

Abstract
Nephrotoxicity is a common complication of cisplatin chemotherapy that limits its clinical use; however, the mechanisms underlying cisplatin-mediated nephrotoxicity are not fully understood. In this study, we investigated the role of anaphylatoxin C5a in the pathogenesis of cisplatin-mediated nephrotoxicity. Our data show that cisplatin-induced renal injury is significantly reduced in C5- or C5aR-deficient mice. However, pretreatment with C5 or C5a restores sensitivity to cisplatin-induced nephrotoxicity in C5-deficient mice. In wild-type mice, administration of cisplatin triggers the increased renal expression of multiple cytokines and caspases. This induction is diminished in C5-deficient mice, which is restored by pretreatment with C5 or C5a proteins. Interestingly, renal injury induced by cisplatin is similar between wild-type and CD59ab double knockout mice, and the formation of membrane attack complexes (MACs) by cisplatin in the kidney is diminished in C5-deficient mice, but not in C5aR-deficient mice. In conclusion, our findings suggest that C5a plays an important role in the pathogenesis of cisplatin nephrotoxicity. Likely, C5a binds to C5aR, leading to induction of proinflammatory cytokines and inflammation. The formation of MACs does not appear to contribute to the nephrotoxicity of cisplatin based on our study results.
Keywords: cytokines, caspase, STAT3, NF-κB
a platinum compound, cisplatin, is one of the most potent chemotherapeutic agents available that is widely used to treat a variety of malignancies, including ovarian, lung, head, and neck cancers, as well as testicular and bladder tumors (25). Unfortunately, at high doses, cisplatin induces cumulative and dose-dependent nephrotoxicity, a major side effect that restricts maximization of therapeutic effects. In clinical practice, approximately one-third of patients experience renal dysfunction after treatment with cisplatin (24). Recently, a number of platinum coordination complexes have been developed and studied to find related compounds that can demonstrate superior efficacy, fewer side effects, less cross-resistance, or improved pharmacological characteristics compared with the cisplatin parent compound. A few related compounds possessing similar or enhanced therapeutic efficacy have been found through preclinical screening; however, these compounds are also associated with nephrotoxicity of varying degrees of severity (10).
Cisplatin-induced nephrotoxicity is a complex process. After administration, cisplatin uptake is greatest within proximal tubular cells of the inner cortex and outer medulla. As a result, these segments are the major sites of cisplatin-induced renal injury, and the loss of tubular cells by necrosis and apoptosis is followed by infiltration of inflammatory cells and fibroproliferative changes. Cisplatin cytotoxicity is likely caused by a combination of multiple mechanisms, involving DNA damage (14), caspase activation (8), mitochondrial dysfunction (28), and formation of reactive oxygen species (13). However, the exact molecular and cellular mechanisms by which cisplatin induces nephrotoxicity remain unclear. Recently, results from multiple studies strongly implicate the importance of inflammatory mechanisms in the pathogenesis of cisplatin-induced nephrotoxicity; specifically through the recruitment of inflammatory cells, such as macrophages and leukocytes (7, 12, 34). It also appears that cisplatin induces increased renal expression of a variety of inflammatory chemokines and cytokines, such as tumor necrosis (TNF)-α, transforming growth factor (TGF)-β, interleukin-1β (IL-1β), and intercellular adhesion molecular (ICAM)-1 (7). Reportedly, cisplatin-induced kidney injury depends on TNF-α, since TNF-α-deficient mice and TNF-α antibody-treated wild-type (WT) mice display resistance to cisplatin-induced kidney damage (23). Importantly, urinary levels of complement terminal complexes (C5b-9), the final end-product of complement activation, is increased in cisplatin-treated patients and is associated with nephrotoxicity (29). These findings, coupled with the important role of complements in immune and inflammatory responses, have led us to hypothesize that activation of the complement system may contribute to cisplatin-induced nephrotoxicity.
The complement system is one of the main effectors employed by the immune system in host defenses, and many by-products generated during complement activation are mediators of inflammation. The complement system consists of ∼30 soluble- and membrane-bound proteins. In plasma, complement proteins interact with one another in one of three different sequential activation cascades known as the classical, alternative, and lectin pathways. Eventually, all three pathways converge, with complement proteins C3 and C5, into one terminal cascade that leads to formation of the membrane attack complex (MAC). The MAC is a macromolecular pore that has the ability to insert itself into cell membranes and lysed bacteria and heterologous cells (22). The small complement fragments generated during complement activation, C3a and C5a, are known anaphylatoxins that induce several biological responses. Uncontrolled complement activation can lead to tissue inflammation or damage, which occurs in many immune-complex-mediated disease such as rheumatoid arthritis, asthma, liver diseases, and renal diseases (1, 4, 18, 22, 31). For example, activation of the complement system has been shown to contribute to the pathogenesis of ischemia-reperfusion renal injury, renal transplantation rejection, immune complex-mediated glomerulonephritis, lupus nephritis, IgA nephropathy, membranoproliferative glomerulonephritis, and proteinuria-mediated renal damage (2, 4–6, 30, 32, 33, 36). In general, activation of the complement system induces MAC formation in tubular cells. The insertion of sublytic amounts of C5b-9 in the cellular membranes of tubular cells results in the production of proinflammatory cytokines that contribute to renal damage (2, 4–6, 30, 32, 33, 36). Conversely, the complement system may also exert protective effects in renal disease; in particular, early activation of the complement system results in clearance of immune complexes (4). At present, the role of the activated complements system in cisplatin-induced renal damage remains unknown.
To investigate whether complement activation contributes to cisplatin-induced nephrotoxicity, C5-deficient (C5KO), C5aR knockout mice (C5aRKO), and CD59ab knockout mice (CD59abKO) were used. Deficiency in C5 or C5a receptor (C5aR) proteins, but not CD59, which is a MAC regulator (21), attenuated cisplatin-induced nephrotoxicity. Moreover, administration of C5 or C5a restored cisplatin-induced nephrotoxicity in C5-deficient mice. These results indicate that C5a is an important pathogenic factor in cisplatin-induced renal injury, suggesting that C5aR blockade may be a novel strategy to prevent cisplatin-induced nephrotoxicity.
MATERIALS AND METHODS
Mice.
Pathogen-free, 6- to 8-wk-old male C5-deficient mice (B10.D2-Hc0H2dH2-T18c/oCnJ, stock number 000461) and corresponding control mice C57BL/10Sn, as well as C5aRKO (C.129S4-C5ar1tm1Cge/J, stock number 006845) on a BALB/c background and BALB/c control mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The CD59ab double knockout mice on a C57BL6 background were generated as described previously (20). All animals were kept in a temperature-controlled environment with a 12:12-h light-dark cycle and were allowed free access to food and water at all times. All animal experiments were conducted in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee.
Materials.
Purified human C5a and C5 proteins were purchased from Complement Technology (Tyler, TX). The C5a and C5 proteins was >90% pure. These proteins were not treated to remove endotoxin contamination during the purification process. However, we checked endotoxin levels in the C5 and C5a protein preparations by using a kit from Sigma (St. Louis, MO). Appreciable endotoxin levels were not detected. Cisplatin was purchased from Sigma.
Treatment of mice with cisplatin, C5, or C5a proteins.
Mice were treated with cisplatin (dissolved in saline, 20 μg/g body wt ip) or saline and killed 72 h postinjection. The kidneys were either immediately fixed with 4% formaldehyde and 2% glutaraldehyde or snap-frozen and kept at −80°C until examination. In some groups, C5KO mice were injected with human C5 (3 μg/g body wt ip) or C5a (1.7 μg/g body wt ip) proteins 10 min before cisplatin administration. The C5 and C5a proteins were dissolved in sterile PBS.
Measurement of serum complement activity with hemolytic assay.
Rabbit erythrocytes were washed in PBS, and a 1% suspension was incubated with an equal volume of mouse anti-rabbit erythrocyte antiserum (1/100 dilution in PBS; 15 min at 37°C). The Ab-sensitized rabbit erythrocytes were washed with veronal buffered saline (VBS) and resuspended at 1% hematocrit. Fifty microliters of the cell suspension plus 50 μl of mouse serum (40% in VBS) were added to 96-well plates in triplicate, and the plates were incubated for 30 min at 37°C. Nonlysed cells were removed by centrifugation, and hemoglobin in the supernatant was measured as OD414. Percent hemolysis in each well was calculated as described previously (19).
Creatinine and blood urea nitrogen assay.
Serum levels of creatinine (Cr) and blood urea nitrogen (BUN) were measured using kits obtained from Drew Scientific (Barrow-in-Furness, UK).
Western blotting.
Anti-STAT3, anti-phospho-STAT3 (Tyr705), and anti-p NF-κB p105 (Ser933) antibodies were obtained from Cell Signaling Technology (Danvers, MA). Renal nuclear protein extracts were purified using an extraction kit (Thermo Scientific; Rockford, IL). The nuclear protein samples were mixed in Laemmli loading buffer, boiled for 5 min, and then subjected to SDS-PAGE (40 mg in each well). After electrophoresis, proteins were transferred onto nitrocellulose membranes and blotted against a primary antibody (1:1,000 dilution) for 16 h. Membranes were washed with TPBS and incubated with a secondary antibody linked with alkaline phosphatase (1:2,500 dilution) (Amersham Pharmacia Biotech, Piscataway, NJ) for 2 h, followed by incubation with enhanced chemifluorescent substrate (Amersham Pharmacia Biotech). Protein bands were visualized by scanning the membrane with PharosFx plus Molecular Imager (Bio-Rad Laboratories, Hercules, CA).
Immunohistochemistry.
Paraffin-embedded sections were cut, deparaffinized, and hydrated by soaking in 100% xylene and descending ethanol, followed by autoclave treatment. Next, sections were incubated in 0.3% H2O2 in PBS to block endogenous peroxidase activity. The sections were incubated with either anti-cleaved caspase 3 (1:100 dilution, Cell Signaling Technology), anti-MPO (Invitrogen, Carlsbad, CA), anti-nitrotyrosine (1:200 dilution, Cayman Chemical, Ann Arbor, MI), or anti-mouse C3b/iC3b/C3c antibody (Cell Sciences, Canton, MA) overnight at 4°C in a moist chamber. Biotinylated secondary antibodies and ABC Reagent were applied. Color development was induced by incubation with a DAB kit (Vector Laboratories, Burlingame, CA) for 3–5 min, and specific staining was visualized by light microscopy.
Periodic acid-Schiff staining.
Following fixation of the kidneys with 10% formalin, renal tissues were sliced and stained with periodic acid-Schiff (PAS) for histological examination. Tubular damage in PAS-stained sections were examined by light microscopy (×200 magnification) and calculated as the percentage of cortical tubules showing epithelial necrosis (0 = normal; 1 = <10%; 2 = 10–25%; 3 = 26–75%; 4 = >75%). Five sections from each sample were randomly selected for scoring by two independent investigators. Five to ten mice were used in each group.
Terminal deoxynucleotidyl transferase-mediated uridine triphosphate nick-end labeling.
Terminal deoxynucleotidyl transferase-mediated uridine triphosphate nick-end labeling (TUNEL) staining was performed using an in situ apoptosis detection kit according to the manufacturer's instructions (Chemicon International, Temecula, CA) and examined by light microscopy.
Malondialdehyde assay.
Malondialdehyde (MDA) formation was used to quantify the tissue. Briefly, tissues were homogenized (100 mg/ml) in 1.15% KCl buffer, and homogenates (200 μl) were then added to a reaction mixture consisting of 1.5 ml of 0.8% thiobarbituric acid, 200 μl of 8.1% sodium dodecyl sulfate, 1.5 ml of 20% acetic acid (pH 3.5), and 600 μl of distilled H2O and heated for 45 min at 90°C. After cooling to room temperature, the samples were centrifuged at 10,000 g for 10 min, and the absorbance of the supernatant at A532 was measured with 1,1,3,3-tetramethoxypropane as an external standard. The level of lipid peroxides was expressed as nanomoles MDA per milligram protein.
Real-time PCR.
Real-time PCR was performed as described previously (3, 9, 15). Briefly, total RNA was isolated from tissue homogenate using TRIzol reagents (Invitrogen) according to the manufacturer's instructions. Total RNA (1 mg) was reverse-transcribed to cDNA using Super-Script II (Invitrogen). Target gene expression was quantified with iTaq Syber green Mix (Bio-Rad) using the Bio-Rad Chromo 4/Opticon system. Amplified samples from each well were analyzed for homogeneity using melting curve analysis. After initial denaturation at 95°C for 2 min, 40 cycles of 95°C for 10 s, followed by 60°C for 30 s, were performed. Relative quantification was calculated using the comparative CT method (2−ΔΔCt method: ΔΔCt = ΔCt sample − ΔCt reference). Lower ΔCT and lower ΔΔCT values reflect relatively greater amounts of gene transcripts. Statistical analyses were carried out for at least eight experimental samples from each set. RT-PCR analyses of the reference gene β-actin mRNA expression were performed in all samples. The results showed that β-actin mRNAs were comparable in all samples. Expression of all candidate gene mRNAs was normalized to β-actin mRNA level during calculation. Primer sequences for TNF-α, IL-1β, ICAM-1, caspase 3, caspase 8, and caspase 9 have been described previously (3, 9, 15).
Statistical analysis.
Data are expressed as means ± SE (n = 5–10). To compare values obtained from two or more groups, Student's t-test or one-way analysis of variance was performed. A value of P < 0.05 was considered significant.
RESULTS
Elevation of complement activity in serum and renal tissues after cisplatin treatment.
Previous studies reported that the final end-product of complement activation is increased in cisplatin-treated patients and is associated with nephrotoxicity (29), suggesting complement is activated after cisplatin treatment in patients. Here, we measured the complement activity in serum and the cleaved C3 fragment deposition in the renal tissues of mice after cisplatin treatment. As shown in Fig. 1A, treatment of mice with cisplatin for 24 and 48 h significantly increased serum complement activity, which was returned to basal levels 72 h posttreatment. Immunohistochemical analyses with the anti-C3b/iC3b/C3c antibody showed that levels of C3b/iC3b/C3c were markedly elevated in the renal tissues of mice treated with cisplatin for 24 and 48 h but not 72 h. Since the anti-C3b/iC3b/C3c antibody only recognizes the cleaved C3 fragments C3b, iC3b, and C3c but not native C3, the findings in Fig. 1B suggest that the complement is activated in renal tissues after cisplatin treatment. In addition, in vitro experiments showed that cisplatin itself did not directly activate complement (data not shown).
Fig. 1.
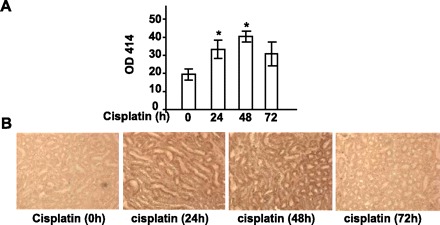
Complement activation during cisplatin-induced nephrotoxicity. A: serum complement activity from the mice treated with cisplatin was measured. Values are means ± SE. P < 0.001 vs. 0 time point. B: representative renal tissues stained with anti-C3b/iC3b/C3c antibody. Enhanced staining was observed in the tissues from 24- and 48-h cisplatin-treated mice.
C5 deficiency protects mice from cisplatin-induced nephrotoxicity.
To investigate the role of C5 in the pathogenesis of cisplatin-induced nephrotoxicity, C5KO mice were used. Basal levels of BUN and creatinine were similar between C5KO and their WT controls (Fig. 2A). No obvious renal histological changes were observed in C5KO mice compared with WT mice. Cisplatin treatment significantly elevated serum BUN and creatinine levels (Fig. 2A) and increased the percentage of damaged tubular cells (Fig. 2B) in WT mice. In contrast, C5KO mice were resistant to cisplatin-induced elevations in serum BUN and creatinine levels and renal tubular damage (Fig. 2). These results suggest that C5 plays a critical role in cisplatin-induced nephrotoxicity.
Fig. 2.
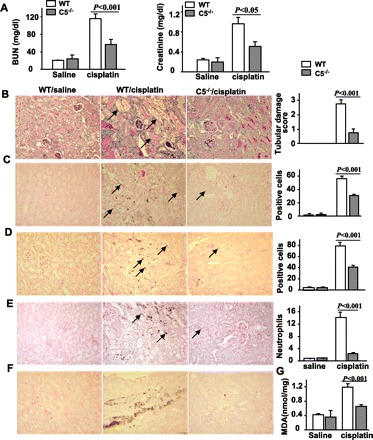
C5 deficiency protects mice from renal injury induced by cisplatin. Wild-type (WT) and C5 knockout (C5KO) mice were injected with cisplatin (20 mg/kg ip) or saline. Blood samples and kidney tissues were collected 72 h after cisplatin administration. A: serum levels of blood urea nitrogen (BUN) and creatinine were measured. B–F: representative renal tissues stained with periodic acid-Schiff (PAS; B), terminal deoxynucleotidyl transferase-mediated uridine triphosphate nick-end labeling (TUNEL; C), anti-cleaved caspase 3 antibody (D), MPO antibody (E), and anti-nitrotyrosine antibody (F) (magnification ×200). Tissue damage scores were quantified and are shown on the right. The number of TUNEL+ cells, cleaved caspase 3+ cells, and neutrophils was randomly counted in 10 fields (magnification ×40) per slide and are shown on the right. G: MDA levels were also measured. Values are means ± SE; n = 5 in saline group and n = 12 in cisplatin group (A); n = 5/group (B–G).
Apoptosis of renal tubular epithelial cells (8), formation of reactive oxygen species (13), and infiltration of inflammatory cells are well-known contributors to cisplatin-induced kidney dysfunction (11). We used TUNEL immunohistochemical staining and cleaved caspase 3 to assess renal tubular epithelial cell apoptosis and MPO, MDA, and nitrotyrosine to assess renal neutrophil infiltration and oxidative/nitrosative stress. In WT mice, cisplatin administration consistently and significantly increased the number of TUNEL-positive apoptotic cells (Fig. 2C), cleaved caspase 3-positive cells (Fig. 2D), and MPO-positive cells (Fig. 2E) compared with C5KO mice. Higher levels of nitrotyrosine staining (Fig. 2F) and MDA (Fig. 2G) were also observed in WT mice. In comparison, C5KO mice exhibited staining that was partially or completed abrogated. These results further suggest that C5 is an important mediator of increased apoptosis, inflammatory cell infiltration, and oxidative/nitrosative stress observed with cisplatin-induced nephrotoxicity.
Administration of C5 or C5a restores cisplatin-induced nephrotoxicity in C5KO mice.
To confirm the role of C5 in cisplatin-induced nephrotoxicity, C5KO mice were injected with human C5 protein before cisplatin administration. As shown in Fig. 3, A and B, serum BUN and creatinine levels, as well as the number of damaged renal tubular cells, were significantly higher after cisplatin administration in C5KO mice pretreated with C5 compared with C5KO mice that did not receive C5 pretreatment. Consistently, C5 pretreatment increased the number of apoptotic cells and infiltrating neutrophils and increased MDA levels in the kidneys of cisplatin-treated C5KO mice (Fig. 3C). Injection of C5 alone did not cause elevation of BUN and creatinine (Fig. 3A) or obvious histological changes in the kidney (data not shown).
Fig. 3.
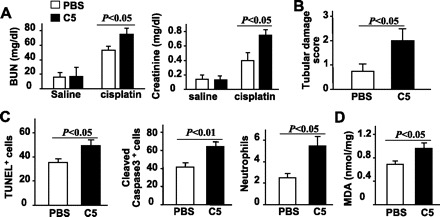
Administration of purified human C5 restores cisplatin-induced nephrotoxicity in C5KO mice. Purified human complement protein C5 (3 μg/g body wt) or PBS was injected intravenously 10 min before saline or cisplatin administration in C5KO mice. Blood samples and renal tissues were collected 72 h after saline or cisplatin administration. A: serum levels of BUN and creatinine were measured. B: tubular damage scores. C: renal tissues were stained with TUNEL, anti-cleaved caspase 3 antibody, or anti-MPO antibody. The number of positive cells was counted. D: MDA levels were measured. Values are means ± SE; n = 5/group.
During complement activation, C5 is cleaved into two smaller fragments, C5a and C5b (1, 31). The C5b fragment participates in the formation of MAC, while C5a functions as an anaphylatoxin. To determine whether C5a contributes to cisplatin-induced renal injury, we injected C5KO mice with C5a before cisplatin administration. Similar to administration with C5, injection of C5a also restored cisplatin-induced nephrotoxicity in C5KO mice. As shown in Fig. 4, BUN and creatinine serum levels, tubular damage, the number of TUNEL-positive cells and neutrophils, and MDA levels were significantly higher in C5KO mice treated with C5a and cisplatin than mice treated with cisplatin alone. Injection of C5a alone had little effect on elevation of BUN and creatinine (Fig. 4A) and histological changes in the kidney (data not shown).
Fig. 4.
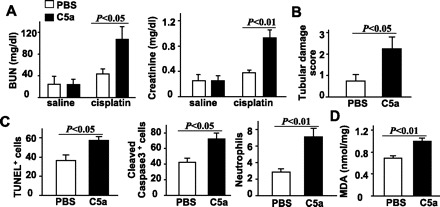
Administration of purified human C5a restores cisplatin-induced nephrotoxicity in C5KO mice. Purified human complement protein C5a (3.5 μg/g body wt) or PBS was injected intravenously 10 min before saline or cisplatin administration in C5KO mice. Blood samples and renal tissues were collected 72 h after saline or cisplatin administration. A: serum levels of BUN and creatinine were measured. B: tissue damage scores. C: kidney tissues were stained with TUNEL, anti-cleaved caspase 3 antibody, or anti-MPO antibody. The number of positive cells was counted. D: MDA levels were measured. Values are means ± SE; n = 5 in saline group and n = 6 in cisplatin group (A); n = 5/group (B and C).
Deficiency of C5a receptor protects against cisplatin-induced renal dysfunction.
The C5a fragment binds to corresponding C5a receptors located on various immune cells. To further identify the downstream pathway involved in C5a, C5aRKO mice were used. At 72 h after cisplatin administration, C5aRKO mice had significantly reduced serum levels of BUN and creatinine compared with WT mice (Fig. 5A). There were also significantly fewer apoptotic cells, less infiltration of neutrophils, lower nitrotyrosine levels, and lower MDA levels in C5aRKO mice compared with WT mice (Fig. 5, B–G). This suggests that the nephrotoxic effects of C5a are committed after binding to C5a receptors, triggering downstream signaling events.
Fig. 5.
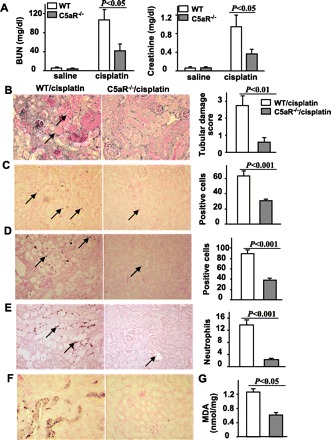
C5aRKO mice are resistant to cisplatin-induced renal dysfunction. WT and C5aRKO mice were injected with cisplatin (20 mg/kg ip). Blood samples and renal tissues were collected 72 h post- cisplatin administration. A: serum levels of BUN and creatinine. B–F: representative renal tissues stained with PAS (B), TUNEL (C), anti-cleaved caspase 3 antibody (D), anti-MPO antibody (E), and anti-nitrotyrosine antibody (F). Magnification ×200. Tissue damage scores were quantified and shown on the right. The number of TUNEL+ cells, cleaved caspase 3+ cells, and neutrophils were randomly counted in 10 fields (magnification ×40) per slide and are shown on the right. G: MDA levels were also measured. Values are means ± SE; n = 4 in saline group and n = 5 in cisplatin group (A); n = 5/group (B–G).
C5a or C5aR deficiency diminishes cisplatin-induced renal gene expression of cytokines and caspases, and activation of p-STAT3 and NF-κB.
It is well documented that cisplatin-induced caspase activation and inflammatory chemokine and cytokine release contribute to cisplatin-induced nephrotoxicity (7, 8, 23). Here, we show that cisplatin administration upregulated expression levels of TNF-α, IL-1β, ICAM-1, caspase 3, caspase 8, and caspase 9 in murine kidneys (Fig. 6A). In C5KO and C5aRKO mice, this upregulation was blunted (Fig. 6, A and B). Administration of purified human C5 or C5a restored the upregulation of these cytokines and caspase genes in cisplatin-treated C5KO mice (Fig. 6, C and D). Injection of C5 or C5a alone (without subsequent cisplatin treatment) had little effect on these markers except for a slight increase in TNF-α expression after C5a injection (Fig. 6, C and D).
Fig. 6.
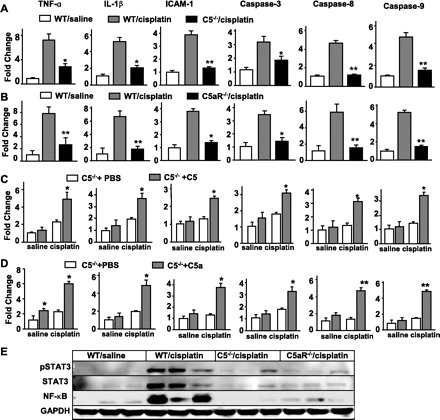
C5 or C5aR deficiency diminishes cisplatin-induced cytokine and caspase gene expression and STAT3/NF-κB activation in murine kidneys. A–D: RT-PCR analyses were conducted to examine expression of cytokines and caspase genes in the kidneys 72 h after cisplatin or cisplatin plus C5 or C5a treatment. Expression levels were normalized to β-actin expression levels and are expressed relative to saline-treated WT mice. Values are means ± SE (n = 4/group). A and B: *P < 0.05, **P < 0.01 compared with corresponding WT controls treated with cisplatin. C and D: *P < 0.05, **P < 0.01 compared with corresponding C5KO mice treated with cisplatin alone. E: nuclear extracts were isolated from the kidneys 72 h after cisplatin administration and used in Western blot analyses.
Additionally, it has been established that activation of C5aR by C5a results in IL-6/TNF induction and subsequent STAT3/NF-κB activation (27). Hence, we examined whether STAT3/NF-κB also becomes activated in the cisplatin-induced renal injury model. As shown in Fig. 6E, cisplatin treatment elevated renal expression of STAT3, p-STAT3, and NF-κB in WT mice, but such induction was not observed in C5KO or C5aRKO mice.
MAC formation does not contribute to cisplatin-induced nephrotoxicity.
The principal regulator of complement MAC assembly on cell membranes is CD59 (21). To exclude involvement of MAC formation in cisplatin-induced renal dysfunction, serum levels of BUN and creatinine were measured 72 h after cisplatin administration in WT and CD59abKO mice. As shown in Fig. 7A, cisplatin treatment elevated serum levels of BUN and creatinine in WT and CD59abKO mice similarly. Immunofluorescence staining for MAC was also performed to assess the formation of MACs in kidneys after cisplatin administration. As expected, WT mice and C5aRKO mice had increased MAC deposition, while C5KO mice had very low MAC deposition (Fig. 7B). No significant MAC deposition was detected in WT mice not treated with cisplatin (Fig. 7B).
Fig. 7.
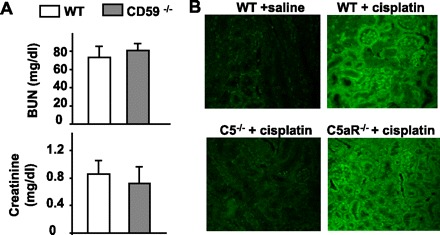
Evidence for the lack of a role for the membrane attack complex (MAC) in cisplatin-induced nephrotoxicity. A: loss of restriction of MAC formation in CD59 KO mice does not enhance cisplatin-induced nephrotoxicity. WT and CD59ab double KO mice were injected with cisplatin (20 mg/kg ip). Blood samples were collected 72 h post-cisplatin administration. Serum levels of BUN and creatinine were measured. Values are means ± SE (n = 5/group). B: representative images of MAC immunofluorescence staining in mouse kidneys 72 h after saline or cisplatin administration (magnification ×200). There was less MAC immunofluorescence staining in C5KO mice compared with WT and C5aRKO mice.
DISCUSSION
For the first time, we demonstrate that C5KO and C5aRKO mice are resistant to cisplatin-induced nephrotoxicity, and administration of purified C5 or C5a restores the sensitivity of C5KO mice to cisplatin-induced nephrotoxicity. Using CD59ab double knockout mice, we have excluded the role of MAC in this process. Moreover, we also demonstrate that C5a or C5aR deficiency diminishes cisplatin-induced cytokine and caspase gene expression, as well as p-STAT3 and NF-κB activation in kidneys from mice. Taken together, these findings indicate that C5a contributes to cisplatin-induced nephrotoxicity through a signaling pathway downstream from C5a receptor binding.
Within the complement system, the C5 protein is a principle mediator of host defenses (22). During activation of the complement cascade, C5a and C5b are generated when C5 is cleaved by its convertase. The C5a fragment is a potent anaphylactic/chemotactic mediator that induces many proinflammatory activities, whereas C5b contributes to formation of the C5b-9 MAC. Increased urinary levels of C5b-9 complexes were found in cisplatin-treated patients and were associated with nephrotoxicity (29). This suggests that injection of cisplatin induces complement activation in patients. Here, we also demonstrate that injection of cisplatin induced complement-mediated renal damage through the C5 receptor pathway, not the formation of MAC. Furthermore, incubation of cisplatin with the serum in vitro did not lead to the complement activation (data not shown). This suggests that cisplatin itself does not directly activate complement and that activation of complement in vivo by injection of cisplatin may result from the secondary cisplatin-induced renal damage or the increased oxidative (MDA)-modified proteins (26). Normally, complement activation would cause complement consumption, leading to decreased serum complement activity. However, we found that the serum complement activity was increased after cisplatin treatment (Fig. 1). At present, the mechanisms by which administration of cisplatin causes complement activation but unexpectedly elevates serum complement activity remain unclear. It is plausible to speculate that injection of cisplatin causes renal damage and production of many proinflammatory cytokines (Fig. 6) (7), which could lead to the increased biosynthesis of the complement components in the liver and subsequently dominate the decreased serum complement levels during activation, leading to the elevated serum complement activity. Inhibition of C5a generation and C5b-9 formation by blocking C5 has been shown to ameliorate or prevent many inflammatory disorders associated with inappropriate complement activation, including renal ischemia-reperfusion injury (2, 6, 35) and glomerulonephritis (17). Here, we demonstrate that C5KO mice are resistant to cisplatin-induced renal injury, suggesting that activation of C5 is involved in the nephrotoxicity of cisplatin. Furthermore, we provide several lines of evidence suggesting that C5a specifically contributes to cisplatin nephrotoxicity. First, administration of purified human C5 or C5a restored cisplatin-induced renal injury in C5KO mice. Second, disruption of the C5aR gene also diminished cisplatin-induced renal injury. The C5a fragment exerts many proinflammatory and immunoregulatory actions after binding to C5aR, which belongs to the G protein-coupled receptor (GPCR) family, and is primarily expressed on the surface of immune cells such as macrophages, neutrophils, and T cells. Our findings show that C5aRKO and C5KO mice are resistant to cisplatin-mediated induction of proinflmammatory cytokines. As administration of C5 or C5a restored inflammation responses in cisplatin-treated C5KO mice, the data collectively suggest that generation of C5a contributes to the induction of inflammation via binding to C5 receptor in cisplatin-induced renal injury, further supporting the conclusion that C5a participates in cisplatin-induced nephrotoxicity. In addition, administration of C5 or C5a alone did not cause nephrotoxic changes (Figs. 3 and 4), suggesting that other insults (e.g., C3bi-driven inflammation) in combination with C5 are required to induce nephrotoxicity.
The C5b fragment is generated when C5 is cleaved by C5 convertase. In turn, C5b contributes to the formation of MAC. It is well known that MAC formation contributes to many inflammatory kidney disorders, such as membranous nephropathy (16) and ischemia-reperfusion injury (2, 36). However, we provide two lines of evidence suggesting that MAC formation may not contribute to cisplatin-induced renal injury. First, CD59 is the major regulator of MAC formation, and cisplatin-induced renal injury was similar in both WT and CD59ab double knockout mice. Second, cisplatin-induced formation of MACs was observed in C5aRKO mice, but not in C5KO mice, while cisplatin-induced renal injury was diminished in both C5KO and C5aRKO mice, further indicating that MAC does not play a role in the renal toxicity of cisplatin.
In summary, our findings suggest that C5a contributes to cisplatin-induced nephrotoxicity through a signaling pathway downstream from C5a receptor binding, which is followed by induction of proinflammatory cytokines and neutrophil infiltration, resulting in renal damage. Additional studies to examine the role of C5a in human cisplatin induced-nephrotoxicity are warranted. The C5aR blockade could be a novel strategy in attenuating cisplatin-induced kidney injury.
GRANTS
This work was supported by the Intramural Program of the National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Acosta J, Qin X, Halperin J. Complement and complement regulatory proteins as potential molecular targets for vascular diseases. Curr Pharm Des 10: 203–211, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. The role of the complement system in ischemia-reperfusion injury. Shock 21: 401–409, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Batkai S, Osei-Hyiaman D, Pan H, El-Assal O, Rajesh M, Mukhopadhyay P, Hong F, Harvey-White J, Jafri A, Hasko G, Huffman JW, Gao B, Kunos G, Pacher P. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J 21: 1788–1800, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger SP, Daha MR. Complement in glomerular injury. Semin Immunopathol 29: 375–384, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown KM, Sacks SH, Sheerin NS. Mechanisms of disease: the complement system in renal injury–new ways of looking at an old foe. Nat Clin Pract Nephrol 3: 277–286, 2007 [DOI] [PubMed] [Google Scholar]
- 6.de Vries B, Kohl J, Leclercq WK, Wolfs TG, van Bijnen AA, Heeringa P, Buurman WA. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J Immunol 170: 3883–3889, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, Edelstein CL. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther 322: 8–15, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int 66: 2202–2213, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D, Moh A, Fu XY, Pacher P, Kunos G, Gao B. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 134: 1148–1158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer 34: 1522–1534, 1998 [DOI] [PubMed] [Google Scholar]
- 11.Li S, Gokden N, Okusa MD, Bhatt R, Portilla D. Anti-inflammatory effect of fibrate protects from cisplatin-induced ARF. Am J Physiol Renal Physiol 289: F469–F480, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Liu M, Chien CC, Burne-Taney M, Molls RR, Racusen LC, Colvin RB, Rabb H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J Am Soc Nephrol 17: 765–774, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Matsushima H, Yonemura K, Ohishi K, Hishida A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J Lab Clin Med 131: 518–526, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest 101: 777–782, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay P, Batkai S, Rajesh M, Czifra N, Harvey-White J, Hasko G, Zsengeller Z, Gerard NP, Liaudet L, Kunos G, Pacher P. Pharmacological inhibition of CB1 cannabinoid receptor protects against doxorubicin-induced cardiotoxicity. J Am Coll Cardiol 50: 528–536, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nangaku M, Shankland SJ, Couser WG. Cellular response to injury in membranous nephropathy. J Am Soc Nephrol 16: 1195–1204, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Pickering MC, Warren J, Rose KL, Carlucci F, Wang Y, Walport MJ, Cook HT, Botto M. Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc Natl Acad Sci USA 103: 9649–9654, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard MT, McMullen MR, Stavitsky AB, Cohen JI, Lin F, Medof ME, Nagy LE. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology 132: 1117–1126, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin X, Dobarro M, Bedford SJ, Ferris S, Miranda PV, Song W, Bronson RT, Visconti PE, Halperin JA. Further characterization of reproductive abnormalities in mCd59b knockout mice: a potential new function of mCd59 in male reproduction. J Immunol 175: 6294–6302, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Qin XHW, Song W, Grubissich L, Hu X, Wu G, Ferris S, Dobarro M, Halperin JA. Generation and phenotyping of mCd59a and mCd59b double-knockout mice. Am J Hematol. In press. [DOI] [PMC free article] [PubMed]
- 21.Qin X, Krumrei N, Grubissich L, Dobarro M, Aktas H, Perez G, Halperin JA. Deficiency of the mouse complement regulatory protein mCd59b results in spontaneous hemolytic anemia with platelet activation and progressive male infertility. Immunity 18: 217–227, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Qin XB, Gao B. The complement system in liver diseases. Cell Mol Immunol 3: 333–340, 2006 [PubMed] [Google Scholar]
- 23.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ries F, Klastersky J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am J Kidney Dis 8: 368–379, 1986 [DOI] [PubMed] [Google Scholar]
- 25.Schrier RW. Cancer therapy and renal injury. J Clin Invest 110: 743–745, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorace JM, Rollins S, Aniagolu JU, Mergner WJ, Cole K, Swartz GM Jr, Green SJ. Role of atheroma liposomes and malondialdehyde-modified low-density lipoproteins in complement activation. Pathobiology 64: 73–78, 1996 [DOI] [PubMed] [Google Scholar]
- 27.Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med 198: 913–923, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugiyama S, Hayakawa M, Kato T, Hanaki Y, Shimizu K, Ozawa T. Adverse effects of anti-tumor drug, cisplatin, on rat kidney mitochondria: disturbances in glutathione peroxidase activity. Biochem Biophys Res Commun 159: 1121–1127, 1989 [DOI] [PubMed] [Google Scholar]
- 29.Tamano M, Ohi H. Complement activation in cisplatin nephropathy. Nephron 81: 442–443, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, Nemenoff RA, Quigg RJ, Holers VM. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ischemia/reperfusion. J Immunol 178: 1819–1828, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Walport Complement MJ. First of two parts. N Engl J Med 344: 1058–1066, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Welch TR. The complement system in renal diseases. Nephron 88: 199–204, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Yamada K, Miwa T, Liu J, Nangaku M, Song WC. Critical protection from renal ischemia reperfusion injury by CD55 and CD59. J Immunol 172: 3869–3875, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Yamate J, Tatsumi M, Nakatsuji S, Kuwamura M, Kotani T, Sakuma S. Immunohistochemical observations on the kinetics of macrophages and myofibroblasts in rat renal interstitial fibrosis induced by cis-diamminedichloroplatinum. J Comp Pathol 112: 27–39, 1995 [DOI] [PubMed] [Google Scholar]
- 35.Zheng X, Zhang X, Feng B, Sun H, Suzuki M, Ichim T, Kubo N, Wong A, Min LR, Budohn ME, Garcia B, Jevnikar AM, Min WP. Gene silencing of complement C5a receptor using siRNA for preventing ischemia/reperfusion injury. Am J Pathol 173: 973–980, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL, Sacks SH. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest 105: 1363–1371, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]