our finding that polymorph I0 is the thermodynamically stable polymorph from the melting point to some temperature below 0 °C is in good agreement with earlier studies.14,17a At lower temperatures, there is a window in which III is the stable form (Figure 5), and form V is the stable polymorph below this temperature interval. This is in accordance with the density rule26 (the most densely packed form has the lowest free energy at absolute zero). This rule holds for polymorph V, but it would also indicate a reversal of the stability order of forms I0 and III at 0 K, which indicates an exception to the density rule. This exception is not entirely unexpected as hydrogen bonding dominates the intermolecular interactions in the Btl polymorphs. The density differences between the Btl forms are significant (V/I, 1.5%; V/III, 4.8%; I/III, 3.3%). Nogami et al.12a reported a transformation of III to the denser form I0 at elevated pressure, which is in line with expectations. However, this is a rare example of a pressure-induced polymorphic phase transition at conditions (300 to 500 MPa) that can be readily achieved with a conventional tablet press. Interestingly, Sekiguchi et al.12b did not note any phase transition when the forms I0, III, and V were compressed at 370, 440, and 490 MPa, respectively. However, these authors observed a transition, V→III, during mechanical grinding, thought to be induced by the heat of friction.
On the basis of thermomicroscopic investigations of eutectic mixtures of barbital polymorphs with methylacetanilide, dinitrosopiperazine, and dimethylpyrone,17a Kofler determined 135 °C as the point of the polymorphic transition V→IV. This is consistent with our ET diagram, which indicates a higher temperature for the V→IV transition than for the transitions V→III, V→I0, and III→I0.
Powder X-ray Diffraction
The forms I0, III, IV, and V are readily distinguishable from one another by their PXRD patterns (Figure 6), and the experimental patterns of I0, III, and V were consistent with the relevant literature data.6,7,11 A special preparation method was required for form IV, which transforms readily to III upon mechanical treatment. Representative diffractograms of form IV could be finally recorded from film preparations prepared on a coverslip, which then was mounted on a silicon holder. Only a rather poor quality diffraction pattern affected by preferred orientation was obtained with this design. Better diffractograms were obtained with melt preparations that were directly produced in a capillary. However, the quality of the obtained PXRD data for IV was not sufficient for the determination of unit cell parameters, but these data matched very well with reflections reported by Huang6a (“form III”; no indices; PDF 00-005-0111, ICDD 2005).
Figure 6.

PXRD patterns of Btl forms (experimental, solid lines; simulated on the basis of single-crystal data from ref (11), dotted lines).
Crystal Structures
The molecules of Btl and analogous barbiturates contain two NH and three C=O functional groups, which are potential bond donor and acceptor sites for hydrogen bonds, respectively. Accordingly, the aggregation of barbiturate molecules in the solid state is directed by intermolecular N–H···O=C interactions. Geometrical constraints arising from the rigidity of the pyrimidinetrione unit mean that only a few N–H···O=C-bonded topology types are frequently observed in this class, most notably four chain types which are denoted C-1 to C-4.15b,15e For the classification of these topologies, one has to distinguish the O2 acceptor function from the two equivalent O4/6 functions (Scheme 1). Known N–H···O=C-bonded structures of Btl polymorphs are the ladder C-4 (form I0), looped chain C-2 (form III), and layer L-1 (form V) (Figure 7). Moreover, certain characteristics in the IR spectrum of polymorph IV suggest the presence of the looped chain type C-1 (see below).
Figure 7.
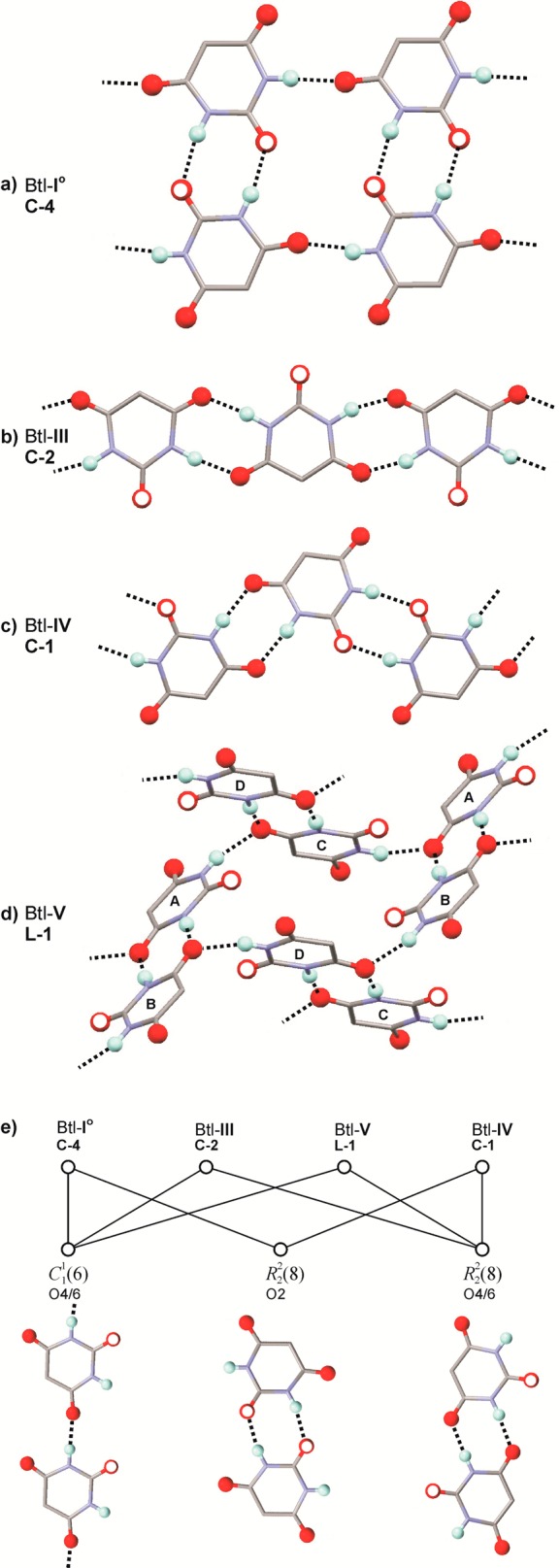
(a–d) N–H···O=C bonded structure types according to ref (15e) in polymorphs of Btl. H atoms engaged in hydrogen bonding and all O atoms are represented as balls, the O2 site is indicated by an additional white circle, and the alkyl chains are omitted for clarity. (e) The common chain and ring motifs of the four types.
The C-4 ladder in the crystal of I0 (space group R3̅; one independent molecule) displays inversion symmetry. Six of these N–H···O=C-bonded structures are arranged around a crystallographic 3̅ axis and three around a 31 axis (Figure S6, Supporting Information). O2 and one of the two topologically equivalent O4/6 carbonyl groups are utilized in hydrogen bonding (Table 3). The C-4 structure displays a sequence of fused R22(8) and R4(16) rings (Table S2, Supporting Information).27 Other examples of the C-4 type were found in polymorphs of barbituric acid,28 butobarbital (soneryl),15a cyclopentobarbital (cyclopal),9 and heptobarbital (rutonal).9
Table 3. Geometrical Parameters for the N–H···O=C Bridges in Polymorphs of Btl Shown in Figures 7a (I0), b (III), and d (V)a.
| form | acceptor molecule | acceptor type | donor molecule | N···O [Å] | H···O [Å] | ∠NHO [deg] |
|---|---|---|---|---|---|---|
| I0 | C2 | 2.888 | 2.02 | 170 | ||
| C4/6 (half) | 2.871 | 1.94 | 173 | |||
| III | C4/6 | 2.876 | 2.03 | 176 | ||
| V | A | C4/6 (half) | B | 2.869 | 1.93 | 173 |
| C | 2.904 | 2.07 | 158 | |||
| B | C4/6 (half) | A | 2.913 | 2.08 | 169 | |
| D | 2.905 | 2.03 | 162 | |||
| C | C4/6 (half) | D | 2.905 | 2.07 | 169 | |
| A | 2.923 | 2.05 | 168 | |||
| D | C4/6 (half) | C | 2.852 | 1.82 | 166 | |
| B | 2.916 | 2.17 | 155 |
The Btl molecule of form III (space group C2/c) lies on a 2-fold axis. Both O4/6 sites per molecule are employed in one N–H···O=C bond, whereas the O2 acceptor function is not used. The molecules are linked via centrosymmetric R22(8) rings into a looped chain of the C-2 type. This type is also present in the acetonitrile solvate of Btl29 and in forms of 5-fluoro-5-phenylbarbituric acid,30 6-oxocyclobarbital,31 and phenobarbital (form III).32 The looped chain topology C-1 is similar to the C-2 type and can be described as a sequence of two kinds of alternating R2(8) rings, with the first linking two molecules via two N–H···O=C4/6 interactions and the second via two N–H···O=C2 bonds.
The space group of polymorph V is P21, and its asymmetric unit contains four molecules (denoted A–D; Figure S6c, Supporting Information). In each molecule, one O4/6 carbonyl group accepts two H-bonds, while O2 and the second O4/6 site are not engaged in hydrogen bonding. The resulting N–H···O=C bonded layer structure L-1 is built up from two independent types of interlinked dimeric building blocks with a central R22(8) ring (A + B and C + D). N–H···O=C-bonded layers of the L-1 type were also found in crystal forms of vinbarbital,33 heptabarbital (medomin),9 and nembutal (form IV).15g The L-1 net is uninodal, e.g., all four independent molecules of form V have the same connectivity. This is illustrated by the comparison to a corresponding Z′ = 1 structure with L-1 topology (Tables S5 and S6, Supporting Information).
The common building blocks of the C-4, C-2, L-1, and C-1 structures were indentified, and their graph set representations27 are given in Tables S2–S6 (Supporting Information). The following formal relationships exist between polymorphs of Btl: (a) the C22(6) chain with O4/6 as H-bond acceptor are present in I0, III, and V; (b) the R2(8) ring with O2 as H-bond acceptor in I0, IV, and V; and (c) the counterpart R22(8) unit with O4/6 in polymorphs III and IV (Figure 7e.) One could speculate whether these units are maintained in a polymorphic phase transition. The hydrogen bond capabilities of Btl have inspired different groups to design cocrystals with other components bearing complementary functional groups.34
FTIR and Raman Spectroscopy
The distinct characteristics of the FT-IR spectra of forms I0, III, IV, and V (Figure 8) make it easy to identify the Btl polymorph in hand. Spectroscopic differences, especially in the N–H stretching vibrations region at around 3200 cm–1 and in the carbonyl bands at around 1700 cm–1 (Table 2) arise from different N–H...O=C interactions. We have recently investigated the general correlation between the IR characteristics of barbiturates and their N–H...O=C bond topologies.35 Our experiments on Btl forms confirmed these earlier assertions about specific IR characteristics associated with the types C-4, C-2, and L-1.
Figure 8.

FT-IR spectra of the Btl forms I0, III, IV, and V.
The IR spectrum of polymorph I0 displays one very strong and sharp N–H vibration at higher wavenumber and a weak N–H vibration at lower wavenumber. A doublet with shoulder found at higher wavenumber is due to the C=O groups. These characteristics are typical for the presence of the C-4 ladder structure and were previously found in IR spectra of heptobarbital (rutonal, forms I and II), phenobarbital (form XI), and cyclopentobarbital (cyclopal, form I).9
The spectrum of form III exhibits three significant N–H vibrations in addition to two strong and one weak C=O vibration, which are similar to the C-2 characteristics in the IR spectra of polymorphs III of phenobarbital15e and III of nembutal.15g In general, the N–H valence vibrations of modification III occur at lower wavenumbers than those of I0, which is commonly interpreted as an indication of stronger hydrogen bonds.14,36 This result is consistent with the solid-state NMR data (see below).
The form IV shows two N–H absorption bands of approximately equal intensity in the range of 3500–3000 cm–1 and two main C=O vibrations between 1800 and 1600 cm–1. Such characteristics are typically associated with the presence of the looped chain C-1. The early work of Brandstätter-Kuhnert and Aepkers contains more evidence linking the C-1 structure to form IV.8b According to these authors, IV is isomorphous with probarbital (ipral, form I) as well as with butobarbital (soneryl, form I), and both were subsequently shown to contain the N–H···O=C bonded C-1 structure by single crystal structure analysis.9,37 In the course of an ongoing, comprehensive study of more than 50 5,5-disubstituted barbiturates, we have recorded the IR spectra of 85 solid forms and found that no less than 45% of these show characteristics that indicate the presence of the C-1 type.35
Polymorph V produces one strong N–H absorption band at ∼2900 cm–1 and three weak absorption bands at lower wavenumbers. The C=O groups produce three distinct absorption bands. These features are similar to IR characteristics9 of polymorphs of heptabarbital (medomin, form I), pentobarbital (nembutal, form IV),15g and vinbarbital (form I),33 which all contain the N–H···O=C bonded L-1 structure.
The FT-Raman spectra of the Btl forms show clear differences over almost the entire spectral range (Figure 9), particularly in the −CH2–CH3 stretching vibrations (at around 3000 cm–1), the −CH2–CH3 deformation bands close to 1465 cm–1, and the C=O vibrations at around 1750 cm–1 (Table 2). The most intense band between 623 and 630 cm–1 arises from the symmetric “breathing” of the pyrimidine ring18 and is suitable for a quick identification of barbiturates based on Raman spectroscopy data. Differences in the low frequency bands between 50 and 200 cm–1 clearly indicate the presence of fundamentally different crystal structures.
Figure 9.

FT-Raman spectra of forms I0, III, IV, and V.
Solid-State NMR Spectroscopy
Figure 10 shows the 75.40 MHz 13C CPMAS spectra of three Btl forms (I0, III, and V), and Table 4 lists the chemical shifts. Unfortunately, the low stability of polymorph IV means it is not feasible to produce a sufficiently large amount of sample for the recording of a 13C CPMAS spectrum. The five chemically distinct carbon atoms and the corresponding signals are marked with numbers as in Scheme 1. Assignment of the signals to the chemically different carbons is easy because of the very different chemical shifts. The small splittings seen for the signals from carbons 2 and 4/6 arise from second-order effects caused by coupling to the quadrupolar 14N nuclei.38 The spectra increase in complexity in the order III < I0 < V, indicating clearly the differing contents of the asymmetric units of the three forms. When the second-order effects mentioned above are ignored, form III gives a single signal for each chemically distinct carbon. Thus, if accidental near-equivalence of resonances is ruled out, the asymmetric unit must be half a molecule, i.e., symmetry relates the two halves of the ring and also the two ethyl groups. In contrast, the spectrum of form I0 shows two signals for C4/6, C7/9, and C8/10, from which it can be deduced that the asymmetric unit is one molecule. The possibility that it is two half-molecules (i.e., halves of two different molecules, each of which has 2-fold symmetry) can be ruled out by the fact that the only single signals are seen for C2 and C5. The spectrum of polymorph V is significantly more complicated. There are many resonances from C7/9 and C8/10, though the other carbons give rise to fewer signals (presumably from accidental near-overlaps). Deconvolution for the C7/9 and C8/10 bands (see Figure S5, Supporting Information) shows that the best fitting comes from the partial overlap of 8 signals each, though it is difficult to obtain unambiguous chemical shifts. It can be concluded that the asymmetric unit of form V consists of 4 molecules. There is clearly much overlapping in the signals for carbons 2, 4, 5, and 6 (though two peaks can be distinguished for C5), indicating that environmental differences are minor in nature. All of the conclusions about asymmetric units are in accord with the crystal structures but can be made independently from NMR.
Figure 10.

Carbon-13 CPMAS spectra at 75.40 MHz of the Btl forms I0, III, and V. The atomic numbering giving the assignments is as in Scheme 1. The samples of III and V used to obtain these spectra were slightly contaminated by the presence of polymorphs I0 and III, respectively. The spectra shown have been “cleaned” by a subtraction procedure.
Table 4. Carbon-13 and Nitrogen-15 Chemical Shifts for Forms I, III, and V.
| form | C4/6a | C2b | C5 | C7/9 | C8/10 | N |
|---|---|---|---|---|---|---|
| III | 178.4/177.7 | 148.0 | 58.6 | 33.3 | 9.9 | –226.7 |
| I0 | 176.5/175.7 | 152.7 | 58.8 | 34.8 | 9.6 | –226.5 |
| 173.4/172.7 | 31.8 | 9.0 | –228.5 | |||
| V | 180.3/179.7 | 149.0 | 58.4 | 34.4–32.0c | 10.3–9.0d | –223.5e |
| 172.1/171.4 | 57.9 | –227.1e |
The doublet splittings for I0 and III are caused by second-order effects of coupling to one 14N nucleus. This also affects the splittings of V. The true chemical shifts (in ppm) lie closer to the more intense (low-frequency) components.
The apparent triplet splitting arises from the second-order effects of coupling to two 14N nuclei.
In principle, there are 8 signals. Four maxima of differing intensities can be observed.
In principle, there are 8 signals. Three maxima of differing intensities, together with a shoulder can be observed. Deconvolution results are consistent with the existence of 8 signals (see Figure S5 in the Supporting Information).
The signals are in an approximately 3:5 intensity ratio. In principle, there should be 8 resonances.
The chemical shifts for C2 and C4/6 give immediate information about hydrogen bonding since it causes high-frequency shifts. It is evident that there is hydrogen bonding at C2 for Btl-I0, but not for form III or V. It can also be deduced that polymorph III is hydrogen bonded at C4/6, whereas only half of the C4/6 carbons are hydrogen bonded for modifications I0 and V. It seems that this hydrogen bonding is particularly strong in the case of form V. These conclusions are also borne out by the crystal structures (see Table 3). In particular, half of the C4/6 carbons which are hydrogen bonded for Btl-V are involved in two such bonds each, thus causing the substantial high-frequency shift. Moreover, in this case the range of hydrogen bond lengths (Table 3) is relatively small (the 8 distances between heavy atoms are between 2.852 and 2.923 Å), so the lack of resolution is not surprising.
The 15N spectra are displayed in Figure 11, and the shifts are listed in Table 4. As expected, III gives rise to a single resonance, whereas two signals are seen for form I0 (representing nitrogens hydrogen-bonded to C2 and C4/6). Somewhat unexpectedly, polymorph V also produces only two lines, which is hard to explain in detail. The signals for all the forms fall within a relatively small chemical shift range (ca. 5 ppm), which is in accord with the fact that all of the nitrogens are directly involved in hydrogen bonding and that the range of distances between the participating nitrogen and oxygen atoms (Table 3) is only 0.07 Å.
Figure 11.

Nitrogen-15 CPMAS spectra at 30.39 MHz for (left to right) barbital forms III, I0, and V.
The three Btl polymorphs provide an excellent example of NMR crystallography,39 e.g., of the way in which NMR signals give information about intermolecular hydrogen bonds and unit-cell symmetry.
Conclusions
The polymorphism of barbital involves many interesting phenomena, and this system highlights the problems in the generation of reliable and consistent data for solid-state forms exhibiting complex thermodynamic relationships. Even though the best accessible polymorphs (I0, III, and V) have been studied extensively in the past, the available literature data did not permit one to establish a clear and consistent picture of their thermodynamic and kinetic stabilities. The energy–temperature diagram derived in this study now allows the clear identification of the relative stability of each form. A remarkable characteristic of this system is that the lowest melting polymorph V (i.e., the least stable at Tfus) exhibits the highest density and becomes the most stable form at absolute zero. Form V is therefore the polymorph with the lowest entropy and is enantiotropically related to all other forms. Modification I0 was identified as the thermodynamically stable form at room temperature and above, and it exhibits the highest entropy. Polymorph III can be regarded as the “kinetic form,” which is primarily obtained by solvent crystallization. Therefore and because of its high kinetic stability, this polymorph is present in most commercial products. As the energetic differences between I0 and III are small, these two forms show similar solubility in water so that it may be irrelevant which of them is used for the preparation of drug products. For industrial purposes (processing), their differences in other parameters such as density and morphology may be more relevant, and their different solid-state spectra are definitely relevant for identification assays employing spectroscopic techniques. This concerns, for example, the common infrared spectroscopy test in pharmacopoeias. Interestingly, barbital is one of only a few compounds where the European Pharmacopoeia specifies the determination of a mixed melting point as an identity test. Other highlights of this polymorphic system are (a) the exception to the density rule regarding forms I0 and III, (b) the observation12a of a pressure induced transformation III→I0, and (c) the formation of concomitant polymorphs40 by sublimation and in solvent crystallization processes.
The high thermal stability of Btl enables crystallization approaches such as melt crystallization and sublimation, which are rarely applied in polymorph screening41 programs in contrast to solvent based crystallization techniques. As has been demonstrated in many past and recent studies, these methods are very successful in generating new metastable polymorphs,15b,15g producing single crystals for crystal structure analysis42,43 or suitable seeds for the subsequent crystallization of a specific polymorph from solvents.44 Moreover, microscopic melt crystallization techniques can be successfully employed in cocrystal screens45,46 and allow the quick evaluation of the characteristics of a binary system, i.e., to ascertain whether two compounds form only a simple eutectic, a solid solution or a molecular compound (which is usually meant by the term cocrystal). Many pharmaceutically relevant coformers melt without or with only weak thermal decomposition, and the lack of solvent effects may simplify experimental efforts in cocrystal screens compared to solvent based approaches. Additionally, microscopic melt crystallization techniques require only a small amount of substance and may enhance the chance for detecting and isolating such binary crystals. Two forms of Btl (II and VI) were detected by melt crystallization experiments in the presence of isomorphic additives.8 This concept of “isomorphic seeding”9 is an intriguing alternative route for exploring the polymorph landscape and may give access to new metastable forms, which do not appear in other crystallization experiments. If applicable, melt crystallization and sublimation (vapor deposition) offer valuable routes to different solid state forms in addition to solvent based polymorph or cocrystal screening practices, which in industry are increasingly performed with automated high throughput (HT) screening47 technologies. Presumably, such HT-technologies would detect the most important polymorphs (I0, III, and V) of Btl as well as the five (or even additional) solvates, which we found in a quick solvent screen. However, the identification of polymorphs is only one step in the solid form selection process and cannot provide a crucial solid understanding of the thermodynamics and phase transition processes of those forms, which may occur under storage, stress, and/or manufacturing conditions.
This study confirms the previously established classification,9 linking certain characteristics in the IR spectrum of a solid barbiturate form to its specific hydrogen-bonded structure, and this scheme was also used to determine the hydrogen-bonded structure of Btl-IV. Moreover, it was shown that Raman spectroscopy is well suited to discriminate between the different forms. The solid-state NMR spectra of the Btl polymorphs I0, III, and V provide a neat teaching example for demonstrating how solid-state NMR signals relate to molecular and crystal symmetry.
We think that most of the contradictions concerning the stability of the relevant polymorphs of Btl have been eliminated as a result of this study. The investigated compound had been in use for more than 100 years, and it was surprising to find that the existing literature did not contain consistent data on its polymorphism. A similar situation may exist with regard to many other small organic molecules that are used in important applications and exhibit polymorphic behavior of similar complexity.
Acknowledgments
T.G. gratefully acknowledges financial support from the Lise Meitner programme of the Austrian Science Fund (FWF, M 1135-117). We are grateful to Dr. D. Toebbens for assistance with the PXRD pattern of form IV and Dr. Ram K. R. Jetti for his crystallographic input at a very early stage of the project. The U.K. EPSRC is thanked for access to the Solid-State NMR Research Service based at Durham.
Supporting Information Available
Solvent crystallization scheme; table assigning the Btl forms to the appellations used in the literature; photomicrographs of Btl forms crystallized from solvents (characteristic morphologies); DSC curve showing melting endotherms of four polymorphs; scheme demonstrating the energetic phase transformation data for the Btl polymorphs; deconvolution of the methyl 13C NMR signal for Btl-V; graph set representations of N–H···O=C bonded structures C-4, C-2, C-1, and L-1; a table, listing performed solvent crystallization experiments; an enlarged image of Figure 2. This material is available free of charge via the Internet at http://pubs.acs.org.
This article is part 13 of a series of articles with the title Polymorphic Drug Substances of the European Pharmacopoeia.
The authors declare no competing financial interest.
Supplementary Material
References
- The European Pharmacopoeia Onlline (7.2), 7th ed.; Council of Europe, European Directorate for the Quality of Medicines: Strasbourg, France, 2011. [Google Scholar]
- Okamoto M.; Hinman D. J. Physical dependence produced by long duration, low dose chronic barbital treatment. Subst. Alcohol Actions Misuse 1984, 5, 97–103. [PubMed] [Google Scholar]
- Psychotropic Substances-Report 2010: Statistics for 2009; International Narcotics Control Board: Vienna, Austria, 2010.
- Fischer R.; Kofler A. Über den Polymorphismus des Veronals. Arch. Pharm. 1932, 270, 207–214. [Google Scholar]
- Lindpaintner E. Mikroskopische Untersuchungen an polymorphen Substanzen. Mikrochim. Acta 1939, 27, 21–41. [Google Scholar]
- a Huang T.-Y. The identification of derivatives of barbituric acid by x-ray analysis II. Acta Pharm. Int. 1951, 2, 95–106. [PubMed] [Google Scholar]; b Huang T.-Y. The identification of derivatives of barbituric acid by x-ray analysis VI. Acta Pharm. Int. 1953, 361–363. [PubMed] [Google Scholar]
- Cleverley B.; Williams P. P. Polymorphism in substituted barbituric acid. Tetrahedron 1959, 7, 277–288. [Google Scholar]
- a Brandstätter-Kuhnert M.; Aepkers M. Molekülverbindungen, Mischkristallbildung und neue Polymorphiefälle bei Barbituraten. Microchim. Acta 1962, 50, 1055–1074. [Google Scholar]; b Brandstätter-Kuhnert M.; Aepkers M. Molekülverbindungen, Mischkristallbildung und neue Polymorphiefälle bei Barbituraten. Microchim. Acta 1963, 51, 360–375. [Google Scholar]
- Zencirci N.; Gelbrich T.; Kahlenberg V.; Griesser U. J. Crystallization of metastable polymorphs of phenobarbital by isomorphic seeding. Cryst. Growth Des. 2009, 9, 3444–3456. [Google Scholar]
- Kuhnert-Brandstätter M.; Bachleitner-Hofmann F. Infrared spectroscopy of barbiturates. Arch. Pharm. Ber. Deut. Pharm. Ges. 1971, 304, 580–590. [DOI] [PubMed] [Google Scholar]
- a Craven B. M.; Vizzini E. A.; Rodrigues M. M. The crystal structure of two polymorphs of 5,5′-diethylbarbituric acid (barbital). Acta Crystallogr., Sect. B 1969, 25, 1978–1993. [DOI] [PubMed] [Google Scholar]; b Craven B. M.; Vizzini E. A. The crystal structure of polymorph IV of 5,5-diethylbarbituric acid (barbital). Acta Crystallogr., Sect. B 1971, 27, 1917–1924. [DOI] [PubMed] [Google Scholar]
- a Nogami H.; Nagai T.; Fukoka E.; Yotsuyanagi T. Dissolution kinetics of barbital polymorphs. Chem. Pharm. Bull. 1969, 17, 23–31. [Google Scholar]; b Sekiguchi K.; Kanke M.; Nakamura N. Dissolution behavior of solid drugs. V. Determination of the transition temperature and heat of transition between barbital polymorphs by initial dissolution rate measurements. Chem. Pharm. Bull. 1975, 23, 1347–1352. [Google Scholar]; c Hollenbach K.; Pintje-Hodi K.; Kedvessy G. Polymorphism of barbiturates in powders and tablets. Part 1: Crystal optical studies on, and melting behavior, of barbital. Pharmazie 1979, 34, 164–166. [PubMed] [Google Scholar]; d Hollenbach K.; Mezosi J.; Pintje-Hodi K.; Kedvessy G. Polymorphism of barbiturates in powders and tablets. Part 2: Infrared spectroscopy and x-ray diffractometry of barbital. Pharmazie 1979, 34, 240–242. [PubMed] [Google Scholar]; e Hollenbach K.; Pintje-Hodi K. Polymorphism of barbiturates in powders and tablets. Part 3: Effect of the pressing power on the modification of barbital. Pharmazie 1979, 34, 807–808. [PubMed] [Google Scholar]; f Hollenbach K.; Pintje-Hodi K.; Kedvessy G. Polymorphism of barbiturates in powders and tablets. Part 4: the dissolution of barbital B and Barbital A (modifications I and II) and their release from tablets. Pharmazie 1980, 35, 32–35. [PubMed] [Google Scholar]; g Hollenbach K.; Pintje-Hodi K.; Kedvessy G. Polymorphism of barbiturates in powders and tablets. Part 5: Dissolution of recrystallized barbital substances and their release from tablet. Pharmazie 1980, 35, 95–98. [PubMed] [Google Scholar]; h Hollenbach K.; Pintje-Hodi K.; Kedvessy G. Polymorphism of barbiturates in powders and tablets. Part 6: Properties and behavior of a spray-dried barbital preparation. Pharmazie 1982, 37, 562–565. [PubMed] [Google Scholar]
- Grabowska I.; Kaliszan R. Studies on the polymorphism of barbital. Pol. J. Pharmacol. Pharm. 1976, 28, 529–536. [PubMed] [Google Scholar]
- Burger A.; Ramberger R. On the polymorphism of pharmaceuticals and other molecular crystals. II. Microchim. Acta 1979, 72, 273–316. [Google Scholar]
- a Gelbrich T.; Zencirci N.; Griesser U. J. A polymorph of butobarbital with two distinct hydrogen-bonding motifs. Acta Crystallogr., Sect. C 2007, 63, o751–o753. [DOI] [PubMed] [Google Scholar]; b Zencirci N.; Gelbrich T.; Apperley D. C.; Harris R. K.; Kahlenberg V.; Griesser U. J. Structural features, phase relationships and transformation behavior of the polymorphs I-VI of phenobarbital. Cryst. Growth Des. 2010, 10, 302–313. [Google Scholar]; c Gelbrich T.; Zencirci N.; Griesser U. J. Hydrogen-bond motifs in N-monosubstituted derivatives of barbituric acid: 5-allyl-5-isopropyl-1-methylbarbituric acid (enallylpropymal) and 1,5-di(but-2-enyl)-5-ethylbarbituric acid. Acta Crystallogr., Sect. C 2010, 66, o55–o58. [DOI] [PubMed] [Google Scholar]; d Gelbrich T.; Rossi D.; Griesser U. J. 5,5-Dihydroxybarbituric acid 1,4-dioxane hemisolvate. Acta Crystallogr., Sect. E 2010, 66, o1219. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gelbrich T.; Rossi D.; Häfele C. A.; Griesser U. J. Barbiturates with hydrogen-bonded layer and framework structures. CrystEngComm 2011, 13, 5502–5509. [Google Scholar]; f Gelbrich T.; Rossi D.; Griesser U. J. Tetragonal polymorph of 5,5-dichlorobarbituric acid. Acta Crystallogr., Sect. E 2012, 68, o235–o236. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Rossi D.; Gelbrich T.; Kahlenberg V.; Griesser U. J. Supramolecular constructs and thermodynamic stability of four polymorphs and a co-crystal of pentobarbital (nembutal). CrystEngComm 2012, 14, 2494–2506. [Google Scholar]
- Brandstätter M. Isomorphie und Polymorphie bei Barbitursäurederivaten. Z. Phys. Chem. 1942, 191, 227–240. [Google Scholar]
- a Kofler A. Über die Polymorphie des Veronals. Mikrochemie 1947, 33, 4–7. [PubMed] [Google Scholar]; b Kofler L.; Kofler A.; Brandstätter M. In Thermo-Mikro-Methoden; Verlag Chemie: Weinheim, Germany, 1954; p 543. [Google Scholar]
- Willis J. N. Jr.; Cook R. B.; Jankow R. Raman spectrometry of some common barbiturates. Anal. Chem. 1972, 44, 1228–1234. [DOI] [PubMed] [Google Scholar]
- Kaliszan R.; Halkiewicz J. IR analysis of crystalline samples of some urea derivatives. Pol. J. Pharmacol. Pharm. 1975, 27, 579–587. [PubMed] [Google Scholar]
- McMullan R. K.; Fox R. O. Jr.; Craven B. M. The neutron crystal structure at −75degreesC of polymorph II of 5,5-diethylbarbituric acid (barbital). Acta Crystallogr., Sect. B 1978, 34, 3719–3722. [Google Scholar]
- Caillet J.; Claverie P. On the polymorphism of barbituric acid derivatives. Acta Crystallogr., Sect. B 1980, 36, 2642–2645. [Google Scholar]
- Craven B. M.; Fox R. O. Jr.; Weber H.-P. The charge density in polymorph II of 5,5-diethylbarbituric acid (barbital) at 198 K. Acta Crystallogr., Sect. B 1982, 38, 1942–1952. [Google Scholar]
- Chauvet A.; De Maury G.; Terol A.; Masse J. Etude thermoanalytique de substances psychotropes: V. Barbital et allobarbital. Thermochim. Acta 1986, 97, 143–159. [Google Scholar]
- Fournival J. L.; Rouland J. C.; Céolin R. Sur le Polymorphisme du barbital et le systeme eau-barbital. J. Therm. Anal. 1987, 32, 1547–1557. [Google Scholar]
- Kofler L.; Kofler A.. Thermo-Mikro-Methoden zur Kennzeichnung organischer Stoffe und Stoffgemische; Universitätsverlag Wagner GmbH: Innsbruck, Austria, 1954. [Google Scholar]
- Burger A.; Ramberger R. On the polymorphism of pharmaceuticals and other molecular crystals. I. Microchim. Acta 1979, 72, 259–271. [Google Scholar]
- a Etter M. C.; MacDonald J. C.; Bernstein J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr., Sect. B 1990, 46, 256–262. [DOI] [PubMed] [Google Scholar]; b Bernstein J.; Davis R. E.; Shimoni L.; Chang N.-L. Patterns in hydrogen bonding: functionality and graph set analysis in crystals. Angew. Chem., Int. Ed. 1995, 34, 1555–1573. [Google Scholar]
- Lewis T. C.; Tocher D. A.; Price S. L. An experimental and theoretical search for polymorphs of barbituric acid: the challenges of even limited conformational flexibility. Cryst. Growth Des. 2004, 4, 979–987. [Google Scholar]
- Zencirci N.; Gelbrich T.; Jetti R. K. R.; Kahlenberg V.; Griesser U. J., unpublished results.
- DesMarteau D. D.; Pennington W. T.; Resnati G. Fluorinated barbituric acid derivatives. Acta Crystallogr., Sect. C 1994, 50, 1305–1308. [Google Scholar]
- Chentli-Benchikha F.; Declercq J. P.; Germain G.; Van Meerssche M.; Bouche R.; Draguet-Brughmans M. Structures des oxo-3 et oxo-6 cyclobarbitals. Acta Crystallogr., Sect. B 1977, 33, 2739–2743. [Google Scholar]
- Williams P. Polymorphism of phenobarbitone. II. The crystal structure of modification III. Acta Crystallogr., Sect. B 1974, 30, 12–17. [Google Scholar]
- Craven B. M.; Cusatis C. The crystal structure of 5-ethyl-5-(1-methylbutenyl)-barbituric acid. Acta Crystallogr., Sect. B 1969, 25, 2291–2298. [Google Scholar]
- a Zerkowski J. A.; Seto C. T.; Wierda D. A.; Whitesides G. M. The design of organic structures in the solid state: hydrogen-bonded molecular “tapes”. J. Am. Chem. Soc. 1990, 112, 9025–9026. [Google Scholar]; b Zerkowski J. A.; MacDonald J. C.; Seto C. T.; Wierda D. A.; Whitesides G. M. Design of organic structures in the solid state: molecular tapes based on the network of hydrogen bonds present in the cyanuric acid···melamine complex. J. Am. Chem. Soc. 1994, 116, 2382–2391. [Google Scholar]; c Zerkowski J. A.; Mathias J. P.; Whitesides G. M. New varieties of crystalline architecture produced by small changes in molecular structure in tape complexes of melamines and barbiturates. J. Am. Chem. Soc. 1994, 116, 4305–4315. [Google Scholar]; d Babu N. J.; Reddy L. S.; Nangia A. Amide–N-oxide heterosynthon and amide dimer homosynthon in cocrystals of carboxamide drugs and pyridine N-oxides. Mol. Pharmaceutics 2007, 4, 417–434. [DOI] [PubMed] [Google Scholar]; e Vishweshwar P.; Thaimattam R.; Jaskolski M.; Desiraju G. R. Supramolecular synthons based on N-H···N and C-H···O hydrogen bonds. Crystal engineering of a helical structure with 5,5-diethylbarbituric acid. Chem. Commun. 2002, 1830–1831. [DOI] [PubMed] [Google Scholar]; f Golden E.; Argent S. P.; Blake A. J.; Thébault F.; Champness N. R. Hydrogen-bonded chains formed by 5,5-diethylbarbituric acid and bipyridyl tectons. Supramol. Chem. 2012, 24, 40–47. [Google Scholar]
- Zencirci N.Polymorphism, Isomorphism, Solvate Formation and Structural Classification of Barbiturates. Ph.D. Thesis, University of Innsbruck, 2009. [Google Scholar]
- Lin-Vien D.; Colthup N. B.; Fateley W. G.; Graselli J. G.. The Handbook of Infrared and Raman Characteristic Frequencies or Organic Molecules; Academic Press: San Diego, CA, 1991. [Google Scholar]
- Bideau J. P. Crystalline structure of 5-ethyl-5-butylbarbituric acid. C. R. Acad. Sci., Serie C: Sci. Chim. 1971, 272, 757–760. [Google Scholar]
- Harris R. K.; Olivieri A. C. Quadrupolar effects transferred to spin-12 magic-angle spinning spectra of solids. Prog. NMR Spectrosc. 1992, 24, 435–456. [Google Scholar]
- Harris R. K. NMR crystallography: the use of chemical shifts. Solid State Sci. 2004, 6, 1025–1037. [Google Scholar]
- Bernstein J.; Davey R. J.; Henck J.-O. Concomitant polymorphs. Angew. Chem., Int. Ed. 1999, 38, 3440–3461. [DOI] [PubMed] [Google Scholar]
- Aaltonen J.; Allesø M.; Mirza S.; Koradia V.; Gordon K. C.; Rantanen J. Solid form screening – a review. Eur. J. Pharm. Biopharm. 2009, 71, 23–37. [DOI] [PubMed] [Google Scholar]
- Gelbrich T.; Braun D. E.; Ellern A.; Griesser U. J. Four polymorphs of methyl paraben – structural relationships and relative energy differences. Cryst. Growth Des. 2013, 13, 1206–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmerer A.; Bernstein J.; Griesser U. J.; Kahlenberg V.; Többens D. M.; Lapidus S.; Stephens P. W. A tale of two polymorphic pharmaceuticals: pyrithyldione and propyphenazone and their 1937 co-crystal patent. Chem.—Eur. J. 2011, 17, 13445–13460. [DOI] [PubMed] [Google Scholar]
- Nichols G.; Frampton C. S. Physicochemical characterization of the orthorhombic polymorph of paracetamol crystallized from solution. J. Pharm. Sci. 1998, 87, 684–693. [DOI] [PubMed] [Google Scholar]
- Fucke K.; Myz S. A.; Shakhtshneider T. P; Boldyreva E. V.; Griesser U. J. How good are the crystallisation methods for co-crystals - a comparative study of Piroxicam. New. J. Chem. 2012, 36, 1969–1977. [Google Scholar]
- Berry D. J.; Seaton C. C.; Clegg W.; Harrington R. W.; Coles S. J.; Horton P. N.; Hursthouse M. B.; Storey R.; Jones W.; Friscic T.; Blagden N. Applying hot-stage microscopy to co-crystal screening: A study of nicotinamide with seven active pharmaceutical ingredients. Cryst. Growth Des. 2008, 8, 1697–1712. [Google Scholar]
- Morissette S. L.; Almarsson O.; Peterson M. L.; Remenar J. F.; Read M. J.; Lemmo A. V.; Ellis S.; Cima M. J.; Gardner C. R. High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Delivery Rev. 2004, 56, 275–300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.