

Abstract
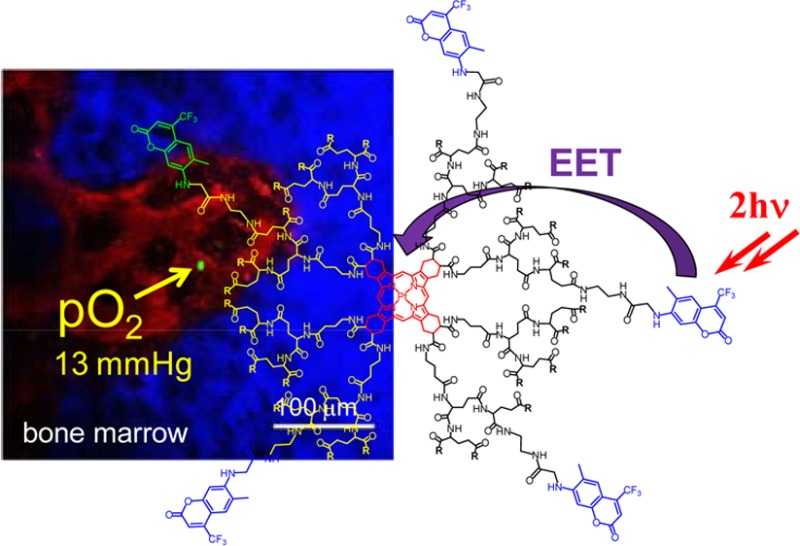
Recent development of two-photon phosphorescence lifetime microscopy (2PLM) of oxygen enabled first noninvasive high-resolution measurements of tissue oxygenation in vivo in 3D, providing valuable physiological information. The so far developed two-photon-enhanced phosphorescent probes comprise antenna-core constructs, in which two-photon absorbing chromophores (antenna) capture and channel excitation energy to a phosphorescent core (metalloporphyrin) via intramolecular excitation energy transfer (EET). These probes allowed demonstration of the methods’ potential; however, they suffer from a number of limitations, such as partial loss of emissivity to competing triplet state deactivation pathways (e.g., electron transfer) and suboptimal sensitivity to oxygen, thereby limiting spatial and temporal resolution of the method. Here we present a new probe, PtTCHP-C307, designed to overcome these limitations. The key improvements include significant increase in the phosphorescence quantum yield, higher efficiency of the antenna-core energy transfer, minimized quenching of the phosphorescence by electron transfer and increased signal dynamic range. For the same excitation flux, the new probe is able to produce up to 6-fold higher signal output than previously reported molecules. Performance of PtTCHP-C307 was demonstrated in vivo in pO2 measurements through the intact mouse skull into the bone marrow, where all blood cells are made from hematopoietic stem cells.
The ability to quantify molecular oxygen (O2) in vivo in real time with high spatial and temporal resolution has long been a much desired objective in biomedical research.1 Oxygen is transported throughout the body by blood, bound to hemoglobin inside red blood cells. After dissociation from the carrier, it reaches its consumption sites, mitochondria, by diffusion, crossing through vessel walls and cellular membranes. While the hemoglobin-bound oxygen can be quantified in vivo using, for example, differences between the absorption spectra of oxy- and deoxyhemoglobin,2 detection of free unbound oxygen is a much more challenging problem. Yet it is the unbound oxygen that is utilized in cellular respiration; and in disease, the balance between the bound and unbound oxygen can be compromised due to impaired delivery and/or consumption. It follows that direct measurements of free oxygen are necessary to quantify tissue metabolism and evaluate physiology of diseased states.3
The phosphorescence quenching method4 has been developed specifically with the above goal in mind and used in numerous in vivo applications: from fiber optic measurements5−7 to wide field imaging,8−11 microscopy,12−14 and tomography.15,16 The method relies on the ability of oxygen to quench emissive triplet states of exogenous probes dissolved in biological fluids.17 Measurements by phosphorescence can be made absolute if probes are able to retain their calibration parameters when placed in biological environment in vivo.18−20 Such probes can be delivered into blood plasma or extravasuclar space, where they report quantitatively on oxygen concentration.17 Due to the presence of large hydrophilic coats, quantitative oxygen probes cannot permeate cellular membranes. On the other hand, a number of recently reported membrane-permeable phosphorescent constructs21−25 successfully accumulate inside cells but can only reflect relative changes in oxygenation, which is due to the intrinsic uncertainty of their calibration in complex intracellular environments.
One recently developed mode of the phosphorescence quenching oximetry is based on the combination of phosphorescence with two-photon excitation, termed two-photon (2P) phosphorescence lifetime microscopy (2PLM).26,27 Increased depth of tissue penetration by infrared light along with confinement of excitation to the vicinity of the laser focus are especially attractive features of this approach, as they minimize production of toxic byproducts of the triplet quenching reaction (e.g., singlet oxygen) and allow high-resolution sampling of oxygenation at depth in tissue. Utility of 2PLM has been recently demonstrated in several tissue imaging applications,28−33 warranting further development and optimization of the method.
Typically, biological oxygen sensors are built around Pt or Pd porphyrins, which possess high emissivity and suitable triplet lifetimes,18,34,35 but very low two-photon absorption (2PA) cross sections.36 To circumvent this problem, we have developed an approach based on antenna-core constructs, in which phosphorescence emission is coupled to 2PA via intramolecular excitation energy transfer (EET).36,37 The antenna-core probes are encapsulated into dendrimers, which control the rate of oxygen diffusion to phosphorescent chromophores and regulate efficiency of quenching. External coating of the dendrimers with poly(ethylene glycol) layers prevents probe aggregation and interactions with endogenous biomolecules. Optimization of such probes requires fine-tuning of several interrelated parameters in order to obtain maximal photon output and desirable analyte sensitivity.37−39
It is important to mention that boosting output of two-photon (2P) phosphorescent probes is especially important in view of their long emission lifetimes (microseconds). In scanning microscopy applications, imaging speed is proportional to the number of photons acquired by the detector per unit time. The latter in turn is proportional to the product of the emission rate and emission quantum yield. Since the triplet (phosphorescence) lifetime is rather long (and the emission rate is low), high phosphorescence quantum yield becomes especially important for fast detection.
The first practical two-photon oxygen probe, PtP-C343, was based on a phosphorescent Pt tetraarylporphyrin (PtP) as a core and Coumarin-343 (C343) moieties, acting as 2PA antennae.27 Although PtP-C343 has been quite successful in demonstrating the methods’ potential,28−33 it has several limitations, such as partial loss of emissivity to competing quenching pathways (e.g., triplet electron transfer) and suboptimal signal dynamic range. Similar and/or other problems have been encountered in several later reported probes,20,40,41 of which one was successfully tested in vivo.20 In this paper, we describe our efforts to overcome the above limitations, focusing on increasing phosphorescence quantum yield, improving efficiency of the antenna-core energy transfer, minimizing quenching of phosphorescence by undesirable intramolecular pathways, and optimizing the signal dynamic range. We disclose a new probe, PtTCHP-C307, whose performance was validated in vivo by performing the pO2 measurement through the intact mouse skull into the bone marrow, where all blood cells are formed from hematopoietic stem cells (HSCs) that are thought to reside in a hypoxic microenvironment.42−44
Experimental Methods
For description of standard procedures, animal protocols and compounds’ synthesis see the Supporting Information. To determine relative two-photon phosphorescence brightness spectra (for a definition of “two-photon brightness”, see Results and Discussion), a setup was constructed for time-resolved phosphorescence measurements under two-photon excitation. The output of a tunable femtosecond laser oscillator (80 MHz repetition rate, Chameleon Ultra II, Coherent) was passed through a half-wave plate and a polarizer for power adjustment and then through an electro-optical modulator (Atseva) to obtain 1 μs-long excitation gates (80 pluses per gate) at 1 kHz frequency. The contrast ratio of the modulator was 1000:1, and the on/off time was <1 ns. The gated beam was focused by a lens (f = 4 cm) at the center of a quartz cuvette (Starna Cells) containing solution of a sample. After the cuvette, the beam was directed at an optical power meter (FieldMaxII-TOP, Coherent), placed immediately after the cuvette compartment.
The sample solution was purged with Ar until phosphorescence lifetime became constant and kept under slight Ar pressure to ensure that no back-diffusion of oxygen occurred during the measurement. The phosphorescence was collected by a convex lens (2.5 cm diameter) placed immediately by the cell compartment at 90° relative to the direction of the excitation beam. The collimated emission was passed through two short-pass filters (RazorEdge SP 694 nm and SP 770 nm, Semrock) to remove scattered laser light and focused by another lens onto the aperture of a photomultiplier module (H7422P-50, Hamamatsu). The output of the PMT was measured across a resistor (2 kΩ) using a data acquisition board (USB-6361, National Instruments) and digitized at 1 MHz frequency. The board was controlled by an in-house program written in C/C++ (Qt, Nokia). Excitation gates (5000–10000) were averaged to obtain a decay, whose integrated intensity was normalized by (i) the overlap integral of the emission spectrum of the sample, quantum efficiency spectrum of the detector and the transmission spectra of the filters; (ii) concentration of the sample; (iii) the square of the refractive index of the solvent;45 and (iv) the square of the excitation flux. The latter was calculated assuming that the cross section of the beam was wavelength-independent. The femtoseconds pulse duration, measured by an aurocorrelator, varied in the range of 120–175 fs through the tunability range of the laser: 700–1050 nm. The average laser power was kept approximately constant throughout the excitation spectrum, typically 100 mW (1.25 nJ per pulse), as measured with the modulator turned off. At the 0.001 duty cycle (gate duration, 1 μs; modulator rep rate, 1 kHz), the average power on the sample with the modulator on was 0.1 mW. The normalized integrated decay intensity was plotted against the excitation wavelength (i.e., central wavelength of the envelope) to give the two-photon phosphorescence brightness spectrum.
In vivo imaging experiments were performed using a custom-built microscope with a video-rate laser-scanning 2P intravital imaging arm and a point-detection 2P phosphorescence lifetime measurement arm.33 The excitation source was a pulsed femtosecond laser (MaiTai, SpectraPhysics, 80 MHz repetition rate). It was tuned to 820 nm and used to visualize the bone by second harmonic generation (SHG) and the bone marrow vasculature by 2P-excited fluorescence of a macromolecular vascular dye (Rhodamine dextran, MW 70 kDa). For excitation of PtTCHP-C307, the laser was tuned to 765 nm, and the beam, focused by a lens (f = 5 cm), was scanned by a galvanometer mirror (6220H, Cambridge Technology) across a 100 μm-wide slit (NT38–560, Edmund Optics), positioned in the focal plane of the lens, to generate a microseconds-long excitation pulse. After passing through the slit, the beam was focused by a water immersion, infinity corrected, near-infrared-coated objective lens (UPLSAPO 60XW, Olympus America Inc.; 60 × 1.2 NA; working distance, 280 μm). The generated phosphorescence was collected by the same objective lens and directed to a photomultiplier tube after passing through several optical filters. The arrival time of individual photons after the pulse excitation was recorded using a custom photon counting circuit. The histogram of the photon arrival times was then analyzed to obtain the phosphorescence lifetime.
Results and Discussion
The state diagram underlying the principles of 2P-enhanced antenna-core phosphorescent probes is shown in Figure 1a. Two-photon excitation first populates a 2P-accessible state of the antenna (AS2P), which may or may not be the same as its first excited singlet state (AS1). State AS2P, if different from AS1, rapidly internally converts to AS1. The antenna chromophore(s) is positioned sufficiently close to the phosphorescent core, so that the excitation energy transfer (EET), presumably via the Förster dipole–dipole mechanism, efficiently populates the excited singlet state of the core (CS1). The following intersystem crossing within the core (CS1→CT1) produces the triplet state (CT1), which either emits phosphorescence or undergoes quenching by molecular oxygen. For imaging applications, it is essential that minimal losses are encountered in the energy cascade leading to the final emissive state (CT1). It is also important that the quantum yield of phosphorescence from the CT1 state is not diminished due to competing deactivation processes [e.g., triplet electron transfer (ET) and subsequent charge recombination (CR),38,46 shown in Figure 1a with dashed lines].
Figure 1.

(a) State energy diagram of the processes occurring in antenna-core two-photon-enhanced phosphorescent probes. EET, excitation energy transfer; ic, internal conversion; isc, intersystem crossing; ET, electron transfer; CR, charge recombination. See text for definition of all involved states. (b) Chromophores used in construction of probe PtTCHP-C307: coumarin-307 (C307) and PtTCHP (2, 3).
The above scheme imposes several requirements on the components of the probe. First, the core chromophore (C) must possess bright oxygen-sensitive phosphorescence. Second, antenna (A) must have a sufficiently high two-photon absorption (2PA) cross section, strong radiative singlet decay, and emission spectrum overlapping with absorption of the core for maximally efficient ETT. Third, the redox potentials of the core and the antenna must be tuned to eliminate the possibility of intramolecular ET between the long-lived triplet state of the core (CT1) and the ground state of the antenna (AS0) with formation of a dark charge separated state (CS). The last but not least, components of the probe must enable efficient synthetic assembly. Selection of the probe components was based on our previous spectroscopic and electrochemical studies.37−39,47
Choice of Chromophores
The majority of phosphorescent probes are based on Pt or Pd complexes of meso-tetraarylporphyrins.17,48 These tetrapyrroles can be readily synthesized with a variety of functional groups; however, inclusion of meso-aryl substituents into the porphyrin macrocycle is known to diminish triplet emissivity.49−52 On the other hand, Pt tetracyclohexenoporphyrin (PtTCHP, 2; Figure 1b), which also has eight functionalizable carboxylic groups, is free of that problem. The phosphorescence quantum yield of 2 in deoxygenated organic solvents (e.g., in dimethylacetamide, DMA) reaches as high as 0.43 (compared to 0.07 for Pt tetraphenylporphyrin), and its triplet lifetime of ∼100 μs (in deoxygenated solutions at 22 °C) ensures excellent oxygen sensitivity (Table 1).
Table 1. Photophysical Data for Probe Components.
| absorption | emission |
|||
|---|---|---|---|---|
| compound | solvent | λmax (nm) (lg ε) | λmax (nm) | emission type (ϕ/τ) (s)b,c |
| 2 | DMA | 382 (5.51)a, | 647 | p, 0.43/99 × 10–6 |
| 535 (4.85)a | ||||
| 3 | DMA | 382, 535 | 647 | p, 0.42/98 × 10–6 |
| C307 | DMF | 399 | 488 | f, 0.91/5.1 × 10–9 |
| 5 | DMF | 376 | 498 | f, 0.01 |
| 5-NH3+CF3CO2– | H2O | 363 | 509 | f, <0.01 |
| 6 | EtOH | 387 | 484 | f, 0.74 |
| 8 | EtOH | 387 (4.27) | 485 | f, 0.93/5.0 × 10–9 |
| 9d | H2O | 375 | 493 | f, 0.80/5.3 × 10–9 |
Measured in CH2Cl2.
Emission quantum yields (ϕ) and lifetimes (τ) of phosphorescence were measured in deoxygenated solutions.
Quantum yields were determined relative to the fluorescence of Rhodamine 6G (φfl = 0.95 in EtOH).53 When measured against fluorescence of Rhodamine 6G, free-base tetraarylporphyrin in benzene, commonly used as a standard in porphyrin spectroscopy, exhibits fluorescence quantum yield of 0.055 and not 0.11 as reported previously.54
Compound 9 here refers to compound 8 (Scheme S2 of the Supporting Information) with Boc protection removed.
2 was synthesized by insertion of Pt into the corresponding free-base porphyrin (Scheme S1 of the Supporting Information), which appeared as an intermediate in an earlier published synthesis of meso-unsubstituted tetrabenzoporphyrins.47 Hydrolysis of the peripheral ester groups yielded octaacid 3, suitable for subsequent functionalization.
In our earlier studies, we have found that “good” 2P dyes usually are also very potent quenchers of porphyrin triplet states38,39 by way of ET/CR processes (Figure 1a). Among tested chromophores, Coumarin-343 (C343) was found to be the least efficient quencher, and it was paired with Pt meso-tetra-4-alkoxyphenylporphyrin (PtP) for construction of PtP-C343.27 However, diffusion-based quenching experiments (see ref (39) for experimental details) revealed that C343 strongly quenches phosphorescence of 2, indicating that the reduction potential of PtTCHP is lower than that of PtP.55 Therefore, we turned our attention to antenna dyes more prone to oxidation.
Coumarin 307 (C307; Figure 1b) is a commercially available bright fluorescent dye, whose photophysical properties (Figure 2, panels a and b, Table 1) have been thoroughly studied.57−60 2PA of C307 (Figure 2c) is somewhat weaker than that of C343 (see the Supporting Information for 2PA spectrum), but its fluorescence overlaps well with absorption of 2 (Figure 2d), ensuring efficient EET. Due to the presence of electron-withdrawing CF3 group, the oxidation potential of C307 is relatively high [i.e., 1.41 V (measured vs NHE)].59 Indeed, phosphorescence of porphyrin 2 was found to be completely insensitive to C307, even when the latter was present in millimolar quantities. In contrast, C343 caused noticeable quenching of the phosphorescence of 2 already at micromolar levels.
Figure 2.

(a) Absorption and (b) normalized emission spectra of porphyrin 2 and C307 in DMF. For emission: λex = 531 nm (2) and 365 nm (C307). See the Supporting Information for details of photophysical measurements. (c) 2PA spectrum of C307 in DMF. This spectrum was measured by the relative fluorescence method, as described previously.56 (d) Emission spectrum of C307 and absorption spectrum of 2, normalized by the peak emission (488 nm) value for C307 and the peak absorption at the Q-band maximum (535 nm) for 2, respectively.
It has been reported in the literature that fluorescence of 7-aminocoumarins depends strongly on the degree of substitution in the amino group.58 For example, alkylation of C307 with tBu-bromoacetate (Scheme S2 of the Supporting Information) gave derivative 4, whose fluorescence quantum yield was drastically reduced compared to that of parent coumarin C307 (ϕfl = 0.01 vs 0.91, Table 1). To preserve bright fluorescence and at the same time enable functionalization, an analogue of C307 was synthesized (coumarin 8) with monoalkylated amino group (Scheme 1), having a substituent terminated by protected carboxylic acid residue. Coumarin 6 was obtained by Pechmann condensation,61,62 followed by monoalkylation with tBu-bromoacetate, cleavage of tBu ester, and amidation with mono-Boc-protected ethylenediamine. Important for our application, the deprotected (acid) form of 8 retained its high fluorescence quantum yield in water (ϕfl = 0.8).
Scheme 1. Synthesis of Probe PtTCHP-C307.

Reagents and conditions: (a) ZnCl2, CF3COCH2CO2CH2CH3, ethanol, reflux, overnight (74%); (b) BrCH2CO2tBu, DMF, 80 °C, overnight (65%); (c) H2N(CH2)2NHCO2tBu, HBTU/DIPEA, DMF, rt, overnight (70%); (i) H2N(CH2)3CO2Et·HCl, HBTU/DIPEA, DMF, rt, overnight (85%); (ii) KOH, THF/CH3OH/H2O, rt, (80%); (iii) TFA, rt, 2 h; (iv) HBTU/DIPEA, DMF, rt, overnight (92%); (v) KOH, THF/CH3OH/H2O, rt (71%); (vi) (a) HBTU/DIPEA, DMF, rt, 5 h; and (vii) mPEG-NH2 (av MW ≈ 1000), HBTU/DIPEA, DMF, rt, overnight (85% for 2 steps).
Excitation Energy Transfer (EET)
The efficiency of the EET between C307 and PtTCHP was evaluated based on the Förster theory, using the spectroscopic data for coumarin 8 and porphyrin 2 in DMF (Figure 3a) and assuming random orientation of the transition dipole moment vectors (orientation factor κ2 = 2/3). It is important to note that the acceptor in our case is a highly symmetrical metalloporphyrin that has not one but two orthogonally polarized degenerate transitions, Qx and Qy, near 540 nm. Consequently, the value of κ2 is expected to be higher than 2/3, as both Qx and Qy can interact with the donor (C307). At the same time, the oscillator strength for each of the transitions should be one-half of that measured for the Q-band of PtTCHP. The overall effect of this arrangement should be a larger apparent EET efficiency than for a single acceptor transition dipole moment. Nevertheless, even in this simplified estimate, the Förster radius R0 was found to be quite large at R0 = 44.9 Å, suggesting that the EET between coumarin and porphyrin in a covalent assembly, where distances between the chromophores do not exceed 1–2 nm, should be very efficient.
Figure 3.
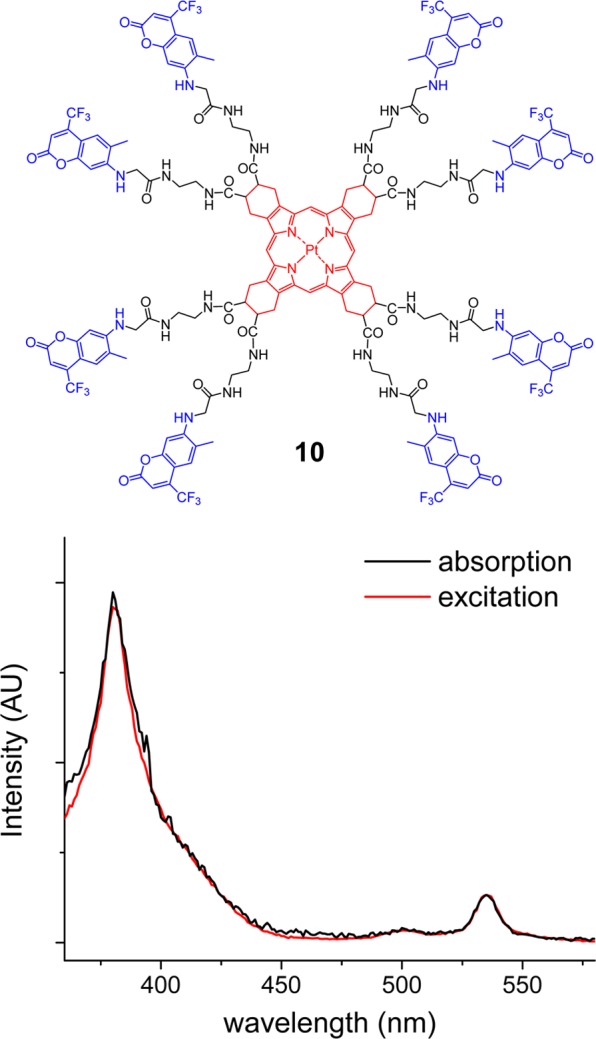
Structure and absorption and excitation spectra of model compound 10 in DMF. The excitation spectrum is recorded for the emission maximum at 647 nm.
For experimental evaluation of EET, we first constructed model compound 10 (Figure 3), in which eight coumarin moieties are attached directly to the peripheral carboxyl groups on the porphyrin via short ethylenediamine linkers (Scheme S1 of the Supporting Information). This molecule was much easier to synthesize than the final dendritic oxygen probe (see below), while it permitted testing of the key photophysical processes underlying the probes’ function.
Nearly complete lack of the coumarin fluorescence upon excitation of 10 at λex = 440 nm revealed that the excitation energy in this model molecule is fully directed from the donor chromophore into nonradiative channels. Almost a complete match between the absorption and excitation spectra (λem = 647 nm, Figure 3) indicated that fluorescence quenching was indeed due to a very efficient EET (>99%). For this analysis, the excitation and the absorption spectra were normalized by the maxima of the lowest-allowed transition [i.e., CS0→CS1-Q-band of PtTCHP (λmax = 535 nm)]. It is known that in Pt porphyrins S1→T1 intersystem crossing occurs with nearly unity efficiency.63,64 Therefore, all the energy that reached the CS1 level could be considered utilized in the formation of the emissive CT1 state (Figure 1). Importantly, concentrations of the samples for recording of the excitation spectra were kept very low, such that the absorption maxima in the Soret band region (λmax = 380 nm) were below 0.05 o.d. (The molar extinction of the porphyrin near 400 nm is very high, ε > 3 × 105 M–1 cm–1). This precaution was necessary in order to avoid inner filter effects that otherwise may have significantly distorted the excitation spectra.
The phosphorescence quantum yield of 10 was found to be almost identical to that of porphyrin 2, establishing that the triplet ET in 10 is not an effective quenching pathway despite the presence of eight coumarin moieties directly linked to the porphyrin core. Taken together, these measurements confirmed that PtTCHP and C307 indeed are suitable as components for construction of a practical two-photon phosphorescent oxygen probe.
Two-Photon-Enhanced Phosphorescent Probe PtTCHP-C307
The structure and the synthesis of probe PtTCHP-C307 (15) are shown in Scheme 1. In the first step, the carboxyl groups on porphyrin 3 were amended by 4-aminobutyrate linkers to facilitate coupling of bulky dendrons. This strategy proved very efficient previously in the synthesis of other dendritic probes,19 serving to decrease the steric interference between the adjacent coupling sites on the porphyrin.
Pt porphyrin 11 was dendrimerized with eight Gen 2 glutamic dendrons. The role of the dendrimer has been discussed extensively in our previous publications.17,18,36 In brief, dendritic encapsulation prevents aggregation of porphyrins, enhances their solubility, and enables control over the rate of oxygen diffusion to the excited state triplet chromophore. In addition, positioning of 2P antenna at the periphery of the dendrimer keeps antenna chromophores at a distance from the core, further decreasing probability of the phosphorescence quenching by triplet ET.27,37 The choice of polyglutamic dendrimers in the present case was motivated by their relatively moderate shielding efficiency.65 One of our goals was to achieve optimal dynamic range of phosphorescence lifetimes throughout the range of physiological oxygen concentrations (0–160 mmHg). While no shielding of the porphyrin would result in a nearly complete quenching and consequently very low photon output already at ambient oxygen concentrations, too much shielding would narrow the dynamic range of lifetimes and thus decrease oxygen sensitivity. On the basis of our previous studies27,65,66 and considering ∼80–100 μs triplet lifetime of PtTCHP’s (at zero-oxygen), moderate shielding by glutamic dendrimers seemed more appropriate than by aryl-glycine (AG) dendrons used in the synthesis of PtP-C343.27
Gen 2 glutamic dendrons65 were linked to the porphyrin core using standard HBTU/DIPEA coupling method, followed by hydrolysis of the peripheral ester groups. The resulting Pt porphyrin-dendrimer acid (14) exhibits very high aqueous solubility, resembling in this regard other polyglutamic porphyrin-dendrimers, but having much brighter phosphorescence. C307 antenna fragments were linked divergently to the termini of 14, and the remaining carboxyl groups were modified with monomethoxypolyetheleneglycol amines (mPEG-NH2, av MW 1000) in a single-pot reaction, using the same HBTU/DIPEA coupling method. After purification of 15 by size-exclusion chromatography (Biorad SX-1, THF), UV–vis absorption spectra revealed that each probe molecule on average contains 4–5 residues of C307. MALDI analysis of the final material showed distribution of masses with the center mass corresponding to the dendrimer bearing 27 out of ∼33 possible PEG residues. Similar PEGylation efficiency has been seen previously in other dendritic probes.19
The photophysical constants of the new probe PtTCHP-C307 (15) and its precursor 14 are summarized in Table 2 and Figure 4.
Table 2. Photophysical Data for Porphyrin-Dendrimer 14 and Probe 15.
| absorption | emission |
|||
|---|---|---|---|---|
| compound | solvent | λmax (nm) | λmax (nm) | ϕphos; τ (s)a |
| 14 | DMF | 383, 536 | 647 | 0.24; 98 × 10–6 |
| 14 | H2O | 380, 534 | 647 | 0.20; 81 × 10–6 |
| 15 | DMF | 383, 536 | 647 | 0.25; 78 × 10–6 |
| 15 | H2O | 380, 535 | 647 | 0.20; 88 × 10–6 |
Measured in deoxygenated solutions at 22 °C.
Figure 4.

(a) Absorption and (b) emission spectra of probe 15 (phosphate buffer, pH 7.2). (b) Phosphorescence spectrum was recorded using deoxygenated solution, λex = 430 nm. (c) Relative two-photon brightness spectra of compounds 14, 15, and PtP-C34327 in deoxygenated aqueous solutions. (d) Stern–Volmer oxygen quenching plots (τ–1 vs pO2, where τ is the phosphorescence lifetime) of 15 in phosphate buffer (pH 7.2), in the same buffer containing BSA (2%) and in mouse blood plasma.
The absorption spectrum of 15 (Figure 4a) closely resembles superposition of a weighted spectrum of C307 and PtTCHP, indicating no significant ground or excited state electronic interactions between the chromophores. Similar to 10, excitation of the antenna in 15 at 430 nm (off the Soret peak of PtTCHP) in deoxygenated solution produced strong porphyrin phosphorescence (Figure 4b) but only a small residual coumarin fluorescence (ϕfl < 0.01), suggesting that the efficiency of EET in 15 is quite high. Indeed, comparison of the absorption and excitation spectra of 15 in the same manner as the described for 10 revealed that EET in the probe molecule occurs with ∼97% efficiency.
The phosphorescence quantum yields of both 15 and 14 were found to be practically the same but somewhat lower than the quantum yield of the core PtTCHP (i.e., 0.25 vs 0.43 in deoxygenated DMF). In water, additional reduction in the emission yield was observed similar to other phosphorescent porphyrins.18,19 Nevertheless, both 15 and 14 remain by far the brightest emitters among known oxygen probes.
To evaluate performance of probe 15 under 2P excitation, we compared its phosphorescence output to that of the “nonenhanced” dendritic porphyrin 14 as well as to the previously published probe PtP-C343.27 Integrated phosphorescence intensity under 2P excitation plotted against excitation wavelength gave the 2P phosphorescence brightness spectra (Figure 4c), directly suitable for comparison between different probes (see Experimental for details). Importantly, the signal was confirmed to exhibit quadratic power dependence (i.e., slope of the log–log plot = 2.00 ± 0.05) at each excitation wavelength. 2P brightness here is defined as a parameter proportional to the product of the 2P excitation cross section and emission quantum yield.67 Note that the latter may not be constant throughout the excitation range. For example, for 2P-enhanced probes, such as 15, the emission quantum yield is the product of the quantum yields of all processes connecting the initially populated AS2P state to the final emissive state CT1 (Figure 1). In the region where 2PA is dominated by the antenna, this includes the quantum yield of the EET.
As expected, “nonenhanced” porphyrin 14 shows negligible 2PA in the Soret band region (S0→S2), where its linear absorption is extremely strong (linear spectrum of 14 is shown in Figure 4c by a dashed line). In accordance with the parity selection rules, for centrosymmetrical chromophores (such as PtTCHP), electrical-dipole-allowed transitions (g→u) are not accessible for two-photon excitation,68,69 which instead may occur into states of g symmetry (g→g). Noteworthy is a small peak at 995 nm, which coincides with the vibronic Q01 band in the linear absorption spectrum and, therefore, may be due to an unsymmetrical vibration.
The enhancement of the excitation efficiency in the range of 730–900 nm for 15, which corresponds to the 2P absorption of C307 antenna, is apparent from Figure 4c. For example, at 750 nm, the increase in the signal due to the absorption of C307 and EET is ∼14-fold compared to “nonenhanced” porphyrin 14. Notably, moving excitation to shorter wavelengths results in lowering of the power dependence order due to the increasing contribution of linear S0→T1 absorption. In Pt porphyrins, spin-forbidden S0→T1 transitions may gain significant dipole strength due to a very strong spin–orbit coupling,38,52 the effect also responsible for their high triplet emissivity.
Compared to the previously designed probe PtP-C343, 15 shows substantial increase in the signal intensity in the near-Soret band region. In all so-far published neuroimaging studies,28−30,32 PtP-C343 was excited at 840 nm (i.e., away from its 2PA maximum (near 920 nm). Nevertheless, even such off-peak excitation was adequate to sample oxygenation of regions as deep as 400–500 μm under the tissue surface. To that end, when excited at 840 nm probe 15 produces ∼3 times stronger phosphorescence than PtP-C343, and at 760 nm its output is more than 6 times higher than that of PtP-C343. It is worth noting that the gain in the signal comes mainly from the increase in the phosphorescence quantum yield, which is the key parameter in 2PLM and other scanning microscopy applications. Higher 2PA cross section helps produce the emissive excited state using less laser power, but under saturation conditions, signal output is determined solely by the emission yield.27,70,71
Oxygen sensitivity of 15 was measured in a standard phosphate-buffered solution as well as in a solution containing bovine serum albumin (BSA, 2%) and directly in the mouse blood plasma. Albumin is present in the blood plasma in ∼2% concentration and is extremely effective in binding various organic molecules, including porphyrins and other dyes. Binding to BSA dramatically changes oxygen quenching properties of phosphorescent probes,4,65 while lack of such changes can be considered a good indicator that the probe is not interacting with biological solutes; in vivo its calibration will be retained. PEGylation of dendrimers suppresses interactions with albumin and other biological macromolecules.18 It can be seen in Figure 4d that the Stern–Volmer oxygen quenching plot of 15 is completely insensitive to the presence of BSA. Furthermore, oxygen quenching parameters of the probe are unchanged in intact blood plasma, and such insensitivity to the environment is sustained at body temperatures (see the Supporting Information for probe calibration plots at 36.5 °C). Collectively, these data suggest that the probe can be used for quantitative oxygen measurements in vivo. The Stern–Volmer oxygen quenching constant of 15 was found to be ∼1200 mmHg–1 s–1 at 36.5 °C, which is ∼10 times higher than that of PtP-C343 under similar conditions. Therefore, 15 has much larger dynamic range (r) of lifetimes (r = τ0/τair, where τ0 and τair are the phosphorescence lifetimes at zero-oxygen and at air equilibrium, respectively) than PtP-C343 (rPtTCHP-C307 ≈ 18 vs RPtP-C343 ≈ 3) and higher oxygen sensitivity. In fact, the lifetime and the quantum yield of 15 are optimal for measurements in the pO2 range of 0–60 mmHg, but at higher pO2’s its phosphorescence becomes rather strongly quenched. Luckily, in the majority of biological settings tissue pO2 does not exceed 50 mmHg.
Application of Probe PtTCHP-C307 in Vivo
As a test-bed application, the new probe 15 was used to measure intravascular pO2 in bone marrow of a live mouse.
Physiological environment of the bone marrow is believed to play a critical role in the formation of all blood cells (leukocytes and erythrocytes) from stem and progenitor cells, the process called hematopoiesis. The hematopoietic stem cells (HSCs) are considered to reside in a relatively hypoxic environment despite the fact that the bone marrow is a highly vascularized tissue,33 with sinusoidal blood vessels occupying 25–30% of the tissue volume and spaced ∼46 μm apart.72 However, until recently,33 no direct measurements of pO2 in the HSC niche have been possible, leaving the question about the role of hypoxia for the HSC development unanswered. Here using two-photon phosphorescence lifetime microscopy and PtP-C307, we were able to show that the sinusoidal blood vessels are indeed poorly oxygenated (Figure 5), supporting the possibility that the HSC may reside in a vascular niche73 and yet be in a deeply hypoxic microenvironment. This result is fully in line with our recent more comprehensive measurements using probe PtP-C343,33 confirming that PtTCHP-C307 is well-suited for high-resolution in vivo oxygen microscopy. Furthermore, higher performance of PtTCHP-C307 enables faster data acquisition and allows for lower excitation energy, important advantages for in vivo imaging.
Figure 5.
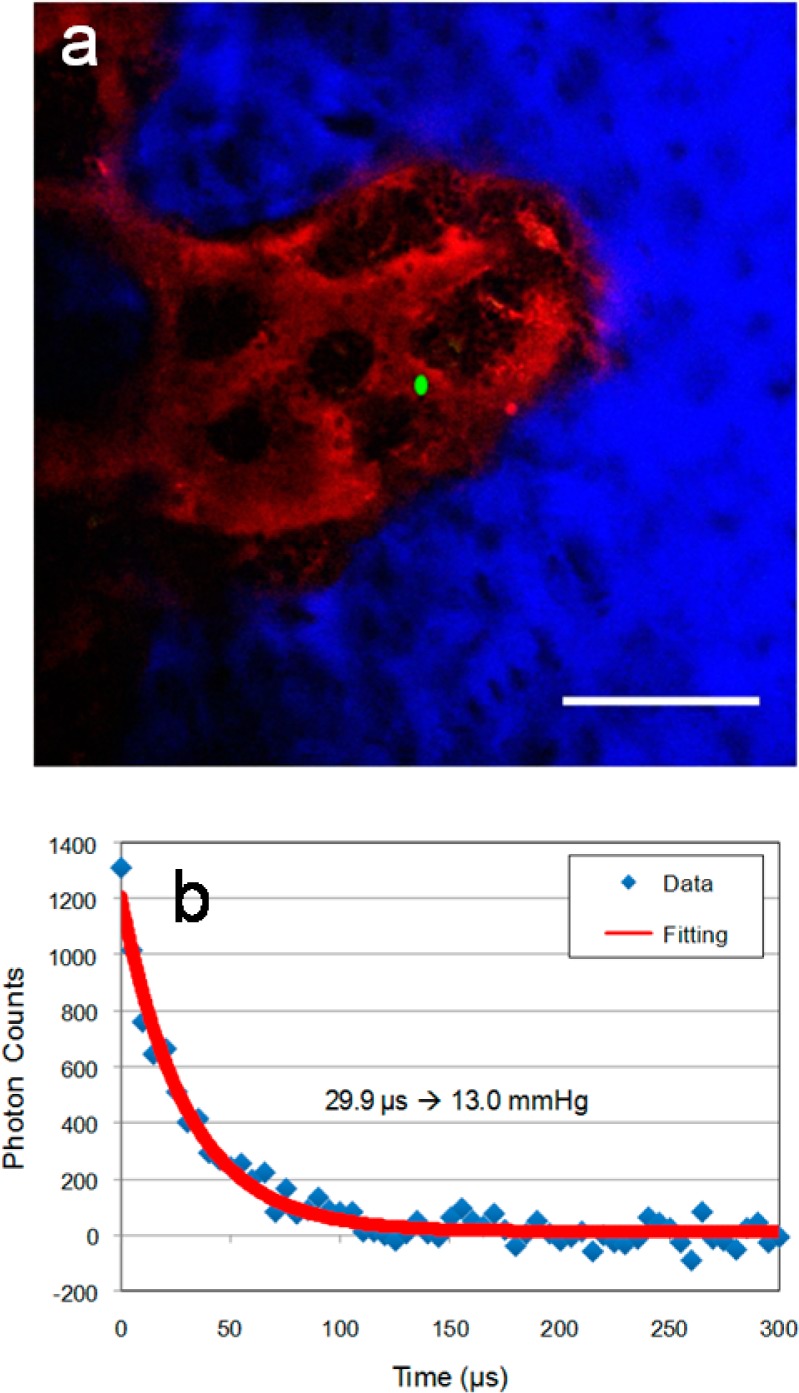
(a) Two-photon intravital image of bone marrow vasculature (red, Rhodamine-Dextran, 70 kDa) and bone (blue, Second Harmonic Generation) in the calvarium of a Nestin-GFP mouse (λex = 820 nm). Phosphorescence lifetime measurement using PtTCHP-C307 was performed at the location shown in green (green dot, λex = 765 nm). (b) Corresponding trace of the phosphorescence decay in location marked in (a). The measurement followed the protocol developed earlier.33 The laser beam was focused ∼80 μm under the bone surface. By scanning the beam across a slit (see Experimental Methods for details), a line trace ∼3.5 μM long was produced with total duration of ∼20 μs. Using such line-excitation allowed us to avoid triplet saturation effects, which may occur in stationary point-excitation.71 Two thousand scans were averaged to produce the trace of phosphorescence. The trace was fit with a single exponential function, and the pO2 value was generated by using an in vitro obtained calibration curve (37 °C and pH 7.2). Scale bar ∼100 μm.
Acknowledgments
Support of the Penn Medicine Neuroscience Center and the Penn Abramson Cancer Center are gratefully acknowledged. Fluorescence lifetime measurements were performed in the Regional Laser and Biomedical Technology Laboratories (RLBL) (NIH/NCRR Grant P41RR001348) with assistance of Dr. Thomas Troxler. Ms. Abigail Cember and Dr. Tomoyasu Mani are acknowledged for their help in measuring photophysical properties of probe PtTCHP-C307. The authors are grateful to Mr. Erich Beuerman and Prof. Mikhail Drobizhev of the Department of Physics of Montana State University for measurements of the two-photon absorption spectra of coumarines C307 and C343.
Supporting Information Available
Additional experimental details, description, and schemes of synthesis, NMR, and MALDI-TOF data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pittman R. N. Acta Physiol. 2011, 202, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobsis F. F. Science 1977, 198, 1264. [DOI] [PubMed] [Google Scholar]
- Wilson D. F. Am. J. Physiol.: Heart Circ. Physiol. 2008, 294, H11. [DOI] [PubMed] [Google Scholar]
- Vanderkooi J. M.; Maniara G.; Green T. J.; Wilson D. F. J. Biol. Chem. 1987, 262, 5476. [PubMed] [Google Scholar]
- Pawlowski M.; Wilson D. F. Adv. Exp. Med. Biol. 1992, 316, 179. [DOI] [PubMed] [Google Scholar]
- Mik E. G.; Johannes T.; Ince C. Am. J. Physiol. Renal Physiol. 2008, 294, F676. [DOI] [PubMed] [Google Scholar]
- Yu J.; Ramadeen A.; Tsui A. K. Y.; Hu X.; Zou L.; Wilson D. F.; Esipova T. V.; Vinogradov S. A.; Leong-Poi H.; Zamiri N.; Mazer C. D.; Dorian P.; Hare G. M. T. Anaesthesia 2013, 68, 723. [DOI] [PubMed] [Google Scholar]
- Rumsey W. L.; Vanderkooi J. M.; Wilson D. F. Science 1988, 241, 1649. [DOI] [PubMed] [Google Scholar]
- Vinogradov S. A.; Lo L.-W.; Jenkins W. T.; Evans S. M.; Koch C.; Wilson D. F. Biophys. J. 1996, 70, 1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakadžić S.; Yuan S.; Dilekoz E.; Ruvinskaya S.; Vinogradov S. A.; Ayata C.; Boas D. A. Appl. Opt. 2009, 48, D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonat R. D.; Kight A. C. Ann. Biomed. Eng. 2003, 31, 1084. [DOI] [PubMed] [Google Scholar]
- Hogan M. C. J. Appl. Physiol. 1999, 86, 720. [DOI] [PubMed] [Google Scholar]
- Golub A. S.; Pittman R. N. Am. J. Physiol.: Heart Circ. Physiol. 2008, 294, H2905. [DOI] [PubMed] [Google Scholar]
- Yaseen M. A.; Srinivasan V. J.; Sakadzic S.; Wu W.; Ruvinskaya S.; Vinogradov S. A.; Boas D. A. Opt. Express 2009, 17, 22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apreleva S. V.; Wilson D. F.; Vinogradov S. A. Appl. Opt. 2006, 45, 8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R. X.; Davis S. C.; Demers J. L. H.; Glaser A. K.; Gladstone D. J.; Esipova T. V.; Vinogradov S. A.; Pogue B. W. J. Biomed. Opt. 2013, 18, 50503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov S. A.; Wilson D. F.. Porphyrin-dendrimers as biological oxygen sensors. In Designing Dendrimers; Capagna S., Ceroni P., Eds.; Wiley: New York, 2012. [Google Scholar]
- Lebedev A. Y.; Cheprakov A. V.; Sakadžić S.; Boas D. A.; Wilson D. F.; Vinogradov S. A. ACS Appl. Mater. Interfaces 2009, 1, 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esipova T. V.; Karagodov A.; Miller J.; Wilson D. F.; Busch T. M.; Vinogradov S. A. Anal. Chem. 2011, 83, 8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N. W.; Verbridge S. S.; Williams R. M.; Chen J.; Kim J. Y.; Schmehl R.; Farnum C. E.; Zipfel W. R.; Fischbach C.; Stroock A. D. Biomaterials 2012, 33, 2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. E. K.; Ulbrich E. E.; Kim G.; Hah H.; Strollo C.; Fan W. Z.; Gurjar R.; Koo S. M.; Kopelman R. Anal. Chem. 2010, 82, 8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev R. I.; Zhdanov A. V.; Ponomarev G. V.; Yashunski D. V.; Papkovsky D. B. Anal. Biochem. 2010, 398, 24. [DOI] [PubMed] [Google Scholar]
- Coogan M. P.; Court J. B.; Gray V. L.; Hayes A. J.; Lloyd S. H.; Millet C. O.; Pope S. J. A.; Lloyd D. Photochem. Photobiol. Sci. 2010, 9, 103. [DOI] [PubMed] [Google Scholar]
- Napp J.; Behnke T.; Fischer L.; Wurth C.; Wottawa M.; Katschinski D. M.; Alves F.; Resch-Genger U.; Schaferling M. Anal. Chem. 2011, 83, 9039. [DOI] [PubMed] [Google Scholar]
- Kondrashina A. V.; Dmitriev R. I.; Borisov S. M.; Klimant I.; O’Brien I.; Nolan Y. M.; Zhdanov A. V.; Papkovsky D. B. Adv. Funct. Mater. 2012, 22, 4931. [Google Scholar]
- Estrada A. D.; Ponticorvo A.; Ford T. N.; Dunn A. K. Opt. Lett. 2008, 33, 1038. [DOI] [PubMed] [Google Scholar]
- Finikova O. S.; Lebedev A. Y.; Aprelev A.; Troxler T.; Gao F.; Garnacho C.; Muro S.; Hochstrasser R. M.; Vinogradov S. A. ChemPhysChem 2008, 9, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakadžić S.; Roussakis E.; Yaseen M. A.; Mandeville E. T.; Srinivasan V. J.; Arai K.; Ruvinskaya S.; Devor A.; Lo E. H.; Vinogradov S. A.; Boas D. A. Nat. Methods 2010, 7, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq J.; Parpaleix A.; Roussakis E.; Ducros M.; Houssen Y. G.; Vinogradov S. A.; Charpak S. Nat. Med. 2011, 17, 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor A.; Sakadžić S.; Saisan P. A.; Yaseen M. A.; Roussakis E.; Srinivasan V. J.; Vinogradov S. A.; Rosen B. R.; Buxton R. B.; Dale A. M.; Boas D. A. J. Neurosci. 2011, 31, 13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmi S. M. S.; Salvaggio A. J.; Estrada A. D.; Hemati M. A.; Shaydyuk N. K.; Roussakis E.; Jones T. A.; Vinogradov S. A.; Dunn A. K. Biomed. Opt. Express 2013, 4, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpaleix A.; Houssen Y. G.; Charpak S. Nat. Med. 2013, 19, 241. [DOI] [PubMed] [Google Scholar]
- Spencer J. A.; Ferraro F.; Roussakis E.; Klein A.; Wu J. W.; Runnels J. M.; Zaher W.; Mortensen L. J.; Alt C.; Turcotte R.; Yusuf R.; Cote D.; Vinogradov S. A.; Scadden D. T.; Lin C. P. Nature 2014, 508, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood D.; Gouterman M. J. Mol. Spectrosc. 1970, 35, 359. [Google Scholar]
- Papkovsky D. B.; O’Riordan T. C. J. Fluores. 2005, 15, 569. [DOI] [PubMed] [Google Scholar]
- Brinas R. P.; Troxler T.; Hochstrasser R. M.; Vinogradov S. A. J. Am. Chem. Soc. 2005, 127, 11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. Y.; Troxler T.; Vinogradov S. A. J. Porphyrins Phthalocyanines 2008, 12, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finikova O. S.; Troxler T.; Senes A.; DeGrado W. F.; Hochstrasser R. M.; Vinogradov S. A. J. Phys. Chem. A 2007, 111, 6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finikova O. S.; Chen P.; Ou Z.; Kadish K. M.; Vinogradov S. A. J. Photochem. Photobiol., A 2008, 198, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. F.; Bull B.; Christensen K.; McNeill J. Angew. Chem., Int. Ed. 2009, 48, 2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon C. M.; Karnas E.; Bawendi M. G.; Nocera D. G. Inorg. Chem. 2013, 52, 10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T.; Takubo K.; Semenza G. L. Cell Stem Cell 2011, 9, 298. [DOI] [PubMed] [Google Scholar]
- Mohyeldin A.; Garzón-Muvdi T.; Quiñones-Hinojosa A. Cell Stem Cell 2010, 7, 150. [DOI] [PubMed] [Google Scholar]
- Lee K. E.; Simon M. C. Curr. Opin. Cell Biol. 2012, 24, 232. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Plenum Press: New York, 2006. [Google Scholar]
- Mani T.; Niedzwiedzki D. M.; Vinogradov S. A. J. Phys. Chem. A 2012, 116, 3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finikova O. S.; Cheprakov A. V.; Vinogradov S. A. J. Org. Chem. 2005, 70, 9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaranta M.; Borisov S. M.; Klimant I. Bioanal. Rev. 2012, 4, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. Y.; Filatov M. A.; Cheprakov A. V.; Vinogradov S. A. J. Phys. Chem. A 2008, 112, 7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer J. R.; Shelton A. H.; Parthasarathy A.; Ghiviriga I.; Reynolds J. R.; Schanze K. S. Chem. Mater. 2011, 23, 5296. [Google Scholar]
- Knyukshto V. N.; Shul’ga A. M.; Sagun E. I.; Zen’kevich E. I. Opt. Spectrosc. 2002, 92, 53. [Google Scholar]
- Knyukshto V. N.; Shul’ga A. M.; Sagun E. I.; Zen’kevich E. I. Opt. Spectrosc. 2006, 100, 590. [Google Scholar]
- Kubin R. F.; Fletcher A. N. J. Lumin. 1982, 27, 455. [Google Scholar]
- Seybold P. G.; Gouterman M. J. Mol. Spectrosc. 1969, 31, 1. [Google Scholar]
- Chen P.; Finikova O. S.; Ou Z. P.; Vinogradov S. A.; Kadish K. M. Inorg. Chem. 2012, 51, 6200. [DOI] [PubMed] [Google Scholar]
- Makarov N. S.; Drobizhev M.; Rebane A. Opt. Express 2008, 16, 4029. [DOI] [PubMed] [Google Scholar]
- Deshpande A. V.; Kumar U. J. Fluoresc. 2006, 16, 679. [DOI] [PubMed] [Google Scholar]
- Jones G.; Jackson W. R.; Choi C. Y.; Bergmark W. R. J. Phys. Chem. 1985, 89, 294. [Google Scholar]
- Seidel C. A. M.; Schulz A.; Sauer M. H. M. J. Phys. Chem. 1996, 100, 5541. [Google Scholar]
- Yu P. M.; Valerii A. S.; Reznichenko A. V.; Myachin A. Y.; Bakhareva S. S.; Dolotov S. M.; Kopylova T. N.; Ponomarenko E. P. Quantum Electron. 2003, 33, 803. [Google Scholar]
- Bissell E. R.; Larson D. K.; Croudace M. C. J. Chem. Eng. Data 1981, 26, 348. [Google Scholar]
- Subhas Bose D.; Rudradas A. P.; Hari Babu M. Tetrahedron Lett. 2002, 43, 9195. [Google Scholar]
- Kobayashi T.; Straub K. D.; Rentzepis P. M. Photochem. Photobiol. 1979, 29, 925. [Google Scholar]
- Kim D. H.; Holten D.; Gouterman M.; Buchler J. W. J. Am. Chem. Soc. 1984, 106, 4015. [Google Scholar]
- Rozhkov V.; Wilson D.; Vinogradov S. Macromolecules 2002, 35, 1991. [Google Scholar]
- Vinogradov S. A.; Lo L. W.; Wilson D. F. Chem.—Eur. J. 1999, 5, 1338. [Google Scholar]
- Xu C.; Webb W. W. J. Opt. Soc. Am. B 1996, 13, 481. [Google Scholar]
- Belfield K. D.; Yao S.; Bondar M. V.; Belfield K. D.; Yao S.; Bondar M. V.. Two-Photon Absorbing Photonic Materials: From Fundamentals to Applications. Photoresponsive Polymers I; Springer-Verlag: Berlin, 2008; Vol 213, p 97.
- Drobizhev M.; Karotki A.; Kruk M.; Rebane A. Chem. Phys. Lett. 2002, 355, 175. [Google Scholar]
- Denk W.; Piston D. W.; Webb W. W.. Multiphoton Molecular Excitation in Laser-Scanning Microscopy. In Handbook of Biological Confocal Microscopy, 3 ed.; Pawley J. B., Ed.; Springer Science & Business Media, LLC: New York, 2006; pp 535. [Google Scholar]
- Sinks L. E.; Robbins G. P.; Roussakis E.; Troxler T.; Hammer D. A.; Vinogradov S. A. J. Phys. Chem. B 2010, 114, 14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y.; Bruns I.; Scheiermann C.; Ahmed J.; Pinho S.; Zhang D. C.; Mizoguchi T.; Wei Q. Z.; Lucas D.; Ito K.; Mar J. C.; Bergman A.; Frenette P. S. Nature 2013, 502, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison S. J.; Scadden D. T. Nature 2014, 505, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.