

Abstract
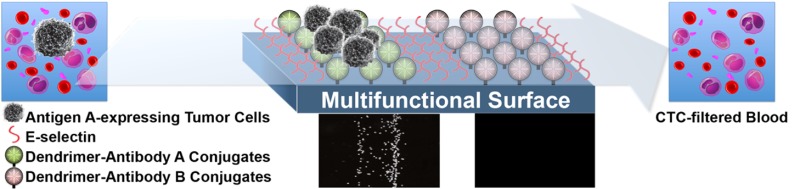
Effective quantification and in situ identification of circulating tumor cells (CTCs) in blood are still elusive because of the extreme rarity and heterogeneity of the cells. In our previous studies, we developed a novel platform that captures tumor cells at significantly improved efficiency in vitro using a unique biomimetic combination of two physiological processes: E-selectin-induced cell rolling and poly(amidoamine) (PAMAM) dendrimer-mediated strong multivalent binding. Herein, we have engineered a novel multifunctional surface, on the basis of the biomimetic cell capture, through optimized incorporation of multiple antibodies directed to cancer cell-specific surface markers, such as epithelial cell adhesion molecule (EpCAM), human epidermal growth factor receptor-2 (HER-2), and prostate specific antigen (PSA). The surfaces were tested using a series of tumor cells, MDA-PCa-2b, MCF-7, and MDA-MB-361, both in mixture in vitro and after being spiked into human blood. Our multifunctional surface demonstrated highly efficient capture of tumor cells in human blood, achieving up to 82% capture efficiency (∼10-fold enhancement than a surface with the antibodies alone) and up to 90% purity. Furthermore, the multipatterned antibodies allowed differential capturing of the tumor cells. These results support that our multifunctional surface has great potential as an effective platform that accommodates virtually any antibodies, which will likely lead to clinically significant, differential detection of CTCs that are rare and highly heterogeneous.
The detection and enumeration of CTCs in blood have been reported to correlate with cancer progression and patient survival,1 providing an effective tool for the diagnosis and prognosis of cancer metastasis.2−5 Despite the recent vigorous research efforts and progress in this field, the sensitive and selective detection of CTCs with clinically sufficient purity still remains a technical challenge because of the rarity of CTCs in blood (one CTC in the background of 106–109 hematologic cells).6,7 One of the most commonly used methods for CTC detection is to differentiate the tumor cells using their surface markers that are not expressed by normal hematologic cells.8−10 These surface markers include EpCAM,11 HER-2,12,14 PSA,13 epidermal growth factor receptor (EGFR),15 and carcinoembryonic antigen (CEA).16
However, the detection and enrichment of CTCs based on a single cancer cell marker, most commonly EpCAM, often encounter major challenges because of the phenotypic heterogeneity among CTCs4 and their biological plasticity during the metastatic process, known as the epithelial-mesenchymal-transition (EMT).17 While most of the currently available detection methods including the FDA-approved CellSearch target EpCAM, it has been reported that approximately 20–30% of tumors such as sarcoma and melanoma express low-to-no EpCAM.18 Furthermore, because CTCs frequently lose their epithelial nature upon EMT, resulting in down-regulated EpCAM expression,19 detection solely based on aEpCAM is insufficient to capture the CTCs.17,19 Capturing using HER-2 also has limitations since HER-2 is overexpressed by only 20–30% of breast and prostate cancers,12 resulting in vast variations in detection sensitivity.20
Attempts to address these issues include a few proof-of-concept studies using antibody cocktails that have demonstrated enhanced capture efficiencies, compared to a single antibody-based approach. Various combinations of antibodies have been used, including mixtures of EpCAM/cytokeratin (CK),21 EpCAM/HER-2/EGFR,5 and EpCAM/c-Met/folate binding receptor/N-Cadherin/CD318/HER-2/Muc-1/EGFR.22 Although the antibody cocktail-based detection successfully showed enhanced capture efficiencies, it still has limitations, such as low purity of CTCs (approximately 14%) captured among the contaminating leukocytes5 and necessity of postcapture analysis for identification of the captured cells. Recently, we demonstrated a novel, surface engineering approach to achieve enhanced detection of tumor cells by employing a unique combination of two physiological phenomena: cell rolling and multivalent binding.23−25 E-selectin-mediated cell rolling served as an effective way of recruiting flowing cells to the capture surface, and tumor cell-specific binding was substantially improved by incorporation of PAMAM dendrimer-mediated strong multivalent binding (over 1 million fold improvement in dissociation constant). This configuration resulted in a novel CTC detection surface which significantly enhanced capture efficiency up to 7-fold when compared to the surfaces immobilized with aEpCAM alone.24
In this study, we have designed a biomimetic surface that accommodates patterned multiple antibodies to capture heterogeneous populations of tumor cells in a differential manner, in addition to increasing sensitivity through the biomimetic combination of cell rolling and multivalent binding, as illustrated in Figure 1. To assess the feasibility of this design, antibodies against three cancer-specific biomarkers (EpCAM, HER-2, and PSA) were selected and immobilized in pattern via PAMAM dendrimers on epoxy-functionalized glass surfaces, followed by addition of E-selectin. After optimization, the functionalized surfaces with multiple antibodies were validated using model CTCs, such as prostate cancer (MDA-PCa-2b) and breast cancer (MDA-MB-361 and MCF-7) cells, in mixture as well as after being spiked into human blood to demonstrate their clinical translatability.
Figure 1.

Schematic illustration of preparation of a capture surface functionalized with multiple antibodies through dendrimers. (a) Multiple antibodies against cancer cell surface markers were immobilized on a dendrimer-functionalized slide. (b) The surface was then backfilled with E-selectin, followed by (c) flow chamber assembly for flow experiments. Chemical reaction scheme used for the surface functionalization with PEG and G7 PAMAM dendrimers were drawn in the bottom panel.
Results and Discussion
Our design rationale was to achieve tumor cell-specific capture patterns based on binding preferences of the tumor cells to the three antibodies (aEpCAM, aHER-2, and aPSA). We first assessed the binding patterns of the three individual cell lines that were employed as CTC models, by observing their binding behaviors with each antibody using a flow chamber. The antibodies and E-selectin were immobilized on epoxy-functionalized substrates, as reported in our earlier study.23 The protein-immobilized surfaces were characterized using X-ray photoelectron spectroscopy (XPS, Table S1 in Supporting Information) and energy filtered transmission electron microscopy (TEM)-carbon mapping analysis (Figures S1 and S2 in Supporting Information). As shown in Supporting Information, Figure S3, MDA-PCa-2b cells were bound to all three antibody-surfaces, and MDA-MB-361 cells were bound to both aHER-2 and aEpCAM. MCF-7 cells were only bound to the aEpCAM stripes. On E-selectin-immobilized surfaces, all cell lines in addition to HL-60, a leukocyte model, exhibited stable rolling at various velocities (0.1–6.5 μm/s) under 0.2 dyn/cm2 of shear stress (Figure S3 in Supporting Information).
We then hypothesized that the different binding behaviors of the tumor cells could be translated into one substrate, which could be substantially augmented by introduction of cell rolling and multivalent binding. First, the widths of aEpCAM and E-selectin-immobilized stripes were optimized at 500 μm and 2 mm, respectively, based on the measured capture efficiencies of varied dimensions (Figure 1 and Figures S4 and S5 in Supporting Information). Next, the multivalent binding effect was engineered into the surface by immobilizing the three antibodies via PAMAM dendrimers. The nanoscale topography and carbon distribution of the surfaces were characterized using energy filtered TEM (Figures S1 and S2 in Supporting Information).
The dendrimer-coated surfaces exhibited significantly enhanced capture efficiencies in all conditions, as expected based on our previous study using aEpCAM.24 Figure 2a shows a set of representative data using MDA-PCa-2b cells where significant enhancement was achieved by dendrimer-mediated multivalent binding. Addition of E-selectin to induce cell rolling further enhanced surface capture efficiency. As shown in Figure 2a, the micropatterned surface with the three dendrimer-antibody conjugates and E-selectin (defined as a “multifunctional surface” throughout this paper) showed the dramatically enhanced capture efficiencies for MDA-PCa-2b cells by up to ∼60-fold when compared to the surfaces with the corresponding antibodies. After the rolling cells were washed off using EGTA-PBS buffer, the clear cell binding patterns on the multifunctional surface (aEpCAM-coated regions in the images) was observed at a low magnification without postlabeling or treatment (Figure 2b). Cell accumulations at the interfaces between the dendrimer-antibody- and E-selectin-coated regions were also observed. The quantitative comparison to the capture solely based on individual antibodies revealed that the multifunctional surfaces are most effective in capture efficiency across all CTC model cells tested, achieving up to 150-fold enhancement (Figure 2b) at up to 81% capture efficiency (Figure 2d). Cell rolling (the left-hand side of the images) and stationary binding (the right-hand side of the images) of the cells at the interface of E-selectin and antibodies, respectively, were observed on the multifunctional surfaces under flow (direction from left to right) as shown in Supporting Information, Figure S6. Being further supported by Supporting Information, Movie S1, these data clearly show the capture mechanism of our multifunctional surfaces, that is, cell recruitment to the surface via E-selectin and subsequent specific cell capture on the dendrimer-antibody-stripes, resulting in the substantially enhanced detection sensitivity.
Figure 2.

Enhanced capture efficiencies of the multifunctional surfaces by the combined effect of cell rolling and multivalent binding. (a) Fold enhancement in capture efficiency of the surface functionalized with the three antibodies, measured using MDA-PCa-2b cells. The surface with E-selectin and G7 PAMAM dendrimers shows substantially enhanced capture efficiencies up to 60-fold, compared to the surface with antibodies only. (b) Bright field microscopic images of the captured MDA-PCa-2b, MDA-MB-361, and MCF-7 cells on the surfaces functionalized with aEpCAM. The numbers of the captured cells clearly increase with addition of the dendrimers and E-selectin without an increase of nonspecific capture (Scale bar = 200 μm). (c) Fold increase in capture efficiencies of the surfaces with the three antibodies, G7 PAMAM dendrimers, and E-selectin, compared to the surfaces functionalized with the antibodies alone. The three cancer cell lines, depending on their surface marker expressions, all demonstrate enhanced capture efficiencies on the multifunctional surfaces, as high as 150 fold. (d) Quantitative capture efficiencies of each of the antibodies on the multifunctional surface toward various cancer cells. Although there is a degree of variations, the surface marker-dependent capturing achieves up to 81% capture efficiency. *No fold enhancements because of negligible binding without E-selectin. The Error bars: standard error (n = 4).
The capture mechanism through sequential rolling and stationary, multivalent binding allows the multifunctional surfaces to differentiate “live” cells from their mixtures according to their surface markers. To identify each cell line in the mixture throughout the analytical process, the individual cell lines were labeled with cell-permanent dyes that are used for cell viability assays: MDA-PCa-2b with Calcein AM (green), MDA-MB-361 with Cell Alive Blue dye (blue), and MCF-7 with Cell Alive Red dye (red). The schematic diagram of the differential capture of the cells on the patterned surface is shown in Figure 3a, and the quantitative measurements are summarized in Figure 3c. For example, MDA-PCa-2b cells, the only PSA-positive cell line among the three CTC models, bound primarily on aPSA-stripe at 91–100% purity from the cell mixtures with PSA-negative cells. All of the captured cancer cells using the multifunctional surfaces were fluorescent, indicating that the bound cells were still alive (Figure 3b). It was confirmed that the combination of cell rolling and multivalent binding enhanced the capture efficiency over all three CTC models in vitro (up to 82%, Figure 3c). Interestingly, MCF-7 cells showed noticeable binding to the aHER-2-coated surface after addition of E-selectin (Figure S6c in Supporting Information), which was not observed on the same surface without E-selectin (Figures S3c and S7 in Supporting Information). This suggests that the enhanced recruitment of the cells to the capture surface via E-selectin helps improve the capture efficiency of the antibodies even though the cells express a low level of the corresponding surface receptors.
Figure 3.
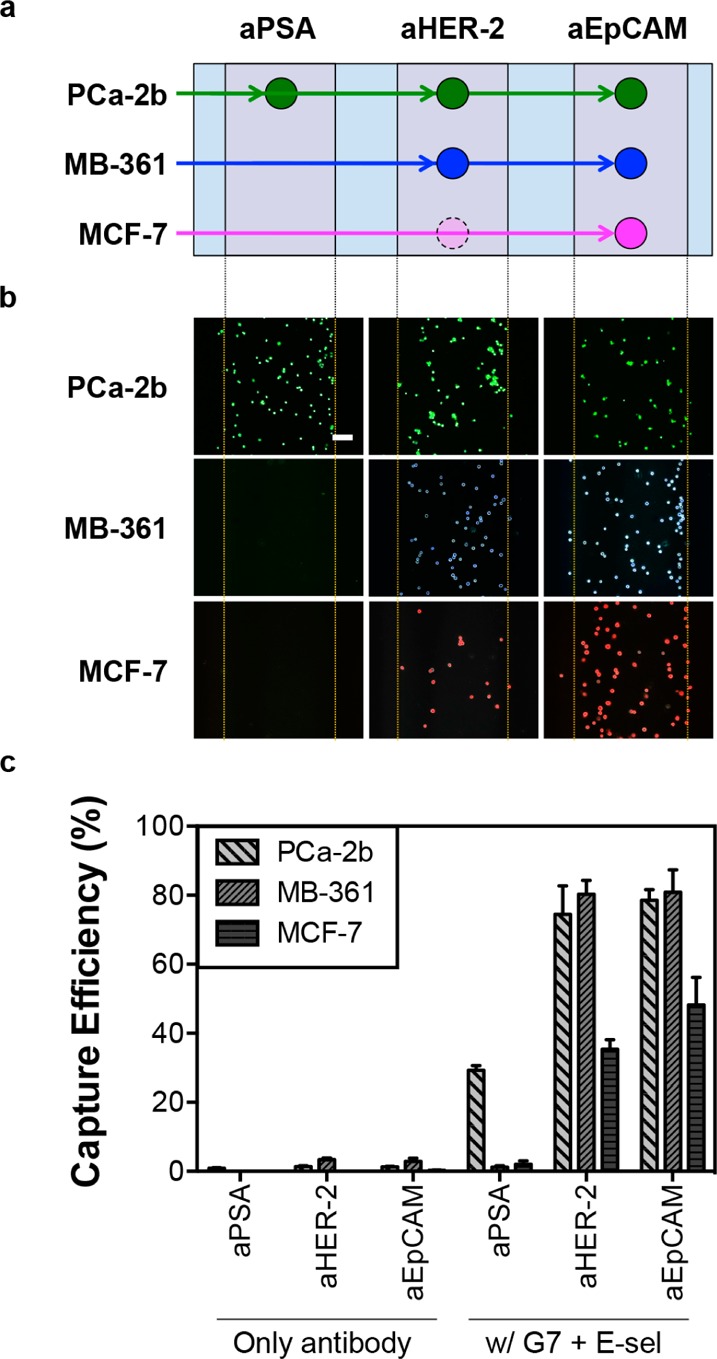
Antibody-dependent surface capture of tumor cells with enhanced capture efficiencies. (a) Schematic illustration of the surface marker-dependent cell capture using aPSA, aHER-2, and aEpCAM. The dotted, faded circle of MCF-7 cells for aHER-2 indicates the lower capture efficiency due to low HER-2 expression of MCF-7 cells. (b) The capture patterns of the three cell lines, visualized in color using fluorescently labeled cells: Calcein AM (green) for MDA-PCa-2b, Alive cell tracker Blue (blue) for MDA-MB-361, and Alive cell tracker Red (red) for MCF-7 (Scale bar = 100 μm). (c) Enhanced capture efficiencies of the cell lines on the three antibody-coated domains by the combined effect of cell rolling and multivalent binding (up to 82%). Error bars: standard error (n = 4).
The capture efficiency of the multifunctional surfaces was then finally validated using cancer cell-spiked human blood. To measure the tumor cell binding in clinically relevant conditions, the tumor cells were spiked into human blood withdrawn from healthy donors, which represents approximately 1 tumor cell mixed with 10 thousand leukocytes and 10 million red blood cells per 1 mm3. Mononuclear cells including tumor cells in a buffy coat were separated from whole blood using density gradient centrifugation for subsequent analysis. The hematological cells and fluorescence-labeled cancer cells in a buffy coat exhibited the rolling responses on the E-selectin stripes (Movies S3 and S4 in Supporting Information). The rolling leukocytes (impurity) were efficiently removed by washing with EGTA-supplemented buffer, resulting in separation of a highly pure population of the captured tumor cells (50–90% purity of tumor cells captured among the contaminating leukocytes, Figure 4a). Note that the typical purity reported in the literatures is only between 0.1 and 14%.5,26,27 The cancer cells from the blood samples were effectively captured as they maintained the surface marker-dependent binding, similar to the in vitro results (Figure 4b and Supporting Information, Figure S9). All the capture efficiencies in the blood samples were substantially enhanced by up to 12 folds, which is attributed to the combination effect of the two biomimetic approaches, compared to those of antibody-stripes without E-selectin and dendrimers (Figure 4b and Supporting Information, Figure S9).
Figure 4.

Validation of the multifunctional surfaces using cancer cell-spiked blood specimens. (a) A set of representative images of the captured tumor cells from blood samples on the multifunctional stripes with aEpCAM. Note the left blood cells (nonfluorescence cells) on the multifunctional surface were between 10% and 50%, which indicated that the purity of the captured cancer cells is between 50% and 90% (scale bar = 100 μm). (b) Detection sensitivity of the multifunctional surfaces was significantly higher, by up to 20-fold, than that of the surfaces with only antibody stripes. The surface marker-dependent bindings of cancer cell models were also observed. Error bars: standard error (n = 4).
As shown in the results presented herein, surface micropatterning with E-selectin and multiple antibodies demonstrates great potentials to effectively enrich tumor cells in cell mixtures and in blood samples. The fast association and dissociation kinetics between E-selectin and its ligands on cells can facilitate the recruitment of the cancer cell-containing cell population during flow across the micropatterned surface (Movie S3 in Supporting Information).23,28 The cell recruitment and subsequent rolling along E-selectin harness the selective accumulation of cancer cells on the adjacent antibody-coated stripes (Supporting Information Figure S6). As a result, MCF-7 cells, despite their relatively low expression of HER-2,29 exhibited stable binding to aHER-2 when the surface sensitivity was augmented by addition of E-selectin (Figure 3c and Supporting Information, Figure S6c). This clearly displays the benefit of E-selectin-induced rolling for cell recruitment. After rolling on E-selectin, the densely immobilized antibodies through dendrimers can exploit multivalent binding effect,24 which allows significantly increased strength of surface binding with cells and thereby enhanced detection sensitivity using blood samples (over 10-fold enhancement than the surface with individual antibodies at up to 82% capture efficiency, Figure 4).
Our results indicate that the combination of cell rolling and multivalent binding, along with incorporation of the multiple antibodies, is critical to enhance the detection specificity and in situ CTC identification. One can argue that E-selectin decreases specificity by increasing the number of the bound cells that are not targeted, e.g. binding of other PSA-negative cells such as MCF-7 cells (Figure 3c) and leukocytes (Figure 4a)30 on the aPSA stripes. However, we have shown that nonspecifically bound cells on E-selectin can be easily removed using EGTA-supplemented PBS buffer because the interaction between E-selectin and cells is Ca2+-dependent (Movie S2 in Supporting Information).25 In addition, the ultralow dissociation constants achieved through dendrimer-mediated multivalent binding allow the specifically bound cells to withstand the harsh washing step that may be needed to further increase purity of the capture cells.24 Compared to other detection methods that typically achieve the purity of the collected CTCs in the range of 1–20%,8 the high purity (50–90%, Figure 4) of the captured cells from blood samples on our biomimetic surfaces clearly shows the enhanced detection specificity of this biomimetic system. This also enables identification at low magnification without postlabeling by reading the capture patterns of the bound cells (Figure 2b). It is noteworthy that a significant progress has been recently made in the CTC capture devices using various approaches such as those based on graphene/gold patterns and silicone nanostructures coated with polymers in an integrated manner.31−36 Liu et al. reported a microfluidic system that integrated an aEpCAM-immobilized substrate and nonlaminar flow induced by incorporated microstructures, achieving approximately 90% capture efficiency and over 50% capture purity from breast cancer cell-spiked human blood.35 Another example is a polymer nanofiber-embedded device developed by the Tseng group where high capture efficiency (75%) at a highly purity (without quantitative values reported) of prostate cancer cells in the presence of human blood was demonstrated.36 However, it is technically difficult to conduct fair comparison of those devices to ours because the capture efficiency can substantially vary depending on the experimental conditions, such as flow rate, tumor cell concentrations, and types of capturing agents (antibody, peptide, or aptamer). Nonetheless, our system is unique in that it utilizes the biomimetic combination of cell rolling and multivalent binding, achieving high capture efficiency and specificity from both cell suspensions and human blood.
The versatile, multifunctional platform for tumor cell detection has potential advantages. The results shown here can be easily expanded to incorporate a variety of surface markers, such as CD24/44, CD146, CEA, and others as needed via simple chemical conjugation reactions. The E-selectin-based approach also allows to capture cells using those surface markers with low-level expression, which is particularly important, given that many tumor cells undergo phenotypic changes in terms of surface marker expression upon EMT and yet such metastatic/post-EMT cells still highly express E-selectin ligands.37,38 Another advantage includes that cell screening based on multiple cancer cell markers could provide additional pathological information on cancer progress. The expression of specific cancer markers in an early phase could be used as an indicator of later-stage expression of other markers. For example, HER-2 expression confers androgen-independent growth to prostate cancer cells in vitro through the activation of the androgen receptor in a ligand-independent way.39 The HER-2-activated androgen receptor could affect the transcription of its downstream target, PSA, and eventually promote the prostate cancer progress.39 Therefore, simultaneous screening of HER-2 and PSA using this highly sensitive multifunctional surface presented in this study could be potentially used to monitor the progress of prostate cancer. Furthermore, the pathological information on an individual cancer patient obtained using the multifunctional surfaces could be also used as an indicator for his/her drug responsiveness to the personalized treatment (e.g., cancer immunotherapy), which can provide clinically valuable information to further enhance the therapeutic efficiency. This clinical validation of our multifunctional CTC capture platform is the subject of our future clinical studies.
Taken together, this study demonstrates that our biomimetic, multifunctional surface shows substantially enhanced capture efficiency of the tumor cells at high purity, both in culture media and human blood, compared to the surfaces coated with antibodies only. Versatility and modularity of this surface platform allow expansion to multiple antibodies, resulting in the enhanced sensitivity (up to 82% capture efficiency) and specificity (up to 90% capture purity) of cell capture. Additionally, we have shown that the surface micropatterning can be applicable to differentiate the captured cancer cells and their cell surface markers in situ. This multiple antibody-immobilized dendrimer platform along with high detection sensitivity and specificity paves the way for potential use of this system as a diagnostic and prognostic tool for monitoring cancer progress and responses to therapies of metastatic cancer patients.
Methods
Materials
Antihuman epithelial-cell-adhesion-molecule (EpCAM)/TROP1 antibody (aEpCAM), antihuman epidermal growth factor receptor-2 (HER-2)/TROP1 antibody (aHER-2), antihuman prostate specific antigen (PSA)/TROP1 antibody (aPSA), and recombinant human E-selectin (E-selectin) were all purchased from R&D systems (Minneapolis, MN). Epoxy-functionalized glass surfaces (SuperEpoxy2) were purchased from TeleChem International, Inc. (Sunnyvale, CA). PAMAM dendrimers (generation 7), bovine serum albumin (BSA), Calcein AM, and all other chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and used without further purification unless otherwise specified.
Surface Functionalization by Immobilization of Adhesive Proteins
A polydimethylsiloxane (PDMS) gasket with well-defined micropatterning was used to define the area of an epoxy functionalized glass slide, as depicted in Figure 1. Surface immobilization of Generation 7 (G7) PAMAM dendrimer and subsequent conjugation with antibodies were performed, as previously reported.24 For antibody conjugation, antibody solutions of aEpCAM (5 μg/mL), aHER-2 (5 μg/mL) and aPSA (10 μg/mL) in PBS were used. The micropatterned antibodies were treated with FITC-conjugated BSA (1 mg/mL in PBS buffer) for 1 h to define the dendrimer-antibody-coated regions. After the PDMS gasket was removed, the surface with the antibody stripes was incubated with 0.2 mL of E-selectin at a concentration of 5 μg/mL in PBS for 4 h. The volumes of all reagent solutions except E-selectin were fixed at 20 μL. All incubation processes were carried at room temperature with constant gentle shaking, and between all preparation steps, the surfaces were washed with DDI water and PBS three times to remove the residual chemicals from the surfaces. Potential nonspecific binding of both protein-coated and uncoated regions was blocked by a final incubation with 1% (w/v) BSA or 1 μg/mL methoxy PEG-NH2 (Nektar Therapeutics, Huntsville, AL) solution. The functionalized surfaces were kept at 4 °C, and the experiments using the surfaces were performed within 24 h after the surface preparation.
Fluorescence Labeling for Viable Cells
Cells at a concentration of 1 × 106 cells/mL (5 mL) were seeded onto a 25 cm2 T flask 1 day before the experiment. To label the viable cells with fluorescence, the MDA-PCa-2b, MDA-MB-361, and MCF-7 cells were treated with 4 μM Calcein AM, 5 μM Alive cell track It Blue (AAT Bioquest, Inc., Sunnyvale, CA), and 5 μM Alive cell track It Red (AAT Bioquest, Inc.), respectively, at 37 °C in dark for 30 min. The labeled cells were trypsinized to make their suspensions at a predetermined concentration in FBS-supplemented cell culture media or whole blood withdrawn from healthy donors. The prepared cell suspensions were kept on ice throughout the subsequent experiments.
Blood preparation using Ficoll-Paque Plus
The heparin-treated blood was kept at 4 °C in a refrigerator and the experiments were performed within 48 h after drawing. Fluorescence-labeled cancer cells were spiked to 3 mL of whole blood as a final concentration of 1 × 105 tumor cells/mL blood. Mononuclear cells including tumor cells in buffy coat was separated from whole blood using Ficoll-Paque Plus (Stemcell Technologies Inc., Vancouver, Canada) as described in section 7 of Supporting Information. Briefly, the blood samples loaded with Ficoll for separation were centrifuged at 20 °C for 20 min at 1,500× g with brake function off. After the buffy coat was washed twice with the FBS/heparin-included PBS buffer via centrifuge, the recovered cells were suspended with 3 mL of the complete cell culture media and used for subsequent experiments. Studies using human blood were reviewed and approved by UIC institutional review board (IRB) (protocol 2012-0139).
Observation of Cellular Responses Using Flow Chamber
A typical flow chamber experiment was performed as we reported earlier.23 Suspensions of HL-60 and fluorescence-labeled cancer cells (MDA-PCa-2b, MDA-MB-361, MCF-7) were injected into a rectangular flow chamber with a gasket (30 mm (L) × 10 mm (W) × 0.15 mm (D), Glycotech, Gaithersburg, MD) using a syringe pump (New Era pump Systems Inc., Farmingdale, NY). The number of captured cells on each of the antibody-immobilized stripes defined using FITC-BSA was counted using the images taken after one cycle, consisting of forward flow (pushing) for 5 min and backward flow (withdrawing) for 5 min at 50 μL/min (0.2 dyn/cm2). The surface was washed using PBS for 10 min at 200 μL/min (0.9 dyn/cm2). To remove the leukocyte cells from the E-selectin-regions, the surface was washed using EGTA/Mg2+ in PBS buffer for 3 min at 200 μL/min (0.9 dyn/cm2).25 On the basis of the known number of cancer cells perfused into the flow chamber, the numbers of captured cells per antibody-stripe were calculated and converted into capture efficiency (%). All cells on the surface were monitored using an Olympus IX70 inverted microscope (IX 70-S1F2, Olympus America, Inc., Center Valley, PA) with fluorescence light, and images were recorded using a 10× objective and a CCD camera (QImaging Retiga 1300B, Olympus America, Inc.). The number of cells on the surfaces was counted, based on the images taken in independent observations/measurements using ImageJ (NIH).
Acknowledgments
This work was supported by National Science Foundation (NSF) under Grant CBET-0931472, University of Illinois Cancer Center, and the Office of Technology Managements of the University of Illinois at Chicago (UIC). This investigation was conducted in a facility constructed with support from grant C06RR15482 from the NCRR NIH. The authors thank Dr. Shawn Oppegard for his help in fabricating the PDMS gaskets with the customized channels and helpful discussion throughout the work. J.H.M. was partially supported by Dean’s scholarship and Chancellor’s Graduate Fellowship, both from UIC. Energy filtered transmission electron microscopy was conducted at the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Division of Scientific User Facilities, Office of Basic Energy Sciences, US Department of Energy.
Supporting Information Available
Details regarding the surface characterization using X-ray photoelectron spectroscopy and transmission electron microscopy, additional cellular responses of cancer cells on the functionalized surfaces under flow, the details regarding the optimization of surface patterns, additional graphs of the cells on the multifunctional surfaces, and details regarding the preparation of blood samples. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.H.M and S.H. planned and designed the experiments. J.H.M., K.A.G., and J.C. conducted the experiments and analyzed the data. J.H.M., R.E.M., and S.H. carried out clinical procedures and recruitment. J.H.M., K.A.G., and S.H. wrote the manuscript. S.H. supervised and directed the overall project. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): SH serves as President for Capio Biosciences, Inc., a company in which he holds financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cristofanilli M.; Budd G. T.; Ellis M. J.; Stopeck A.; Matera J.; Miller M. C.; Reuben J. M.; Doyle G. V.; Allard W. J.; Terstappen L. W.; Hayes D. F. N. Engl. J. Med. 2004, 351, 781–791. [DOI] [PubMed] [Google Scholar]
- de Bono J. S.; Scher H. I.; Montgomery R. B.; Parker C.; Miller M. C.; Tissing H.; Doyle G. V.; Terstappen L. W. W. M.; Pienta K. J.; Raghavan D. Clin. Cancer Res. 2008, 14, 6302–6309. [DOI] [PubMed] [Google Scholar]
- Hayes D. F.; Cristofanilli M.; Budd G. T.; Ellis M. J.; Stopeck A.; Miller M. C.; Matera J.; Allard W. J.; Doyle G. V.; Terstappen L. W. W. M. Clin. Cancer Res. 2006, 12, 4218–4224. [DOI] [PubMed] [Google Scholar]
- Attard G.; Swermenhuis J. F.; Olmos D.; Reid A. H. M.; Vickers E.; A’Hern R.; Levink R.; Coumans F.; Moreira J.; Riisnaes R.; Oommen N. B.; Hawche G.; Jameson C.; Thompson E.; Sipkema R.; Carden C. P.; Parker C.; Dearnaley D.; Kaye S. B.; Cooper C. S.; Molina A.; Cox M. E.; Terstappen L. W. M. M.; de Bono J. S. Cancer Res. 2009, 69, 2912–2918. [DOI] [PubMed] [Google Scholar]
- Yu M.; Bardia A.; Wittner B. S.; Stott S. L.; Smas M. E.; Ting D. T.; Isakoff S. J.; Ciciliano J. C.; Wells M. N.; Shah A. M.; Concannon K. F.; Donaldson M. C.; Sequist L. V.; Brachtel E.; Sgroi D.; Baselga J.; Ramaswamy S.; Toner M.; Haber D. A.; Maheswaran S. Science 2013, 339, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth T. R. Aus. Med. J. 1869, 15, 146–149. [Google Scholar]
- Allard W. J.; Matera J.; Miller M. C.; Repollet M.; Connelly M. C.; Rao C.; Tibbe A. G.; Uhr J. W.; Terstappen L. W. Clin. Cancer Res. 2004, 10, 6897–6904. [DOI] [PubMed] [Google Scholar]
- Nagrath S.; Sequist L. V.; Maheswaran S.; Bell D. W.; Irimia D.; Ulkus L.; Smith M. R.; Kwak E. L.; Digumarthy S.; Muzikansky A.; Ryan P.; Balis U. J.; Tompkins R. G.; Haber D. A.; Toner M. Nature 2007, 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix-Panabieres C.; Pantel K. Clin. Chem. 2013, 59, 110–118. [DOI] [PubMed] [Google Scholar]
- Myung J. H.; Gajjar K. A.; Han Y. E.; Hong S. P. Polym. Chem. 2012, 3, 2336–2341. [Google Scholar]
- Osta W. A.; Chen Y.; Mikhitarian K.; Mitas M.; Salem M.; Hannun Y. A.; Cole D. J.; Gillanders W. E. Cancer Res. 2004, 64, 5818–5824. [DOI] [PubMed] [Google Scholar]
- Thierry B.; Kurkuri M.; Shi J. Y.; Lwin L. E.; Palms D. Biomicrofluidics 2010, 4, 32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis M.; Kallergi G.; Ntoulia M.; Perraki M.; Apostolaki S.; Kafousi M.; Chlouverakis G.; Stathopoulos E.; Lianidou E.; Georgoulias V.; Mavroudis D. Clin. Cancer Res. 2008, 14, 2593–2600. [DOI] [PubMed] [Google Scholar]
- King M. R.; Western L. T.; Rana K.; Liesveld J. L. J. Bionic Eng. 2009, 6, 311–317. [Google Scholar]
- Maheswaran S.; Sequist L. V.; Nagrath S.; Ulkus L.; Brannigan B.; Collura C. V.; Inserra E.; Diederichs S.; Iafrate A. J.; Bell D. W.; Digumarthy S.; Muzikansky A.; Irimia D.; Settleman J.; Tompkins R. G.; Lynch T. J.; Toner M.; Haber D. A. N. Engl. J. Med. 2008, 359, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S.; Kagan D.; Hu C. M.; Campuzano S.; Lobo-Castanon M. J.; Lim N.; Kang D. Y.; Zimmerman M.; Zhang L.; Wang J. Angew. Chem., Int. Ed. Engl. 2011, 50, 4161–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorges T. M.; Tinhofer I.; Drosch M.; Rose L.; Zollner T. M.; Krahn T.; von Ahsen O. BMC Cancer 2012, 12, 178 10.1186/1471-2407-12-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momburg F.; Moldenhauer G.; Hammerling G. J.; Moller P. Cancer Res. 1987, 47, 2883–2891. [PubMed] [Google Scholar]
- Sieuwerts A. M.; Kraan J.; Bolt J.; van der Spoel P.; Elstrodt F.; Schutte M.; Martens J. W.; Gratama J. W.; Sleijfer S.; Foekens J. A. J. Natl. Cancer Inst. 2009, 101, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkin E. B.; Weinstein M. C.; Winer E. P.; Kuntz K. M.; Schnitt S. J.; Weeks J. C. J. Clin. Oncol. 2004, 22, 854–863. [DOI] [PubMed] [Google Scholar]
- Deng G.; Herrler M.; Burgess D.; Manna E.; Krag D.; Burke J. F. Breast Cancer Res. 2008, 10, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot C. V.; Bischoff F. Z.; Mayer J. A.; Wong K. L.; Pham T.; Bottsford-Miller J.; Stone R. L.; Lin Y. G.; Jaladurgam P.; Roh J. W.; Goodman B. W.; Merritt W. M.; Pircher T. J.; Mikolajczyk S. D.; Nick A. M.; Celestino J.; Eng C.; Ellis L. M.; Deavers M. T.; Sood A. K. Cancer Discovery 2011, 1, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J. H.; Launiere C. A.; Eddington D. T.; Hong S. Langmuir 2010, 26, 8589–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J. H.; Gajjar K. A.; Saric J.; Eddington D. T.; Hong S. Angew. Chem., Int. Ed. Engl. 2011, 50, 11769–11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launiere C.; Gaskill M.; Czaplewski G.; Myung J. H.; Hong S.; Eddington D. T. Anal. Chem. 2012, 84, 4022–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott S. L.; Hsu C. H.; Tsukrov D. I.; Yu M.; Miyamoto D. T.; Waltman B. A.; Rothenberg S. M.; Shah A. M.; Smas M. E.; Korir G. K.; Floyd F. P. Jr.; Gilman A. J.; Lord J. B.; Winokur D.; Springer S.; Irimia D.; Nagrath S.; Sequist L. V.; Lee R. J.; Isselbacher K. J.; Maheswaran S.; Haber D. A.; Toner M. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18392–18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Ting D. T.; Stott S. L.; Wittner B. S.; Ozsolak F.; Paul S.; Ciciliano J. C.; Smas M. E.; Winokur D.; Gilman A. J.; Ulman M. J.; Xega K.; Contino G.; Alagesan B.; Brannigan B. W.; Milos P. M.; Ryan D. P.; Sequist L. V.; Bardeesy N.; Ramaswamy S.; Toner M.; Maheswaran S.; Haber D. A. Nature 2012, 487, 510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J. H.; Gajjar K. A.; Pearson R. M.; Launiere C. A.; Eddington D. T.; Hong S. Anal. Chem. 2011, 83, 1078–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtidis A.; Srinivasaiah R.; Carkner R. D.; Brosnan M. J.; Conklin D. S. Breast Cancer Res. 2009, 11, R16doi:10.1186/bcr2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gala J. L.; Heusterspreute M.; Loric S.; Hanon F.; Tombal B.; Van Cangh P.; De Nayer P.; Philippe M. Clin. Chem. 1998, 44, 472–481. [PubMed] [Google Scholar]
- Hou S.; Zhao H.; Zhao L.; Shen Q.; Wei K. S.; Suh D. Y.; Nakao A.; Garcia M. A.; Song M.; Lee T.; Xiong B.; Luo S. C.; Tseng H. R.; Yu H. H. Adv. Mater. 2013, 25, 1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H. J.; Kim T. H.; Zhang Z.; Azizi E.; Pham T. M.; Paoletti C.; Lin J.; Ramnath N.; Wicha M. S.; Hayes D. F.; Simeone D. M.; Nagrath S. Nat. Nanotechnol. 2013, 8, 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Deng Y.; Tai Q.; Cheng B.; Zhao L.; Shen Q.; He R.; Hong L.; Liu W.; Guo S.; Liu K.; Tseng H. R.; Xiong B.; Zhao X. Z. Adv. Mater. 2012, 24, 2756–2760. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Xu L.; Zhao L.; Wu D.; Fan Y.; Zhou Y.; Ouyang W. H.; Xu X.; Zhang Z.; Song M.; Lee T.; Garcia M. A.; Xiong B.; Hou S.; Tseng H. R.; Fang X. Adv. Mater. 2013, 25, 2368–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. B.; Zhang W.; Huang F.; Feng H. T.; Shu W. L.; Xu X. P.; Chen Y. Biosens Bioelectron. 2013, 47, 113–119. [DOI] [PubMed] [Google Scholar]
- Zhao L. B.; Lu Y. T.; Li F. Q.; Wu K.; Hou S.; Yu J. H.; Shen Q. L.; Wu D. X.; Song M.; OuYang W. H.; Luo Z.; Lee T.; Fang X. H.; Shao C.; Xu X.; Garcia M. A.; Chung L. W. K.; Rettig M.; Tseng H. R.; Posadas E. M. Adv. Mater. 2013, 25, 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubli H.; Borsig L. Semin. Cancer Biol. 2010, 20, 169–177. [DOI] [PubMed] [Google Scholar]
- Sakuma K.; Aoki M.; Kannagi R. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7776–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoretti S.; Montironi R.; Manola J.; Altimari A.; Tam C.; Bubley G.; Balk S.; Thomas G.; Kaplan I.; Hlatky L.; Hahnfeldt P.; Kantoff P.; Loda M. J. Natl. Cancer Inst. 2000, 92, 1918–1925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.