

Conspectus
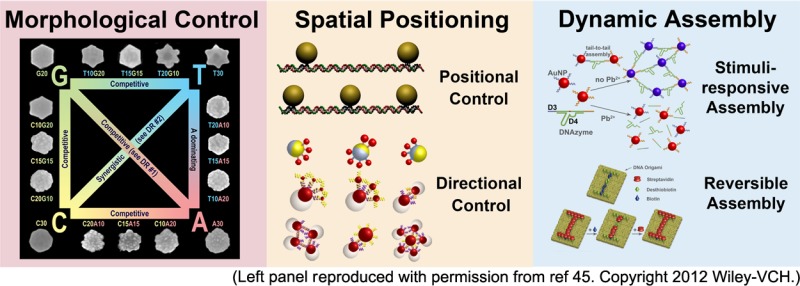
Several properties of nanomaterials, such as morphologies (e.g., shapes and surface structures) and distance dependent properties (e.g., plasmonic and quantum confinement effects), make nanomaterials uniquely qualified as potential choices for future applications from catalysis to biomedicine. To realize the full potential of these nanomaterials, it is important to demonstrate fine control of the morphology of individual nanoparticles, as well as precise spatial control of the position, orientation, and distances between multiple nanoparticles. In addition, dynamic control of nanomaterial assembly in response to multiple stimuli, with minimal or no error, and the reversibility of the assemblies are also required. In this Account, we summarize recent progress of using DNA as a powerful programmable tool to realize the above goals. First, inspired by the discovery of genetic codes in biology, we have discovered DNA sequence combinations to control different morphologies of nanoparticles during their growth process and have shown that these effects are synergistic or competitive, depending on the sequence combination. The DNA, which guides the growth of the nanomaterial, is stable and retains its biorecognition ability. Second, by taking advantage of different reactivities of phosphorothioate and phosphodiester backbone, we have placed phosphorothioate at selective positions on different DNA nanostructures including DNA tetrahedrons. Bifunctional linkers have been used to conjugate phosphorothioate on one end and bind nanoparticles or proteins on the other end. In doing so, precise control of distances between two or more nanoparticles or proteins with nanometer resolution can be achieved. Furthermore, by developing facile methods to functionalize two hemispheres of Janus nanoparticles with two different DNA sequences regioselectively, we have demonstrated directional control of nanomaterial assembly, where DNA strands with specific hybridization serve as orthogonal linkers. Third, by using functional DNA that includes DNAzyme, aptamer, and aptazyme, dynamic control of assemblies of gold nanoparticles, quantum dots, carbon nanotubes, and iron oxide nanoparticles in response to one or more stimuli cooperatively have been achieved, resulting in colorimetric, fluorescent, electrochemical, and magnetic resonance signals for a wide range of targets, such as metal ions, small molecules, proteins, and intact cells. Fourth, by mimicking biology, we have employed DNAzymes as proofreading units to remove errors in nanoparticle assembly and further used DNAzyme cascade reactions to modify or repair DNA sequences involved in the assembly. Finally, by taking advantage of different affinities of biotin and desthiobiotin toward streptavidin, we have demonstrated reversible assembly of proteins on DNA origami.
1. Introduction
Nanomaterials have shown enormous promise as the next-generation materials in many areas of applications ranging from catalysis to photonics, environmental detection, biomedical diagnostics and therapy.1−4 Such promise arises from unique properties conferred by these nanomaterials, such as their plasmonic and quantum confinement effects that largely depend on morphologies (e.g., shapes and surface structures)5 and interparticle distances.6,7 Therefore, to realize the full potential of these nanomaterials, it is important to control the morphologies of individual nanomaterials, as well as positioning and orientation of these nanomaterials in three-dimensional space. Furthermore, it is also important to exert selective and dynamic control over the assembly disassembly process in response to needs or stimuli.
To meet these requirements, directed-assembly methods are often used, but these methods rely on control of external variables, such as concentration, reaction time, pH, and solution temperature. In contrast, Nature produces high quality materials under almost constant external variables. For example, seashells and pearls from the ocean are produced from the same material under the same ambient conditions, yet form different final structures in response to biological signals. Learning from Nature would allow us to perform directed-assembly of different types of materials in response to multiple stimuli. In addition, the ability of Nature to generate “perfect” or error-free materials should be investigated as well.8,9 Finally, while many directed-assemblies performed in the laboratory are irreversible, biological assembly processes are often reversible, allowing the building of dynamic structures to perform their biological functions. By mimicking these biological processes in Nature, we can achieve similar controls for abiological materials at the nanoscale.
As the genetic storage material in biological systems, DNA stands out as an excellent material for providing both spatial and dynamic control, because of its predictable base-pair interactions and its programmable sequence. Recent progress has shown that DNA can form various geometries and motifs beyond the standard double helix and then assemble into 1D, 2D, and 3D structures.10−16 These DNA nanostructures can be further conjugated to nanomaterials through modified functional groups on the DNA, such as amine, sulfhydryl, or carboxylate groups.17−20 Furthermore, DNA has been found to serve as enzymes (called deoxyribozymes or DNAzymes) and recognition molecules like antibodies (called aptamers). This class of DNA, called functional DNA (fDNA), can be identified through a process called Systematic Evolution of Ligands by Exponential Enrichment (SELEX), in which fDNA is isolated from a library of ∼1015 random DNA sequences. This process allows fDNA to be highly selective for almost any targets ranging from metal ions, small organic molecules, to proteins, cancer cells, bacteria, and viruses.21−24 Therefore, these fDNA molecules are ideal choice for stimuli-responsive control of nanoassembly, as described in several other reviews.25−27
In this Account, we summarize our recent research progress of using these DNA molecules to provide morphological control of nanoparticles, spatial control of nanoassemblies, and dynamic control of the nanomaterial assembly and disassembly process in response to chemical or biological stimuli. Extensions of these methods for preparing error-free materials and achieving reversible assembly are also described.
2. Discovery of DNA “Genetic Codes” To Control Nanoparticle Morphology
The discovery of three-letter genetic codes, which consists of combinations of DNA nucleotides for the synthesis of proteins, is a foundation for modern biology. We hypothesized that a similar exploration of DNA sequence combinations could be used for controlling the morphology of nanomaterials. This discovery could have transformative potentials not only in advancing the fundamental knowledge of biomolecular interactions with nanomaterials, but also producing nanomaterials with novel morphology for different applications including catalysis, environmental detection and medical diagnosis and therapy.1−4
DNA molecules have been widely used as templates or scaffolds for assembly of nanoparticles (NPs) for various applications.28−32 The majority of the work reported so far, however, has involved NP functionalization with DNA after the NPs are synthesized,33−37 thereby the DNA could not influence the NP morphology. A few recent studies using DNA in NP synthesis reported encouraging findings that different DNA sequences can influence both the structure and property of the nanomaterials.38−43 However, few studies have comprehensively and systematically reported the effects of varying DNA sequences on the growth of NPs, and few rules have been summarized and applied toward the synthesis and fine control of NP morphology. To bridge this knowledge gap, we have used a seed-mediated growth method to investigate the influence of different DNA sequence combinations on the growth of NPs into different morphologies, upon preincubation of the NP seeds with different DNA sequences. In our initial study, we found that reduction of Au in the presence of a single stranded DNA (ssDNA) containing 30 repeating units of either cytosine (C30) or adenine (A30) transformed spherical AuNP seeds into stable spiky nanoflowers.44 In contrast, Au NP growth in the presence of T30 resulted in only larger spherical AuNPs (Figure 1a).44 Since the nanoflowers displayed excellent scattering properties, they were used for dark-field imaging of cancer cells, showing higher imaging contrast than corresponding spherical AuNPs. More interestingly, the DNA on the Au nanoflowers exhibited enhanced stability against dithiothreitol replacement in comparison to thiolated DNA on AuNP surface, probably due to partial embedding of the DNA into the nanoflowers during the growth process. Despite being partially embedded, the DNA on the surface retain its biorecognition and hybridization ability.44
Figure 1.

(a) Shape-controlled growth of AuNP from spherical seeds into Au nanoflowers and Au nanospheres in the presence of A30, C30, or T30 DNA molecules. Enhanced stability of the DNA on the Au nanoflowers can be attributed to partially imbedded DNA in the Au nanostructures. (b) Scheme summarizing different AuNP morphologies grown from Au prism seeds in the presence of different DNA sequences and the relationship between different sequence combinations in governing different morphologies (the sizes are on average 180 nm). (Reproduced with permission from ref 45. Copyright 2012 Wiley-VCH.) (c) Table summarizing the rules of different DNA bases and their combinations in controlling the AuNP morphology from Au nanoprism seeds.
Encouraged by these results, we further explored the effect of sequence combinations of DNA to investigate if there is any synergy between different DNA bases. Instead of using spherical AuNPs, we chose triangular Au nanoprism as the seed, because it contains corners, edges, and faces that may interact with different DNA bases and their combinations differently. This system produced a variety of morphologies such as smooth hexagonal plates, smooth six-pointed stars, round flat plates, and round rough plates, in the presence of G20, T30, C30, and A30, respectively (Figure 1b).45 The morphologies of AuNPs obtained were found to be independent of the length of the DNA strands, suggesting the important role of the nucleobases in controlling the shape. More excitingly, we found that there is a clear semiquantitative transition from one morphology (e.g., G20) to another (e.g., T30) when using different sequence combinations under identical experimental conditions. For example, a transition from smooth hexagon plates to smooth six-pointed stars was observed with increasing T to G ratio, that is, from G20 to T10G20, T15G15, T20G10, and T30 (Figure 1b). Similar transitions in other sequence combinations were also observed. These results suggest that DNA has the ability to control and encode the synthesis of NPs to form specific morphologies based on the DNA sequence, similar to the genetic codes for protein translation. Based on these results, a set of rules that govern the morphology and shape transition of the NPs for homogeneous and mixed DNA strands has been established (Figure 1c).
3. Using DNA for Spatial Control of Nanoscale Assembly
While it is significant to synthesize NPs with different morphologies, they are just building blocks for nanodevices. To achieve the assembly desired for performing specific functions, precise control of distances and orientation between two different NPs are critical.7,31,46 Here, we summarize our efforts for precise spatial control of nanomaterial assembly in different dimensions using DNA.
3.1. Precise Distance Control Using Phosphorothioate DNA and Bifunctional Linkers
A major trait of using DNA for assembly is the ability to control the distance between two NPs, due to the programmability of DNA through sequence-specific hybridization, and the rigidity of the double-stranded (ds) structure with a persistence length of ∼50 nm, and even longer when three-way or four-way junctions are employed. These properties have resulted in formation of NP assemblies with defined order and distance in 1D, 2D, and 3D structures.10−16 However, most of these methods require careful preparation and inefficient purification methods for obtaining monofunctionalized NPs, making it difficult to scale up for practical applications. Furthermore, it is also difficult to place the NPs at any desirable position on DNA nanostructures without affecting the stability. To overcome these limitations, we took advantage of phosphorothioate modified DNA and synthesized a bifunctional linker that can selectively conjugate to phosphorothioate backbone on one end (e.g., iodoacetamide) and bind to NPs selectively on the other end (e.g., maleimide, hydroxysuccinamide, or biotin) (Figure 2a).47,48 The conjugation was found to have high yields up to 90%. By introducing the phosphorothioate modifications at specific locations on a DNA, the placement of NPs at any position on DNA structures can be accomplished. Since the distance between two adjacent phosphodiesters on the DNA double helix is 0.34 nm, this method may in principle achieve sub-nanometer resolution. Using this method, assemblies of two AuNPs on dsDNA were demonstrated with DNA strands modified with phosphorothioate groups at 80, 70, or 60 base pairs apart (Figure 2b).47 The average distances between AuNPs, determined to be 27.2, 23.8, and 20.4 nm, respectively, based on scanning electron microscopy (SEM) images, matched well with the calculated distances. In addition to assembling NPs, this method has been used to attach different proteins with precise spatial control in 1D DNA double helix49 and 3D DNA tetrahedron.50 In the latter work, the vertices of the tetrahedron were modified with phosphorothioates and subsequently functionalized with biotins to conjugate with streptavidins (Figure 2c).50 This study demonstrated that our method can be used to attach proteins onto 3D structures that are known to be delicate and not very stable. Since the phosphorothioate modification occurs on the backbone, this functionalization method has little effect on the integrity and stability of the 3D structure compared to other methods. With proper linker choices, this method can be widely applied to attach any proteins or nanomaterials at any position of DNA structures with precisely defined distances.
Figure 2.

(a) Reaction of phosphorothioate modified DNA backbone with different iodoacetamide bilinkers. (b) SEM images showing sets of AuNP pairs with defined distances on phosphorothioated dsDNA (scale bar = 200 nm). (Reproduced with permission from ref (47). Copyright 2007 Wiley-VCH.) (c) Scheme of phosphorothioated DNA tetrahedron functionalized with biotin and streptavidin. (Reproduced with permission from ref (50). Copyright 2011 Wiley-VCH.)
3.2. Directional Control of NP Assembly Using DNA-Functionalized Janus Nanoparticles
In addition to distance control, the direction and orientation of building blocks are also important in nanoassembly. One of the ways to achieve directional control of nanoparticle assembly is the use of Janus nanoparticles (JNPs), which contain regions of distinct functionalities that are spatially separated from each other, thereby allowing directional interaction.51 Commonly used approaches to synthesize Janus particles include phase separation methods like emulsion polymerization, seeded growth, and microfluidics-assisted fabrication, as well as surface-based masking techniques and self-assembly of block polymers.51 Despite the promising roles of JNPs in directional control, however, it is difficult to functionalize JNP regioselectively in high yields. Several groups have reported the synthesis of metallic nanoparticles with anisotropic DNA functionalization by using microsized beads as masks for directional attachment of DNA28,52,53 and in situ phase separation synthesis of asymmetric DNA-functionalized NPs.54 We have functionalized JNPs with two orthogonal DNA sequences on each side of JNPs, via two methods: the surface template method55 and the colloidal ligand-competition method.56 The surface-template method is more modular and tunable in terms of synthesis of larger nanoparticles, while the colloidal ligand-competition method produces high yield JNPs with smaller sizes.
In the first method, the JNP was fabricated from electron beam-assisted evaporation of Au onto a monolayer of polystyrene (PS) beads where only half hemisphere of the PS beads was coated with Au.57,58 The Au hemisphere was first functionalized with thiolated DNA, D1. The regioselective functionalization of JNP was observed when Au nanospheres (AuNSs) containing the complementary strand, D1′, selectively assembled only onto the Au side of the JNP but not the PS side.55 No assembly was observed when noncomplementary DNA strands were used. Using this method, AuNSs of different sizes ranging from 15 to 80 nm with complementary DNA strands were assembled on the Au hemisphere of a 160 nm JNP, with well-controlled structure and yield (Figure 3a). Consistent with theoretical prediction, the number of AuNSs on each JNP decreased with increasing sizes of AuNS. In addition, a second DNA was introduced to the PS surface of the JNP through a bifunctional linker that connected the surface amine group on the JNP with thiolated DNA strands, D2. The second functional group enabled JNPs to be functionalized with two different types of AuNSs orthogonally and regioselectively (Figure 3a).
Figure 3.

(a) (Top) JNPs functionalized with two different DNA strands with spatial control showing regioselective assemblies. (Bottom) SEM figures show assembly of JNPs with AuNSs of 15–80 nm in size (scale bar = 200 nm). (Reproduced with permission from ref (55). Copyright 2012 American Chemical Society.) (b) Anisotropic nanoparticles prepared in a one-pot synthesis and the various nanostructures formed through DNA hybridization (scale bar = 25 nm). (Reproduced with permission from ref (56). Copyright 2013 American Chemical Society.)
Despite the modular and tunable features of the surface template method, it is still difficult to scale up the method, due to the limited surface area of the template when the JNPs are manufactured. To address this issue, we introduced a facile colloidal synthesis method that could form anisotropic NPs using ligand competition.56 In this method, AuNPs were incubated with a mixture of hydrophobic ligands (thiolated lipids), hydrophilic ligands (thiolated DNA strands) and amphiphilic polymers in DMF/H2O. Upon heating for 2 h and cooling to room temperature, the anisotropic NPs formed had partial polymer attachment (Figure 3b). The ligand on the Au surface that was exposed to the solvent can be exchanged with any other thiolated DNA for directional assembly. In addition, a second functionality was introduced on the polymer surface using amine functionalized DNA to conjugate to the carboxylic groups. In addition to the spatial control offered by DNA, the use of DNA-modified JNPs allows specific and regioselective assembly of NPs.
4. Using Functional DNA for Dynamic Control of Nanoscale Assembly
To achieve fully functional nanodevices, the assembly in nanoscale requires not only precise spatial control, but also dynamic control of the assemblies in response to stimuli. Furthermore, the assembly process needs to be error-free to ensure the consistent performance of these materials or devices. Finally, the assembly process is preferred to be reversible so that malfunctional components can be replaced modularly or new components can be introduced. Nature has used numerous reversible assembly processes to synthesize mostly error-free materials that simultaneously respond to multiple internal stimuli under ambient conditions. Inspired by Nature, we have mimicked many of these bioassembly processes using fDNA.
4.1. Dynamic Control in Response to a Stimulus
As mentioned in the Introduction, a number of fDNA sequences that respond to a wide range of targets have been identified.25 By linking the fDNA with nanomaterials, we have demonstrated a number of directed assembly processes in response to the target molecules as stimuli.59 The first demonstration is the use of a Pb2+-specific DNAzyme that contains a substrate strand, D3, with a single RNA base as the cleavage site, and an enzyme strand, D4. The AuNPs assembled through hybridization between D3 and complementary DNA strands on the AuNPs would appear purple due to plasmon coupling (Figure 4a).60 In the presence of a metal cofactor, like Pb2+ for 8–17 DNAzyme, the D3 will be cleaved into two shorter DNA fragments, whose melting temperatures of the hybridization with the complementary DNA strands on the AuNPs are below room temperature. As a result, dehybridization of the DNA fragments from D4 caused the disassembly of AuNPs and purple-to-red color change, making it a simple colorimetric sensor for Pb2+. This method can be generally applied to other DNAzymes, such as a uranyl-selective DNAzyme.61 Furthermore, assembly of AuNPs can also be controlled in response to stimuli, as demonstrated with the use of Cu2+-catalyzed DNA ligation.62
Figure 4.

(a) AuNP–DNAzyme conjugates responsive to the absence and presence of Pb2+, leading to assembled (blue) and disassembled (red) nanostructures, respectively. (b) AuNP–aptamer conjugates responding to adenosine. (c) Multiplex detection with assemblies of AuNPs and QDs, with 525 nm emission for adenosine and 585 nm emission for cocaine. (d) Aptazyme responsive to both adenosine and Pb2+. (Reproduced with permission from ref (64). Copyright 2007 American Chemical Society.) (e) Two cooperatively triggered disassemblies of NPs in the presence of adenosine and/or cocaine. (f) Time dependent changes of the ratio of extinction at 522 nm over 700 nm for the two designs in (e) in the presence of adenosine (Ade) and/or cocaine (Coc). (Reproduced with permission from ref (69). Copyright 2006 Wiley-VCH.)
In addition to metal ions, other molecules, such as organic molecules and proteins, can be used as triggers of assembly or disassembly of NPs, through the use of aptamers. For example, DNA-functionalized AuNPs can be assembled using a linker strand that contains an aptamer sequence for adenosine, D5 (Figure 4b). Upon binding of adenosine, the hybridization between D5 and the DNA strands on the AuNPs is weakened, resulting in disassembly of the AuNPs and a color change from blue to red.63 This method to trigger the release of AuNPs was further extended to other nanomaterials, such as quantum dots (QDs),64 carbon nanotubes (CNTs),65 and iron oxide NPs,66 resulting in fluorescent and magnetic resonance imaging signal changes for sensing of different targets. One of such examples is shown in Figure 4c,64 where two QDs of different emission wavelength (525 nm, QD525 and 585 nm, QD585) were functionalized with adenosine and cocaine aptamers, respectively. The QD525 emission was initially quenched due to energy transfer with nearby AuNPs. In the presence of adenosine, the AuNPs bound to the QD525 would be released, resulting in an increase of 525 nm emission. Similarly, 585 nm fluorescence would increase in the presence of cocaine. Because of the narrow emissions of QDs, simultaneous detection of both cocaine and adenosine was demonstrated.
4.2. Dynamic Control in Response to Multiple Stimuli
In biological processes, multiple events can be triggered simultaneously, often with cooperativity. Similarly, NP assembly can be designed to respond to multiple stimuli. An application of such design is to increase the selectivity for sensing, as it relies on the cooperation between two or more targets.67 One such example is the use of aptazymes, consisting of an aptamer and a DNAzyme, whose activity can be modulated in the presence of the aptamer target. As shown in Figure 4d, an adenosine aptamer sequence was inserted into a Pb2+-specific DNAzyme, inactivating the DNAzyme even in the presence of Pb2+. Once adenosine is added, the aptamer sequence would fold to regenerate the catalytic site,68 restoring the Pb2+-dependent DNAzyme activity. When conjugated to AuNPs, the color change observed from disassembly of AuNPs only occurred when both Pb2+ and adenosine were present.
In addition to aptazymes, two aptamers can be designed to trigger disassembly in response to either one or both of the targets cooperatively (Figure 4e).69 In this design where AuNPs were linked by a DNA strand with two aptamer sequences, the disassembly process occurred in the presence of either target of the aptamers. On the other hand, when both aptamer sequences were used to link the AuNPs, the disassembly process can occur only in the presence of both targets of the aptamers. The color change was observed and quantified using the ratio of absorbance at 522 nm over the absorbance at 700 nm (Figure 4f). These designs were also expanded to respond to K+ ions with a G-quadruplex forming aptamer in addition to either cocaine or adenosine triggers.69
4.3. Proofreading and Error Correction
A major challenge in nanomaterial assembly is that unexpected errors often occur during the assembly process, resulting in defects of obtained materials. A common practice to deal with this issue is to try to optimize the condition to avoid errors in assembly. However, it is extremely difficult to achieve perfect assembly without any error. Another solution to this problem is to design devices that can bypass the defects. While some defects may be tolerable in bulk materials, it can be highly detrimental at the nanoscale, leading to low quality or nonfunctional devices. Similar issues exist in biology, for example, protein synthesis that involves assembly of different amino acids is vulnerable to mistakes by erroneous incorporation of a wrong amino acid. Instead of trying to avoid errors, Nature has elegantly addressed this issue by developing proofreading units that can recognize and remove the errors. Inspired by such concept in biology, we have demonstrated the use of DNAzymes as proofreading units to recognize and remove the erroneous AuNPs containing the incorrect DNA strand D6 from the assembly, while leaving the AuNPs containing the perfect complementary DNA strand D7 intact (Figure 5a).70
Figure 5.

(a) Dynamic control of nanoassemblies where erroneous particles were proofread and removed. (b) A DNA sequence on the AuNP can be replaced with another sequence by using a cleavage DNAzyme (17E) and a ligation DNAzyme (E47). (c) Schematics and AFM images of DNA origami patterned with “I” and “i” using desthiobiotinylated and biotinylated DNA staples (scale bar = 70 nm). (Reproduced with permission from ref (72). Copyright 2013 American Chemical Society.)
In addition to removing erroneous AuNPs, we can further replace the incorrect AuNPs with a correct AuNP.71 This task is achieved by using an enzyme cascade reaction that combines a cleavage DNAzyme (17E) and a ligation DNAzyme (E47). Two DNAzymes work cooperatively to recognize and cleave DNA from D8 or D9, into D10, respectively, followed by ligating D10 to D11. As a result, the erroneous single ribonucleotide in D8 was converted to deoxyribonucleotide in D11. Similarly, the sequence in D9 was modified into D11.
4.4. Controlled Reversible Assembly
Reversible nanoassembly where one component can be replaced with another is important for fine-tuning functions as well as correcting mistakes. While numerous methods have been published in nanomaterial assembly, most of them are irreversible. To demonstrate reversible assembly, we take advantage of the difference in binding affinity between biotin (Kd ≈ 10–15 M) and desthiobiotin (Kd ≈ 10–11 M) toward streptavidin, and have demonstrated the ability to perform reversible assembly of streptavidin that allows conveying of an encrypted message encoded on DNA origami (Figure 5c).72 Upon addition of biotin to the encoded DNA origami that displayed uppercase "I", the streptavidin bound on desthiobiotin would be stripped away by biotin, revealing the lowercase “i” as observed in Figure 5c. The pattern can be reversed to the uppercase “I” with further addition of streptavidin. We have also demonstrated encryption of a message “NANO” in Morse code on DNA origami that can only be revealed with the addition of biotin. This process can further be reset to initial state with the addition of streptavidin.
5. Conclusion and Outlook
Through examples described in this Account, we have demonstrated DNA as a powerful tool to advance different aspects of nanotechnology, first as a versatile capping ligand to systematically control the NP morphology, and then as a template for precise spatial control of NP assembly, including precise distance control, achieved by selective modification of DNA backbone with phosphorothioates, and directional control, accomplished by regioselective functionalization of JNP with DNA strands. Furthermore, by using fDNA such as DNAzyme and aptamer, dynamic control of assemblies of different NPs, including AuNPs, QDs, CNTs, and iron oxide NPs, in response to multiple stimuli with cooperativity has been demonstrated, resulting in colorimetric and fluorescent sensors as well as magnetic resonance imaging contrast agents for a wide range of analytes. The DNAzymes can be further used as proofreading units to remove errors and replace them with correct sequences for assembly. Finally, reversible assembly is demonstrated by taking advantage of different affinities of biotin and its analogue toward streptavidin.
While these examples are encouraging, the detailed mechanism by which the DNA performs these functions remains to be understood. For example, how different DNA sequence combinations fine-tune the NP morphologies is unclear. One factor is the different affinities of the bases (A > C > G > T) for gold,73 but the exact mechanism is much more complicated, including the effects of secondary or even tertiary structure of the DNA. Real-time monitoring of the NP growth and identification of the interaction of DNA with metal nanoparticle are required to elucidate the growth process. Once the mechanism is understood, using DNA to code and predict the growth of NPs would be possible. It would be interesting to discover if the DNA codes can be applied to control the morphology of other nanomaterials, and if similar rules apply. Because these DNA-encoded syntheses are carried in solution with > 95% yield, resulting in uniform shapes, it should be straightforward to scale up the method. However, even though DNA is becoming cheaper, it is still more expensive than most commodity chemicals. Therefore, applications of the methods described in this Account are mostly fundamental understanding of the interactions between biomolecules and nanomaterials as well as application of the fDNA-based sensing and imaging, where the amount of materials needed is small, due to high sensitivity.
The precise spatial control provides an excellent opportunity to investigate functional properties of two or more NPs brought together in a short distance and with defined angle of interactions, such as chiral optical properties for sensing or for negative refractive index materials.46,74,75 Since the method can be applied to control distance of both NPs and biomolecules, it would be interesting to explore assemblies of both proteins and NPs in the same system and their functional properties, such as self-propagating, multistep catalysis and reactions.
Both directional control of assembly using JNPs and dynamic control have a lot of potential for future explorations and applications. The directional control afforded by the regioselective placement of DNA strands on the two hemisphere of the JNPs, combined with dynamic control of NP assemblies in response to multiple stimuli, offers opportunities for 3D assemblies of novel structures and properties.76 The principles demonstrated in the reversible assembly of proteins can be further expanded for reversible assembly of other nanomaterials such as metallic or semiconductor nanoparticles through the conjugation of biotin and desthiobiotin. Such reversible assembly will allow their properties to be reversibly tuned. Finally, although the concepts of proofreading/error correction and reversible assembly have been demonstrated, the method needs to be expanded to other NP systems for broader applications.
Acknowledgments
We wish to thank all Lu group members who made significant contributions to work described in this Account and the U.S. National Science Foundation (DMR-0117792, CTS-0120978, and DMI-0328162) and National Institutes of Health (ES016865) for financial support. L.H.T. was funded at UIUC from NIH National Cancer Institute Alliance for Nano-technology in Cancer “Midwest Cancer Nanotechnology Training Center” Grant R25 CA154015A.
Biographies
Li Huey Tan is currently a Ph.D. student at UIUC. She received her B.Sc. degree in chemistry from Nanyang Technological University, Singapore in 2008. In 2009, she joined Prof. Yi Lu’s group at UIUC. Her current scientific interests are focused on DNA and metal nanoparticle hybrids for self-assembly.
Hang Xing is a Ph.D. student in Prof. Yi Lu’s group in the department of Chemistry at UIUC. He received his B.S. (2005, chemistry) and M.S. (2008, inorganic chemistry) degrees from Nanjing University, China. His current research interests focus on using DNA–nanomaterial conjugates for biomedical applications.
Dr. Yi Lu is Jay and Ann Schenck Professor in the Departments of Chemistry, Biochemistry, Material Science and Engineering, and Bioengineering. He received his B.S. degree from Peking University in 1986 and his Ph.D. degree from UCLA in 1992 under Dr. Joan S. Valentine. After 2 years of postdoctoral research in Dr. Harry B. Gray’s group at Caltech, Dr. Lu started his own independent career at UIUC in 1994. He is also a member of the Center for Biophysics and Computational Biology and Beckman Institute for Advanced Science and Technology. His group interests are in bioinorganic chemistry, biomaterial chemistry, and bioanalytical chemistry.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Lu Z.; Yin Y. Colloidal nanoparticle clusters: functional materials by design. Chem. Soc. Rev. 2012, 41, 6874–6887. [DOI] [PubMed] [Google Scholar]
- Nie Z.; Petukhova A.; Kumacheva E. Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nature Nanotech. 2010, 5, 15–25. [DOI] [PubMed] [Google Scholar]
- Chen X.-J.; Sanchez-Gaytan B. L.; Qian Z.; Park S.-J. Noble metal nanoparticles in DNA detection and delivery. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2012, 4, 273–290. [DOI] [PubMed] [Google Scholar]
- Schroeder A.; Heller D. A.; Winslow M. M.; Dahlman J. E.; Pratt G. W.; Langer R.; Jacks T.; Anderson D. G. Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 2012, 12, 39–50. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Xiong Y.; Lim B.; Skrabalak S. E. Shape-Controlled Synthesis of Metal Nanocrystals: Simple Chemistry Meets Complex Physics?. Angew. Chem., Int. Ed. 2009, 48, 60–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J. A.; Wu C.; Bao K.; Bao J.; Bardhan R.; Halas N. J.; Manoharan V. N.; Nordlander P.; Shvets G.; Capasso F. Self-Assembled Plasmonic Nanoparticle Clusters. Science 2010, 328, 1135–1138. [DOI] [PubMed] [Google Scholar]
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Plasmons in Strongly Coupled Metallic Nanostructures. Chem. Rev. 2011, 111, 3913–3961. [DOI] [PubMed] [Google Scholar]
- Thomas M. J.; Platas A. A.; Hawley D. K. Transcriptional Fidelity and Proofreading by RNA Polymerase II. Cell 1998, 93, 627–637. [DOI] [PubMed] [Google Scholar]
- Blanchard S. C.; Gonzalez R. L.; Kim H. D.; Chu S.; Puglisi J. D. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol. 2004, 11, 1008–1014. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. DNA in a material world. Nature 2003, 421, 427–431. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- He Y.; Ye T.; Su M.; Zhang C.; Ribbe A. E.; Jiang W.; Mao C. Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature 2008, 452, 198–201. [DOI] [PubMed] [Google Scholar]
- Lo P. K.; Karam P.; Aldaye F. A.; McLaughlin C. K.; Hamblin G. D.; Cosa G.; Sleiman H. F. Loading and selective release of cargo in DNA nanotubes with longitudinal variation. Nature Chem. 2010, 2, 319–328. [DOI] [PubMed] [Google Scholar]
- Pinheiro A. V.; Han D. R.; Shih W. M.; Yan H. Challenges and opportunities for structural DNA nanotechnology. Nature Nanotech. 2011, 6, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J. L.; Liu M. H.; Liu Y.; Yan H. Spatially-Interactive Biomolecular Networks Organized by Nucleic Acid Nanostructures. Acc. Chem. Res. 2012, 45, 1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y.; Ong L. L.; Shih W. M.; Yin P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacca B.; Niemeyer C. M. Functionalization of DNA nanostructures with proteins. Chem. Soc. Rev. 2011, 40, 5910–5921. [DOI] [PubMed] [Google Scholar]
- Stadler A.; Chi C.; van der Lelie D.; Gang O. DNA-incorporating nanomaterials in biotechnological applications. Nanomedicine 2010, 5, 319–334. [DOI] [PubMed] [Google Scholar]
- Li L.-L.; Wu P.; Hwang K.; Lu Y. An Exceptionally Simple Strategy for DNA-Functionalized Up-Conversion Nanoparticles as Biocompatible Agents for Nanoassembly, DNA Delivery, and Imaging. J. Am. Chem. Soc. 2013, 135, 2411–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Hwang J.-H.; Kumar S.; Nam J.-M. Tuning and assembling metal nanostructures with DNA. Chem. Commun. 2013, 49, 2597–2609. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 1992, 355, 850–852. [DOI] [PubMed] [Google Scholar]
- Breaker R. R. In Vitro Selection of Catalytic Polynucleotides. Chem. Rev. 1997, 97, 371–390. [DOI] [PubMed] [Google Scholar]
- Tan W.; Donovan M. J.; Jiang J. Aptamers from Cell-Based Selection for Bioanalytical Applications. Chem. Rev. 2013, 113, 2842–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Cao Z.; Lu Y. Functional Nucleic Acid Sensors. Chem. Rev. 2009, 109, 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navani N. K.; Li Y. Nucleic acid aptamers and enzymes as sensors. Curr. Opin. Chem. Biol. 2006, 10, 272–281. [DOI] [PubMed] [Google Scholar]
- Willner I.; Shlyahovsky B.; Zayats M.; Willner B. DNAzymes for sensing, nanobiotechnology and logic gate applications. Chem. Soc. Rev. 2008, 37, 1153–1165. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lu F.; Yager K. G.; van der Lelie D.; Gang O. A general strategy for the DNA-mediated self-assembly of functional nanoparticles into heterogeneous systems. Nature Nanotech. 2013, 8, 865–872. [DOI] [PubMed] [Google Scholar]
- Tan S. J.; Campolongo M. J.; Luo D.; Cheng W. Building plasmonic nanostructures with DNA. Nature Nanotech. 2011, 6, 268–276. [DOI] [PubMed] [Google Scholar]
- Macfarlane R. J.; Lee B.; Jones M. R.; Harris N.; Schatz G. C.; Mirkin C. A. Nanoparticle Superlattice Engineering with DNA. Science 2011, 334, 204–208. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Schreiber R.; Fan Z.; Pardatscher G.; Roller E.-M.; Hogele A.; Simmel F. C.; Govorov A. O.; Liedl T. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 2012, 483, 311–314. [DOI] [PubMed] [Google Scholar]
- Surwade S. P.; Zhou F.; Wei B.; Sun W.; Powell A.; O’Donnell C.; Yin P.; Liu H. Nanoscale Growth and Patterning of Inorganic Oxides Using DNA Nanostructure Templates. J. Am. Chem. Soc. 2013, 135, 6778–6781. [DOI] [PubMed] [Google Scholar]
- Alivisatos A. P.; Johnsson K. P.; Peng X.; Wilson T. E.; Loweth C. J.; Bruchez M. P.; Schultz P. G. Organization of ’nanocrystal molecules’ using DNA. Nature 1996, 382, 609–611. [DOI] [PubMed] [Google Scholar]
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- Wang Z. D.; Lu Y. Functional DNA directed assembly of nanomaterials for biosensing. J. Mater. Chem. 2009, 19, 1788–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A.; Rivera gil P.; Zhang F.; Zanella M.; Parak W. J. Biological applications of gold nanoparticles. Chem. Soc. Rev. 2008, 37, 1896–1908. [DOI] [PubMed] [Google Scholar]
- Li L.-L.; Zhang R.; Yin L.; Zheng K.; Qin W.; Selvin P. R.; Lu Y. Biomimetic Surface Engineering of Lanthanide-Doped Upconversion Nanoparticles as Versatile Bioprobes. Angew. Chem., Int. Ed. 2012, 51, 6121–6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertig M.; Ciacchi L. C.; Seidel R.; Pompe W.; De Vita A. DNA as a selective metallization template. Nano Lett. 2002, 2, 841–844. [Google Scholar]
- Richards C. I.; Choi S.; Hsiang J.-C.; Antoku Y.; Vosch T.; Bongiorno A.; Tzeng Y.-L.; Dickson R. M. Oligonucleotide-Stabilized Ag Nanocluster Fluorophores. J. Am. Chem. Soc. 2008, 130, 5038–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru A.; Dutta S.; Jentzsch E.; Gothelf K.; Mokhir A. Selective dsDNA-Templated Formation of Copper Nanoparticles in Solution. Angew. Chem., Int. Ed. 2010, 49, 5665–5667. [DOI] [PubMed] [Google Scholar]
- Tikhomirov G.; Hoogland S.; Lee P. E.; Fischer A.; Sargent E. H.; Kelley S. O. DNA-based programming of quantum dot valency, self-assembly and luminescence. Nature Nanotech. 2011, 6, 485–490. [DOI] [PubMed] [Google Scholar]
- Choi S.; Dickson R. M.; Yu J. Developing luminescent silver nanodots for biological applications. Chem. Soc. Rev. 2012, 41, 1867–1891. [DOI] [PubMed] [Google Scholar]
- Lim D.-K.; Jeon K.-S.; Hwang J.-H.; Kim H.; Kwon S.; Suh Y. D.; Nam J.-M. Highly uniform and reproducible surface-enhanced Raman scattering from DNA-tailorable nanoparticles with 1-nm interior gap. Nature Nanotech. 2011, 6, 452–460. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zhang J.; Ekman J. M.; Kenis P. J. A.; Lu Y. DNA-Mediated Control of Metal Nanoparticle Shape: One-Pot Synthesis and Cellular Uptake of Highly Stable and Functional Gold Nanoflowers. Nano Lett. 2010, 10, 1886–1891. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Tang L.; Tan L. H.; Li J.; Lu Y. Discovery of the DNA “Genetic Code” for Abiological Gold Nanoparticle Morphologies. Angew. Chem., Int. Ed. 2012, 51, 9078–9082. [DOI] [PubMed] [Google Scholar]
- Xu L.; Kuang H.; Xu C.; Ma W.; Wang L.; Kotov N. A. Regiospecific Plasmonic Assemblies for in Situ Raman Spectroscopy in Live Cells. J. Am. Chem. Soc. 2011, 134, 1699–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Wernette D. P.; Yigit M. V.; Liu J.; Wang Z.; Lu Y. Site-Specific Control of Distances between Gold Nanoparticles Using Phosphorothioate Anchors on DNA and a Short Bifunctional Molecular Fastener. Angew. Chem., Int. Ed. 2007, 46, 9006–9010. [DOI] [PubMed] [Google Scholar]
- Fidanza J. A.; Ozaki H.; McLaughlin L. W. Site-specific labeling of DNA sequences containing phosphorothioate diesters. J. Am. Chem. Soc. 1992, 114, 5509–5517. [Google Scholar]
- Lee J. H.; Wong N. Y.; Tan L. H.; Wang Z.; Lu Y. Controlled Alignment of Multiple Proteins and Nanoparticles with Nanometer Resolution via Backbone-Modified Phosphorothioate DNA and Bifunctional Linkers. J. Am. Chem. Soc. 2010, 132, 8906–8908. [DOI] [PubMed] [Google Scholar]
- Wong N. Y.; Zhang C.; Tan L. H.; Lu Y. Site-Specific Attachment of Proteins onto a 3D DNA Tetrahedron through Backbone-Modified Phosphorothioate DNA. Small 2011, 7, 1427–1430. [DOI] [PubMed] [Google Scholar]
- Walther A.; Müller A. H. E. Janus Particles: Synthesis, Self-Assembly, Physical Properties, and Applications. Chem. Rev. 2013, 113, 5194–5261. [DOI] [PubMed] [Google Scholar]
- Xu X.; Rosi N. L.; Wang Y.; Huo F.; Mirkin C. A. Asymmetric Functionalization of Gold Nanoparticles with Oligonucleotides. J. Am. Chem. Soc. 2006, 128, 9286–9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-W.; Kim J.-H.; Deaton R. DNA-Linked Nanoparticle Building Blocks for Programmable Matter. Angew. Chem., Int. Ed. 2011, 50, 9185–9190. [DOI] [PubMed] [Google Scholar]
- Lee J.-H.; Kim G.-H.; Nam J.-M. Directional Synthesis and Assembly of Bimetallic Nanosnowmen with DNA. J. Am. Chem. Soc. 2012, 134, 5456–5459. [DOI] [PubMed] [Google Scholar]
- Xing H.; Wang Z.; Xu Z.; Wong N. Y.; Xiang Y.; Liu G. L.; Lu Y. DNA-Directed Assembly of Asymmetric Nanoclusters Using Janus Nanoparticles. ACS Nano 2011, 6, 802–809. [DOI] [PubMed] [Google Scholar]
- Tan L. H.; Xing H.; Chen H.; Lu Y. Facile and Efficient Preparation of Anisotropic DNA-Functionalized Gold Nanoparticles and Their Regioselective Assembly. J. Am. Chem. Soc. 2013, 135, 17675–17678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perro A.; Reculusa S.; Ravaine S.; Bourgeat-Lami E.; Duguet E. Design and synthesis of Janus micro- and nanoparticles. J. Mater. Chem. 2005, 15, 3745–3760. [Google Scholar]
- Wu L. Y.; Ross B. M.; Hong S.; Lee L. P. Bioinspired Nanocorals with Decoupled Cellular Targeting and Sensing Functionality. Small 2010, 6, 503–507. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Liu J. Smart Nanomaterials Inspired by Biology: Dynamic Assembly of Error-Free Nanomaterials in Response to Multiple Chemical and Biological Stimuli. Acc. Chem. Res. 2007, 40, 315–323. [DOI] [PubMed] [Google Scholar]
- Liu J. W.; Lu Y. Accelerated color change of gold nanoparticles assembled by DNAzymes for simple and fast colorimetric Pb2+ detection. J. Am. Chem. Soc. 2004, 126, 12298–12305. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Wang Z.; Liu J.; Lu Y. Highly Sensitive and Selective Colorimetric Sensors for Uranyl (UO22+): Development and Comparison of Labeled and Label-Free DNAzyme-Gold Nanoparticle Systems. J. Am. Chem. Soc. 2008, 130, 14217–14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Lu Y. Colorimetric Cu2+ detection with a ligation DNAzyme and nanoparticles. Chem. Commun. 2007, 4872–4874. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. Fast Colorimetric Sensing of Adenosine and Cocaine Based on a General Sensor Design Involving Aptamers and Nanoparticles. Angew. Chem., Int. Ed. 2006, 45, 90–94. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lee J. H.; Lu Y. Quantum Dot Encoding of Aptamer-Linked Nanostructures for One-Pot Simultaneous Detection of Multiple Analytes. Anal. Chem. 2007, 79, 4120–4125. [DOI] [PubMed] [Google Scholar]
- Yim T.-J.; Liu J.; Lu Y.; Kane R. S.; Dordick J. S. Highly Active and Stable DNAzyme–Carbon Nanotube Hybrids. J. Am. Chem. Soc. 2005, 127, 12200–12201. [DOI] [PubMed] [Google Scholar]
- Yigit M. V.; Mazumdar D.; Lu Y. MRI Detection of Thrombin with Aptamer Functionalized Superparamagnetic Iron Oxide Nanoparticles. Bioconjugate Chem. 2008, 19, 412–417. [DOI] [PubMed] [Google Scholar]
- Wang D. Y.; Lai B. H. Y.; Sen D. A General Strategy for Effector-mediated Control of RNA-cleaving Ribozymes and DNA Enzymes. J. Mol. Biol. 2002, 318, 33–43. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. Adenosine-Dependent Assembly of Aptazyme-Functionalized Gold Nanoparticles and Its Application as a Colorimetric Biosensor. Anal. Chem. 2004, 76, 1627–1632. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. Smart Nanomaterials Responsive to Multiple Chemical Stimuli with Controllable Cooperativity. Adv. Mater. 2006, 18, 1667–1671. [Google Scholar]
- Liu J.; Wernette D. P.; Lu Y. Proofreading and Error Removal in a Nanomaterial Assembly. Angew. Chem., Int. Ed. 2005, 44, 7290–7293. [DOI] [PubMed] [Google Scholar]
- Xiang Y.; Wang Z.; Xing H.; Lu Y. Expanding DNAzyme functionality through enzyme cascades with applications in single nucleotide repair and tunable DNA-directed assembly of nanomaterials. Chem. Sci. 2013, 4, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N. Y.; Xing H.; Tan L. H.; Lu Y. Nano-Encrypted Morse Code: A Versatile Approach to Programmable and Reversible Nanoscale Assembly and Disassembly. J. Am. Chem. Soc. 2013, 135, 2931–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura-Suda H.; Petrovykh D. Y.; Tarlov M. J.; Whitman L. J. Base-Dependent Competitive Adsorption of Single-Stranded DNA on Gold. J. Am. Chem. Soc. 2003, 125, 9014–9015. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Park Y.-S.; Li J.; Lu X.; Zhang W.; Zhang X. Negative Refractive Index in Chiral Metamaterials. Phys. Rev. Lett. 2009, 102, 023901. [DOI] [PubMed] [Google Scholar]
- Wu X.; Xu L.; Liu L.; Ma W.; Yin H.; Kuang H.; Wang L.; Xu C.; Kotov N. A. Unexpected Chirality of Nanoparticle Dimers and Ultrasensitive Chiroplasmonic Bioanalysis. J. Am. Chem. Soc. 2013, 135, 18629–18636. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Whitmer J. K.; Jiang S.; Bae S. C.; Luijten E.; Granick S. Supracolloidal Reaction Kinetics of Janus Spheres. Science 2011, 331, 199–202. [DOI] [PubMed] [Google Scholar]