

Abstract
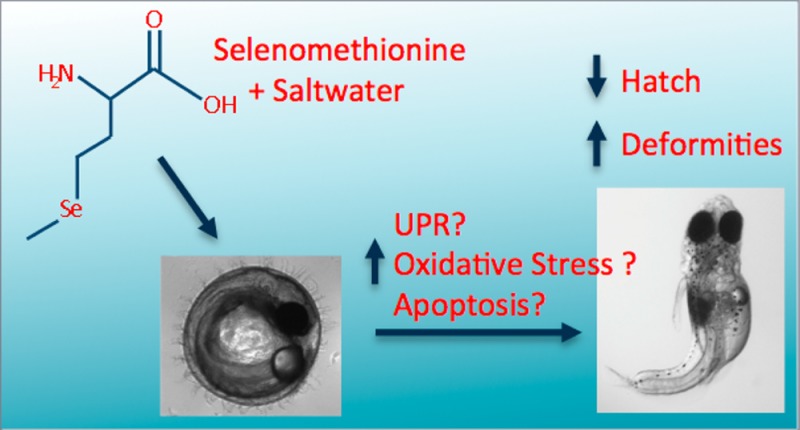
Selenium (Se) is an essential micronutrient that can cause embryotoxicty at levels 7–30 times above essential concentrations. Exposure to hypersaline conditions and 50 μM selenomethionine (SeMet) decreased embryo hatch and depleted glutathione in Japanese medaka embryos without affecting Se accumulation. To better understand the impacts of nonchemical stressors on developmental toxicity of Se in fish, several adverse outcome pathways were evaluated in the Japanese medaka (Oryzias latipes). We treated medaka embryos at 12 h post fertilization with 50 μM SeMet for 12 hours in freshwater or in 13 ppth hypersalinity and evaluated the contributions of oxidative stress, the unfolded protein response and apoptosis to reduced hatch. Exposure to SeMet and hypersalinity decreased embryo hatch to 3.7% ± 1.95, and induced teratogenesis in 100% ± 0 of hatched embryos. In contrast, treatments of freshwater, saltwater, and SeMet in freshwater resulted in 89.8% ± 3.91–86.7% ± 3.87 hatch, and no significant increase in deformities. We found no significant differences in lipid peroxidation, indicating that oxidative stress may not be responsible for the observed toxicity in embryos at this time point (24 h). Although significant changes in apoptosis were not observed, we witnessed up to 100 fold increases in transcripts of the endoplasmic reticulum (ER) chaperone, immunoglobulin binding protein (BiP) and trends toward increasing downstream signals, activating transcription factor 4 (ATF4) and ATF6 indicating potential contributions of the unfolded protein response to the effects of SeMet and hypersaline conditions. These data indicate that multiple adverse outcome pathways may be responsible for the developmental toxicity of Se and salinity, and these pathways may be time dependent.
I. Introduction
Selenium (Se) is an essential micronutrient; levels only 7–30 times greater than required can be toxic.1 This is a concern in aquatic environments, where anthropogenic activities can release large quantities of Se, and include agricultural runoff of irrigation waters in arid regions;2 waste rock from coal, phosphate, and uranium mining;3−5 and combustion waste from coal burning power plants.6 Se usually enters the waterways in its inorganic forms of selenate (Se+4) or selenite (Se+6), which can be taken up by microbes and primary producers and converted into various organic forms, including the amino acid, selenomethionine (SeMet).7 Consumers, such as fish and birds, are exposed to Se primarily in the diet and SeMet has been shown to be the major form of Se in the fish diet.8 One concern for SeMet toxicity is its bioaccumulation potential. SeMet has been demonstrated to move through the food chain by trophic transfer to higher-level organisms.9,10 Following a Se poisoning event at Belews lake, NC, Lemly found Se to have bioaccumulated from 519 times in periphyton to 3975 times in the visceral tissues of fish.10 This is of particular concern for oviparous carnivores, for which maternal offloading may impair development or reproductive success through respective teratogenesis or embryo lethality.10
Because of the potential for biomagnification, traditional water quality measurements of Se concentrations may be ineffective. Recently, the U.S. Environmental Protection Agency has begun advocating tissue concentration measurements for Se monitoring.11 However, even these measurements may not provide an accurate picture of Se effects on an ecosystem, because fish encounter multiple stressors in their environments, which can alter Se toxicity. Recent evidence suggests that hypersalinity may compound Se toxicity.12 This is of particular importance in areas such as the San Joaquin River Valley, CA, and the San Francisco Bay Delta area, where many historically freshwater–waterways are in danger of salinization.13 These areas are often spawning grounds for protected species such as the endangered delta smelt (Hypomesus transpacificus) and threatened steelhead trout (Oncorhynchus mykiss).
The mechanisms behind Se induced teratogenesis and mortality remain unclear. Several studies point to oxidative stress as one mode of action for Se toxicity.14−17 However, oxidative stress is most likely only one factor influencing SeMet toxicity. The unfolded protein response (UPR) is a cellular and molecular response to perturbations in endoplasmic reticulum (ER) homeostasis (See Hetz (2012) for review18). Oxidative stress, calcium disruption, and glycosylation inhibition, can all disrupt protein folding, leading to the accumulation of unfolded proteins in the ER. Protein folding chaperones, such as BiP (immunoglobulin- binding protein; Grp78), initiate the UPR through dissociation from the mediators of the three branches, PERK (protein kinase RNA (PKR)-like ER kinase), IRE1a (inositol- requiring protein-1) and ATF6 (activating transcription factor 6). While the three branches of the response are highly interconnected, they can be generally divided into three categories. The PERK branch is responsible for translational attenuation through Activating Transcription Factor 4 (ATF4). The IRE1a branch is responsible for transcription of ER-Associated Degradation (ERAD) genes through X box protein 1 (XBP1). And finally, ATF6 is responsible for transcription of protein folding enzymes and chaperones. If the response is unable to attenuate the stress, the UPR will initiate cell death, often in the form of programmed cell death (apoptosis).
We have previously demonstrated that 50 μM of SeMet and hypersalinity treatment for 24 h significantly decreased embryo hatch, decreased total reduced glutathione, and increased flavin containing-monooxygenase (FMO) activity in medaka embryos.12 Exposure of Japanese medaka embryos to SeMet under varied salinities did not impact overall Se accumulation but significant differences in toxicity were observed.12 The purpose of the current study was to further elucidate the mechanisms behind SeMet and hypersaline induced embryo mortality after 12 h of SeMet treatment at developmental stages not previously examined. We hypothesized that SeMet would induce oxidative stress, the UPR and apoptosis in Japanese medaka embryos and that hypersaline conditions would potentiate these effects. This research will aid in the development for site specific monitoring for Se in CA.
II. Materials and Methods
2.1. Chemicals and Reagents
Seleno-l-methionine (Purity 98%), 1-butanol, phosphoric acid, thiobarbituric acid and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO). A Milli-Q water purification system (Millipore, Billerica, MA) was used to obtain deionized water. Ethanol (Fisher, Pittsburgh, PA) was of molecular biology grade.
2.2. Embryo Collection and Exposure
Japanese Medaka were cultured at the University of California- Riverside and housed in a 2:3 ratio of males to females in medium-hard water at 27 °C and a photoperiod of 14 h light and 10 h dark. Adults were fed twice daily a diet of live brine shrimp. Embryos were collected 0–1 h following fertilization. Viable embryos were determined based on oil droplet migration to the vegetal pole as outlined by Kirchen and West (1976).19 Nonviable embryos were discarded and viable embryos were placed into 60 × 15 mm Petri dishes containing either freshwater or a makeup of saltwater from the San Joaquin River Valley (20–30 embryos per replicate, and 5–10 replicates per group). Although, water-borne SeMet exposures do not represent the most likely environmental exposure (the primary exposures for SeMet are dietary or via maternal transfer), they are sufficient to study the mechanistic effects of SeMet on medaka embryos. San Joaquin River Valley saltwater was prepared in the lab according to a recipe from Westlands Water District located about 10 km south of Mendota in the San Joaquin River Drainage Basin, CA.20 Salinity was measured with a conductivity meter, and corresponds to about 13‰ and 15.3 g/L suspended solids.
Following 12 h of equilibration in fresh or salt water, the replicates were divided into three groups. The first subset of embryos were frozen −80 °C to represent a time zero control (12hpf). Other embryos were treated with a 50 μM solution of Seleno-l-Methionine (Cat. No. S3132, Sigma-Aldrich) in freshwater or saltwater and exposed for 12 h, then collected and frozen at −80 °C for analysis (24hpf). SeMet concentrations were chosen based on previous research and were intended to represent the upper levels of bioaccumulation measured in embryos.12 Previous studies have also demonstrated uptake of SeMet into the embryo, indicating this system was an effective exposure method.12 The final subset of embryos were left in freshwater or saltwater for 24 h to compare to the SeMet treated 12hpf samples.
2.3. Modified Embryo Larval Toxicity Assay
The medaka embryo-larval toxicity assay was adapted from Farwell et al. (2006).21 Embryos were collected and treated as above with one replicate equal to 15–20 embryos per dish. Following 12 h of SeMet treatment, embryos were rinsed and transferred to new dishes containing freshwater or saltwater. Water was changed every other day and dishes were monitored for mortality with removal of dead embryos. Embryo hatch was monitored for 21 days post fertilization. At hatch, embryos were assessed for deformities and terminated immediately. Percent hatch and percent of hatched embryos with deformities were recorded.
2.4. Analysis of Gene Expression
Total mRNA was isolated from embryos using the Lipid Tissue RNeasy kit (Qiagen, Valencia, CA) following the manufacturers instructions. mRNA quantity and quality was measured using the ND-1000 (NanoDrop, Wilmington, DE). mRNA (1 μg) was converted to cDNA using the Reverse Transcription System (Promega Corporation, Madison, WI), according to the manufacturers instructions.
Primers were designed using IDTDNA PrimerQuest software and optimized using PCR Miner22 (Table 1). As no BAX gene for Japanese medaka has been annotated in the NCBI database, BLAST was used on the medaka genome (http://compbio.dfci.harvard.edu/cgi-bin/tgi/Blast/index.cgi) against BAX from zebrafish (Danio rerio) to develop primers. Similarity between the genes had an E value of 1 × 10−48. EF1α was run as a housekeeping gene. qPCR was performed with the iScript One-step RT-PCR kit with SYBR Green from Bio-Rad (Hercules, CA), omitting the reverse transcriptase, on a MyiQ5 Thermo cycler (Biorad). The samples were denatured and the polymerase activated at 95 °C for 5 min, then 40 cycles of 10s at 95 °C and 30s of 55 °C. Samples were subject to melting curve analysis from 65 to 85 °C in 0.5 °C increments with continuous fluorescence measurement. qPCR was analyzed according to Schmittgen and Livak23 and fold change was calculated against the 12hpf freshwater controls. All data was compared against the 12hpf controls in order represent how the gene expression changed over the 12h time period, and how the treatments affected this change. Rather than observing a discrete point in development, we feel it is necessary to understand how these treatments altered normal development.
Table 1. Primers, Accession Numbers, and Concentrations used for qRT-PCR.
| name | fwd primer (5′-3′) | rev primer (5′-3′) | accession # | conc. |
|---|---|---|---|---|
| EF1-a | CTACATCAAGAAGATCGGCTACAA | CGACAGGGACAGTTCCAATAC | NM_001104662.1 | 2.5 μM |
| CASP3A | CCAAATCCCAGGTCTACTGATG | AGGCAAAGGAGGCAAACTTA | NM_001104670.1 | 5 μM |
| BAX | GCTGGTCATAAAGGCTCTCATC | CCAGATTGCTCGAACCGTAAA | NM_131562.2 | 2.5 μM |
| BiP | GGAGGATTCTGACCTGAAGAAG | GGTGACAGTAGGCTGGTTATC | NM_001278801.1 | 0.5 μM |
| ATF6 | CAAGCCAACTCCAGTCAGTATC | GCCGACTCTCGGTTCTTTATC | NM_001278901.1 | 0.5 μM |
| ATF4 | CTTAGAGGTGAAGGTGCCTATG | TGAGGAAGGAGACCTGTTAGA | XM_004066069.1 | 2.5 μM |
2.5. Analysis of Oxidative Stress
Thiobarbituric Reactive Substances (TBARS) were measured to estimate malondialdehyde (MDA) formation in medaka embryos.24 Embryos (15–20) were weighed and homogenized in 1.15% KCl then centrifuged at 3000 rpm for 5 min at 4 °C. The supernatant was then used in the assay. Samples were run in duplicate on a Wallac Victor2 multilabel plate reader (PerkinElmer, Waltham, MA) with excitation at 535 nm and emission at 585 nm.
2.6. Statistical Analysis
Statistical significance was assessed using a student’s T-Test or 2-way ANOVA in the statistical program R. Statistical significance was determined at p ≤ 0.05, unless otherwise noted. If overall significance was determined following two-way ANOVA, Tukey’s HSD test was performed posthoc. Data was checked for normality and homogeneity of variances. Any non-normal data was log transformed. For data that remained non-normal following log transformation, Kruskal–Wallis tests were performed with Dunn’s test posthoc.
III. Results
3.1. Embryo-Larval Toxicity Assay
While there was no difference in hatch between saltwater and freshwater control embryos, 50 μM SeMet significantly decreased embryo hatch in saltwater treated embryos to 3.7% (Figure 1A). However, SeMet treatment in freshwater did not significantly decrease hatch. The median day to hatch was not significantly affected by SeMet treatment in freshwater or saltwater (Figure 1B). SeMet and hypersaline treatment also significantly increased the deformities in treated embryos (Figure 1C). All SeMet and hypersaline treated embryos had deformities upon hatch. The most common deformities observed were kyphosis, lordosis, craniofacial abnormalities, and yolk sac edema (Figure 1D).
Figure 1.

Effects of combined exposure of SeMet (50 μM) and hypersaline conditions on the development and hatchability of Japanese medaka embryos after exposure at 12 hpf. White bars represent freshwater and black bars represent saltwater. (A) Percent hatch, (B) median day to hatch, (C) percent deformities in hatched embryos, (D) examples of deformities; control is top image, bottom two images demonstrate lordosis, kyphosis, cranio-facial abnormalities, and yolk-sac edema. Each value represents the mean ± standard error (SE) of 5–10 replicates. Statistical significance is indicated by differing letters (Two-way ANOVA, Tukey HSD test, or Kruskal–Wallis, Dunn’s test p ≤ 0.05).
3.2. Oxidative Stress and Apoptosis
There was no significant difference in amount of lipid peroxidation between any of the treatments and no difference between embryos at 12hpf and 24hpf (Figure 2). BAX transcript levels decreased significantly from 12hpf to 24hpf (0.2 fold, p = 0.011), while Caspase 3A levels remained constant (Figure 3A). There was no significant difference in BAX or CASP3A gene expression following treatment with hypersalinity or SeMet (Figure 3B and C).
Figure 2.

Effects of combined exposure of SeMet (50 μM) and hypersaline conditions on lipid peroxidation in Japanese medaka embryos after 12 and 24 hpf. (A) TBARS was measured as nmol/g wet weight tissue in freshwater controls at 12 hpf and 24 hpf. (B) TBARS measured in embryos in freshwater, saltwater, SeMet in freshwater and SeMet in saltwater at 24hpf. Hydrogen peroxide (3% for 3 h) was run as a positive control. Each value represents the mean ± SE of 5–10 replicates Statistical significance is denoted by differing letters at p ≤ 0.05 (One-way ANOVA, Tukey HSD test).
Figure 3.

Effects of combined exposure of SeMet (50 μM) and hypersaline conditions on BAX and CASP3A transcripts in Japanese medaka embryos after 12 and 24 hpf. (A) Change in BAX and CASP3A expression between 12hpf and 24hpf. Expression of (B) BAX and (C) CASP3A in freshwater, saltwater, SeMet in freshwater and SeMet in saltwater. Each value represents the mean ± SE of 5–10 replicates. EF1-α was run as a housekeeping gene. Statistical significance is indicated by differing letters (Two-way ANOVA, Tukey HSD Test p ≤ 0.05).
3.4. Gene Expression of UPR Mediators
UPR gene expression changed from 12hpf to 24 hpf in medaka embryos. ATF6 gene expression decreased significantly from 12hpf to 24 hpf (0.3 fold, p = 0.019), while BiP and ATF4 expression increased significantly (2 fold, p = 0.048, and 4 fold, p = 0.008, respectively). BiP mRNA was increased in both saltwater and freshwater SeMet treatments up to 39 fold over 12 h controls and 107 fold over 12 h controls in the SeMet freshwater and SeMet saltwater treatments, respectively. Though there was a trend toward an increase in ATF6 expresssion with SeMet treatment (p = 0.119), SeMet and hypersalinity did not significantly alter ATF6 expression. Similarly, trends in ATF4 expression indicated a potential difference between freshwater and saltwater treatment (p = 0.07), with SeMet decreasing ATF4 in freshwater, yet increasing it in saltwater.
IV. Discussion
The mechanism of action of SeMet toxicity is not well understood, particularly in the presence of multiple stressors, which may confound regulatory monitoring. We observed a decrease in hatch following treatment with hypersalinity and SeMet, while surviving embryos had deformities. Previous work at this developmental stage and SeMet concentration reported a significant decrease in embryo hatch following 24 h of 50 μM SeMet treatment in freshwater.12 While we did not observe this trend, our results were expected considering the duration of SeMet treatment was half as that previously studied. Overall, our results are consistent with a plethora of other data reporting SeMet’s lethal and teratogenic effects in the field, for example, ref (10).
Japanese medaka are a euryhaline species; adults are able to spawn and embryos hatch in full seawater.25 We observed no significant difference in toxicity between freshwater and hypersaline controls, indicating that our results are not due to osmotic stress alone. Others have found salinity to potentiate SeMet toxicity in embryos.12 The mechanism behind this remains to be elucidated, however, FMO may play a role. FMOs have been shown to oxygenate SeMet, which may contribute to its toxicity.26,12 Several studies have found FMO activity can increase under hypersaline conditions.12,27,28 This increased FMO activity may increase SeMet oxygenation, which in turn may increase its embryo toxicity.
In contrast, studies have shown that SeMet activation can occur following methioninase generation of methylselenol,29 which can subsequently generate oxidative stress in rainbow trout embryos.14 Methylselenol has also been implicated in induction of caspase-mediated apoptosis in cancer cell lines.30 However, considering that neither oxidative stress nor apoptosis was induced in SeMet and hypersaline treated embryos and that FMO has been found to be induced by hypersosmotic conditions, we conclude that FMO activation may be a major contributor to the observed toxicity.
Of interest is that SeMet and hypersalinity did not generate oxidative stress as measured by lipid peroxidation after 12 h of treatment. However, several groups, including ours, have identified oxidative stress as one of the main modes of action of SeMet toxicity.12−15 As lipid peroxidation is an end point for severe oxidative stress, TBARS is not sensitive to small changes in cellular redox.31 Furthermore, in this study, TBARS was measured in whole embryos and did not consider localized effects. Thus, oxidative stress may still be occurring in SeMet and hypersaline treatments, yet it may not be detected by our assays or was not as high as that observed after 24hpf.12 While our results do not eliminate oxidative stress as a mechanism of SeMet induced embryotoxicty, they indicate that other processes may play an important role particularly at the 12 hour time point of exposure. While we observed no difference in whole-embryo lipid peroxidation between 12hpf and 24hpf, many studies demonstrate that the redox status of embryos also undergoes great changes throughout development. Two contradicting reports demonstrate the changes in redox status of medaka embryos throughout development. Wu et al.32 measured changes in oxidative stress in the whole embryo each day post fertilization using Dichloro-dihydro-fluorescein diacetate (DCHFDA; a dye that fluoresces following oxidation) and found overall reactive oxygen species (ROS) increased gradually until hatch. In contrast, another group studying medaka development and silver nanoparticle toxicity, found total ROS decreased throughout development.33 However, in addition to total ROS, both of these studies examined multiple biomarkers for oxidative stress and found no common patterns between them. Hence, the current studies on redox status throughout medaka development confirm that further studies are necessary in order to understand this complex process.
Another adverse outcome pathway that may contribute to SeMet toxicity in medaka at this early life stage is the UPR. Methylselenic acid (MSA) induced the UPR in PC-3 cells, a human prostate cancer cell line34 and we observed increases in BiP expression following SeMet treatment, suggesting that SeMet may be disrupting ER homeostasis. Although not significant, BiP mRNA expression was higher in SeMet and hypersaline treated embryos than in embryos in SeMet and freshwater. This would suggest that a greater UPR is being induced in SeMet and hypersaline conditions, a mechanism that requires further exploration. The trend toward alterations in ATF4 and ATF6 gene expression also indicated a possible role for the PERK and ATF6 branches of the UPR in SeMet toxicity. PERK also activates Nrf2,35,36 a major transcription factor for antioxidant genes during oxidative stress. This provides a link between postulated mechanisms of oxidative stress and the UPR. It must be noted that each branch of the UPR has a different activation step (e.g., ATF6 splicing, PERK phosphorylation)37 according to varying time scales and specific types of ER stress.38 DuRose et al. (2006) compared UPR responses generated by dithriothreitol (DTT; disrupts disulfide bond formation), thapsigargin (Tg, inhibits ER Calcium-dependent ATPase), and tunicamycin (Tm, inhibits protein glycosylation). The ATF6 response to DTT was the most rapid of the three branches, yet it was significantly less sensitive to Tg and Tm.38 In contrast, PERK responded most rapidly to calcium disruption caused by Tg. These responses occurred on different time scales and at different magnitudes; overall, activation of the branches in response to DTT was faster and stronger than to Tm and Tg.38 The varied time scales of each branch of the response to different types of ER stress indicate the necessity to document the UPR throughout development following SeMet and hypersaline treatment. Furthermore, as mentioned above concerning TBARS experiments, these gene expression studies cannot discern between localized effects. Changes in transcription in a few key cells may not result in a distinct difference in fold change in qPCR.
The UPR plays a key physiological role in development.39 The IRE1a-XBP1 pathway has been shown to regulate a variety of developmental processes, including plasma cell differentiation,40 liver development,41 chondrogenesis42 and adipogenesis.43 The PERK/ATF4 branch of the UPR has been found to be involved in osteoblast formation.44 BiP also plays a major role in development as BiP knockout mice are not viable after the peri-implantation stage.45 BiP has a much higher expression during neural development than during adulthood.45 The importance of these responses has been found to be conserved in the medaka.46 Recent work established that ATF6a/b results in embryonic lethality in medaka, as in mice.47 They found the physiological response to be strong at 2 days post fertilization, where it was localized to the brain, otic vesicle and notochord.47 Perturbations in the physiological ER stress response during embryogenesis may result in the inability of the embryo to manage the high demand for protein folding. If the perturbation is not stopped it could result in teratogenesis and embryo lethality. Lordosis, kyphosis, and craniofacial abnormalities were the primary forms of teratogenesis documented in this study. Considering the studies above illustrating the role of the UPR in the development of the notochord and cartilage, it is possible that UPR disruption may be generating the deformities witnessed here.
Apoptosis is one common outcome generated by the UPR.18 We had hypothesized that apoptosis played a significant role leading to embryo lethality generated by SeMet. However, we observed no evidence of apoptosis in our embryos. Neither BAX nor CASP3A gene expression were changed by SeMet treatment. Wu et al.34 demonstrated that 5 μM MSA was able to cause apoptosis following UPR induction in PC-3 cells. However, MSA is highly redox reactive and not a natural form of Se in the fish diet. BiP has been shown to be a negative regulator of apoptosis,18 so the high levels of BiP mRNA induction observed may indicate a repression of apoptosis. Autophagy is another well-documented outcome from UPR,48 and may also contribute to the deformities and reduction of hatch observed.
The comparison of apoptosis taken at 12hpf and 24hpf clearly shows that these important processes fluctuate greatly during development. Indeed, apoptosis has been shown to fluctuate in tissue-specific patterns throughout Japanese medaka development, occurring mostly in the head, spinal column, and tailbud.49 Furthermore, apoptosis plays a key role in neural development.50
The overwhelming evidence for changes in apoptosis, oxidative stress, and the UPR throughout development, indicates that developing Japanese medaka embryos have important windows of susceptibility to SeMet and hypersaline stress. Thus, while the roles of oxidative stress and apoptosis in the developmental toxicity of SeMet may be limited from 12 to 24 h, they may be increased as the oxidation state of the embryos increases, or as apoptosis is more active at later stages of development. It is important to map these processes throughout embryogenesis, so that we may better understand the developmental toxicity of SeMet and hypersaline conditions.
In summary, hypersaline conditions derived from the San Joaquin River Valley, CA, enhanced the toxicity of SeMet in the developing medaka embryo. While the UPR may have played a role, oxidative stress and apoptosis measured in the whole embryo were not associated with SeMet induced mortality and teratogenesis at this early stage. Additional studies will further consider the role of oxidative stress and the UPR throughout medaka development and investigate developmental periods most susceptible to SeMet and hypersaline toxicity.
Figure 4.

Effects of combined exposure of SeMet (50 μM) and hypersaline conditions on BiP, ATF6, and ATF4 transcripts in Japanese medaka embryos after 12 and 24 hpf. (A) Expression of BiP, ATF6, and ATF4 in whole embryos at 12hpf and 24hpf in freshwater. Expression of (B) BiP, (C) ATF6, and (D) ATF4 in 24hpf control embryos and SeMet treated embryos in freshwater and saltwater. Each value represents the mean ± SE of 5–10 replicates. EF1-α was run as a housekeeping gene. Statistical significance is indicated by differing letters (Two-way ANOVA, Tukey HSD Test p ≤ 0.05).
Acknowledgments
We acknowledge Rodney Johnson at the U.S. EPA, Duluth, MN for providing us with the mature Japanese medaka for this study. Funding Source: University of California-Riverside/Agricultural Experiment Station Resource Allocation Program and the University of Washington Superfund Basic Research Program [NIEHS P42ES04696].
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Lemly A. D. A teratogenic deformity index for evaluating impacts of selenium on fish populations. Ecotoxicol. Environ. Saf. 1997, 373259–66. [DOI] [PubMed] [Google Scholar]
- Outridge P. M.; Scheuhammer A. M.; Fox G. A.; Braune B. M.; White L. M.; Gregorich L. J.; Keddy C. An assessment of the potential hazards of environmental selenium for Canadian water birds. Environ. Rev. 1999, 7281–96. [Google Scholar]
- Muscatello J. R.; Bennett P. M.; Himbeault K. T.; Belknap A. M.; Janz D. M. Larval Deformities associated with selenium accumulation in northern pike (Esox lucius) exposed to metal mining effluent. Environ. Sci. Technol. 2006, 40206506–6512. [DOI] [PubMed] [Google Scholar]
- Presser T. S.; Piper D. Z.; Bird K. J.; Skorupa J. P.; Hamilton S. J.; Detwiler S. J.; Huebner M. A.. The phosphoria formation: A model for forecasting global selenium sources to the environment. In Handbook of Exploration and Environmental Geochemistry, Chapter 11; James R. H., Ed.; Elsevier Science B.V., 2004; Vol. 8, pp 299–319. [Google Scholar]
- Ramirez P. Jr.; Rogers B. P. Selenium in a Wyoming grassland community receiving wastewater from an in situ uranium mine. Arch. Environ. Contam. Toxicol. 2002, 424431–6. [DOI] [PubMed] [Google Scholar]
- Wen H.; Carignan J. Reviews on atmospheric selenium: Emissions, speciation and fate. Atmos. Environ. 2007, 41347151–7165. [Google Scholar]
- Fan T. W.; Teh S. J.; Hinton D. E.; Higashi R. M. Selenium biotransformations into proteinaceous forms by foodweb organisms of selenium-laden drainage waters in California. Aquat. Toxicol. 2002, 57, 65–84. [DOI] [PubMed] [Google Scholar]
- Phibbs J.; Franz E.; Hauck D.; Gallego M.; Tse J. J.; Pickering I. J.; Liber K.; Janz D. M. Evaluating the trophic transfer of selenium in aquatic ecosystems using caged fish, X-ray absorption spectroscopy and stable isotope analysis. Ecotoxicol. Environ. Saf. 2011, 7471855–1863. [DOI] [PubMed] [Google Scholar]
- Luoma S. N.; Rainbow P. S. Why is metal bioaccumulation so variable? Biodynamics as a unifying concept. Environ. Sci. Technol. 2005, 3971921–31. [DOI] [PubMed] [Google Scholar]
- Lemly A. D. Symptoms and implications of selenium toxicity in fish: The Belews Lake case example. Aquat. Toxicol. 2002, 57, 39–49. [DOI] [PubMed] [Google Scholar]
- USEPA. Draft Aquatic Life Water Quality Criteria for Selenium; Office of Water and Office of Science and Technology: Washington, DC, 2004; http://water.epa.gov/scitech/swguidance/standards/criteria/aqlife/selenium/upload/complete-2.pdf. [Google Scholar]
- Lavado R.; Shi D.; Schlenk D. Effects of salinity on the toxicity and biotransformation of l-selenomethionine in Japanese medaka (Oryzias latipes) embryos: Mechanisms of oxidative stress. Aquat. Toxicol. 2011, 108, 18–22. [DOI] [PubMed] [Google Scholar]
- Enright C.; Culberson S. D., Salinity trends, variability, and control in the northern reach of the San Francisco Estuary. San Francisco Estuary Watershed Sci. 2010, 7 (2). [Google Scholar]
- Palace V. P.; Spallholz J. E.; Holm J.; Wautier K.; Evans R. E.; Baron C. L., Metabolism of selenomethionine by rainbow trout (Oncorhynchus mykiss) embryos can generate oxidative stress. Ecotoxicol. Environ. Saf. 2004; 58, 17–21. [DOI] [PubMed] [Google Scholar]
- Misra S.; Hamilton C.; Niyogi S. Induction of oxidative stress by selenomethionine in isolated hepatocytes of rainbow trout (Oncorhynchus mykiss). Toxicol. In Vitro 2012, 621–629. [DOI] [PubMed] [Google Scholar]
- Miller L. L.; Wang F.; Palace V. P.; Hontela A. Effects of acute and subchronic exposures to waterborne selenite on the physiological stress response and oxidative stress indicators in juvenile rainbow trout. Aquat. Toxicol. 2007, 83, 263–271. [DOI] [PubMed] [Google Scholar]
- Hoffman D. J.; Heinz G. H.; LeCaptain L. J.; Eisemann J. D.; Pendleton G. W. Toxicity and oxidative stress of different forms of organic selenium and dietary. Arch. Environ. Contam. Toxicol. 1996, 311120–7. [DOI] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13289–102. [DOI] [PubMed] [Google Scholar]
- Kirchen R. V.; West W. R.. The Japanese Medaka: Its Care and Development; Carolina Biological Supply Company, . [Google Scholar]
- Schlenk D.; Zubcov N.; Zubcov E. Effects of salinity on the uptake, biotransformation, and toxicity of dietary seleno-L-methionine to rainbow trout. Toxicol. Sci. 2003, 75, 309–313. [DOI] [PubMed] [Google Scholar]
- Farwell A.; Nero V.; Croft M.; Bal P.; Dixon D. G. Modified Japanese medaka embryo-larval bioassay for rapid determination of developmental abnormalities. Arch. Environ. Contam. Toxicol. 2006, 514600–7. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Fernald R. D. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 2005, 1281047–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T. D.; Livak K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 361101–8. [DOI] [PubMed] [Google Scholar]
- Jentzsch A. M.; Bachmann H.; Furst P.; Biesalski H. K. Improved analysis of malondialdehyde in human body fluids. Free Radical Biol. Med. 1996, 202251–6. [DOI] [PubMed] [Google Scholar]
- Inoue K.; Takei Y. Diverse adaptability in oryzias species to high environmental salinity. Zool. Sci. 2002, 197727–734. [DOI] [PubMed] [Google Scholar]
- Chen G. P.; Ziegler D. M. Liver microsome and flavin-containing monooxygenase catalyzed oxidation of organic selenium compounds. Arch. Biochem. Biophys. 1994, 312, 566–72. [DOI] [PubMed] [Google Scholar]
- Schlenk D.; Peters L. D.; Livingstone D. R. Correlation [corrected] of salinity with flavin-containing monooxygenase activity but not cytochrome P450 activity in the euryhaline fish (Platichthys flesus). Biochem. Pharmacol. 1996, 52, 815–8. [DOI] [PubMed] [Google Scholar]
- El-Alfy A.; Larsen B.; Schlenk D. Effect of cortisol and urea on flavin monooxygenase activity and expression in rainbow trout, Oncorhynchus mykiss. Mar. Environ. Res. 2002, 54, 275–8. [DOI] [PubMed] [Google Scholar]
- Spallholz J. E.; Palace V. P.; Reid T. W. Methioninase and selenomethionine but not Se-methylselenocysteine generate methylselenol and superoxide in an in vitro chemiluminescent assay: Implications for the nutritional carcinostatic activity of selenoamino acids. Biochem. Pharmacol. 2004, 673547–54. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Jiang C.; Lü J. Induction of caspase-mediated apoptosis and cell-cycle G1 arrest by selenium metabolite methylselenol. Mol. Carcinogen. 2002, 343113–120. [DOI] [PubMed] [Google Scholar]
- Hackett C.; Linley-Adams M.; Lloyd B.; Walker V. Plasma malondialdehyde: A poor measure of in vivo lipid peroxidation. Clin. Chem. 1988, 341208. [PubMed] [Google Scholar]
- Wu M.; Shariat-Madar B.; Haron M. H.; Khan I. A.; Dasmahapatra A. K. Ethanol-induced attenuation of oxidative stress is unable to alter mRNA expression pattern of catalase, glutathione reductase, glutathione-S-transferase (GST1A), and superoxide dismutase (SOD3) enzymes in Japanese rice fish (Oryzias latipes) embryogenesis. Comp. Biochem. Physiol., C: Comp. Pharmacol. 2011, 1531159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zhou Q. Dose- and time-related changes in aerobic metabolism, chorionic disruption, and oxidative stress in embryonic medaka (Oryzias latipes): Underlying mechanisms for silver nanoparticle developmental toxicity. Aquat. Toxicol. 2012, 124–125, 238–46. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Zhang H.; Dong Y.; Park Y. M.; Ip C. Endoplasmic reticulum stress signal mediators are targets of selenium action. Cancer. Res. 2005, 65199073–9. [DOI] [PubMed] [Google Scholar]
- Cullinan S. B.; Diehl J. A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 383317–332. [DOI] [PubMed] [Google Scholar]
- Nair S.; Xu C.; Shen G.; Hebbar V.; Gopalakrishnan A.; Hu R.; Jain M. R.; Liew C.; Chan J. Y.; Kong A. N. Toxicogenomics of endoplasmic reticulum stress inducer tunicamycin in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Toxicol. Lett. 2007, 168, 21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribb A. E.; Peyrou M.; Muruganandan S.; Schneider L. The endoplasmic reticulum in xenobiotic toxicity. Drug. Metab. Rev. 2005, 37, 405–442. [DOI] [PubMed] [Google Scholar]
- DuRose J. B.; Tam A. B.; Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol. Biol. Cell 2006, 1773095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo V. H.; Pihan P.; Vidal R. L.; Hetz C. Role of the unfolded protein response in organ physiology: Lessons from mouse models. IUBMB Life 2013, 6512962–75. [DOI] [PubMed] [Google Scholar]
- Reimold A. M.; Iwakoshi N. N.; Manis J.; Vallabhajosyula P.; Szomolanyi-Tsuda E.; Gravallese E. M.; Friend D.; Grusby M. J.; Alt F.; Glimcher L. H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 4126844300–7. [DOI] [PubMed] [Google Scholar]
- Reimold A. M.; Etkin A.; Clauss I.; Perkins A.; Friend D. S.; Zhang J.; Horton H. F.; Scott A.; Orkin S. H.; Byrne M. C.; Grusby M. J.; Glimcher L. H. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 142152–7. [PMC free article] [PubMed] [Google Scholar]
- Han X.; Zhou J.; Zhang P.; Song F.; Jiang R.; Li M.; Xia F.; Guo F. J. IRE1alpha dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development. Cell. Signalling 2013, 25112136–46. [DOI] [PubMed] [Google Scholar]
- Sha H.; He Y.; Chen H.; Wang C.; Zenno A.; Shi H.; Yang X.; Zhang X.; Qi L. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell. Metab. 2009, 96556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A.; Ochiai K.; Kondo S.; Tsumagari K.; Murakami T.; Cavener D. R.; Imaizumi K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem. 2011, 28664809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng W. C.; Lee W. T.; Hsu W. M.; Chang B. E.; Lee H. Role of glucose-regulated Protein 78 in embryonic development and neurological disorders. J. Formosan Med. Assoc. 2011, 1107428–37. [DOI] [PubMed] [Google Scholar]
- Ishikawa T.; Taniguchi Y.; Okada T.; Takeda S.; Mori K. Vertebrate unfolded protein response: Mammalian signaling pathways are conserved in Medaka fish. Cell. Struct. Funct. 2011, 362247–59. [DOI] [PubMed] [Google Scholar]
- Ishikawa T.; Okada T.; Ishikawa-Fujiwara T.; Todo T.; Kamei Y.; Shigenobu S.; Tanaka M.; Saito T. L.; Yoshimura J.; Morishita S.; Toyoda A.; Sakaki Y.; Taniguchi Y.; Takeda S.; Mori K. ATF6alpha/beta-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol. Biol. Cell 2013, 2491387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrook D. M.; Long A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 343286–97. [PubMed] [Google Scholar]
- Iijima N.; Yokoyama T. Apoptosis in the medaka embryo in the early developmental stage. Acta Histochem. Cytochem. 2007, 4011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D.; Honarpour N.; Wang X. Apoptosis in neural development and disease. Annu. Rev. Neurosci. 2000, 23, 73–87. [DOI] [PubMed] [Google Scholar]