

Abstract
The etiology of polycystic ovary syndrome (PCOS) has been difficult to determine because its features are heterogeneous, and its origin may also be heterogeneous. Twin studies suggest that its etiology is strongly heritable and genetic approaches are rapidly uncovering new regions of the genome that appear to confer risk for PCOS. Recent genome-wide association studies (GWAS) in Han Chinese women with PCOS demonstrate 11 genetic loci that are associated with PCOS. The variants identified are in regions that contain genes important for gonadotropin action, genes that are associated with risk for type 2 diabetes and other genes in which the relationship to PCOS is not yet clear. Replication studies have demonstrated that variants at several of these loci also confer risk for PCOS in women of European ethnicity. The strongest loci in Europeans contain genes for DENND1A and THADA, with additional associations in loci containing the LHCGR and FSHR, YAP1 and RAB5/SUOX. The next steps in uncovering the pathophysiology borne out by these loci and variants will include mapping to determine the causal variant and gene, phenotype studies to determine whether these regions are associated with particular features of PCOS and functional studies of the causal variant to determine the direct cause of PCOS based on the underlying genetics. The next years will be very exciting times as groups from around the world come together to further elucidate the genetic origins of PCOS.
Keywords: genome-wide association, gonadotropins, hyperandrogenism
Polycystic ovary syndrome affects 7-10% of reproductive aged women.1-3 The syndrome is composed of a number of features that have been well characterized in physiologic studies. These studies documented the elevated LH:FSH ratio,4, 5 an increased GnRH pulse frequency and GnRH quantity,6 elevated androgen levels,4 elevated insulin levels and evidence of insulin resistance,7, 8 polycystic ovary morphology,9, 10 and high AMH levels.11 In addition, women with polycystic ovary morphology of their ovaries demonstrate higher androgen and insulin levels, even when their menstrual cycles are regular.12 Importantly, the polycystic ovary morphology is found consistently in women with PCOS characterized by oligomenorrhea and hyperandrogenism.4, 13, 14
Although these physiology studies have been critical in defining the spectrum of PCOS, the cardinal features of the syndrome remain controversial based on the heterogeneity of the patient populations. A recent PCOS evidence-based workshop recommended using the Rotterdam criteria to define PCOS, while documenting the features that characterize PCOS in each population or patient.15 This inclusive approach is reasonable because all diagnostic criteria account for specific patient groups that may be seen by endocrinologists, pediatricians, gynecologists or dermatologists,16 and the inciting features of PCOS are unclear. The physiology studies fail to tell us whether it is the elevated androgen levels that led to insulin resistance; insulin resistance that led to hyperandrogenism, polycystic ovary morphology that is an initial feature or whether the increased hypothalamic GnRH secretion drives the disorder. A new approach is necessary to sort out the pathophysiology.
A genetic approach has been taken by a number of groups to identify the etiology of PCOS. Twin studies of heredity provide the most rigorous demonstration that a disorder has a genetic component. In twin studies, the concordance of a disorder is compared between monozygotic twins, who share their entire genome, and dizygotic twins who only share 50% of their genome. In the absence of environmental differences between the twins, which are relatively controlled if they grow up in the same household, the proportion of a disorder that is heritable can be estimated. Using these principles, twin studies suggest that genetic influences explain over 70% of PCOS pathogenesis.17
The initial genetic approaches taken by groups with large patient populations of women with PCOS included linkage and candidate gene approaches. Linkage requires a large family group with multiple affected members sharing a disorder inherited in a Mendelian manner, or may involve parent/affected child trios.18 Linkage identifies areas of the genome shared by affected family members, then requires interrogation of the genes in the shared region to identify mutations that segregate with the disease in that family. The approach has been difficult based on the need to recruit multiple affected family members from large families to improve the power of the study.
Association studies have been easier to complete because they are performed by recruiting cases and unaffected controls. However, they have a number of pitfalls. The candidate gene association approach relies on a hypothesized relationship between a candidate gene and a disorder, which is limited by current knowledge. Candidate gene association studies are also subject to positive study publication bias and population stratification, which occurs when the ethnic background of cases and controls is not well matched, resulting in false positive associations related to underlying differences in the genetic structure of the two populations rather than the disease itself.19 To perform the association testing properly, differences in all of the base pairs in the gene should be examined between cases and controls, correcting for population stratification between cases and controls, and the appropriate statistical analysis must include a correction factor for multiple hypothesis testing based on the number of gene variants and the number of genes examined by the same group.19 In addition, the number of subjects studied must be large and the results must be replicated by independent groups. Previous studies have demonstrated an association between variants in over 70 candidate genes and risk for PCOS. However, the majority of these studies have not applied the three principles described above, including testing all variants in a gene, examining large subject numbers and replicating results in an independent group. Therefore, most of these studies likely represent false associations.20, 21 Nevertheless, there are some strong, replicated candidate gene associations that have been the subject of excellent previous reviews.22 Therefore, the current review will focus on more recent genome-wide association studies (GWASs) of PCOS.
Genetic Risk Variants for PCOS Identified by GWASs
GWASs are made possible by the mapping of the human genome and the HapMap project,23, 24 which catalogued common variation in the genome that is shared by large numbers in the population. The HapMap project also demonstrated that many common variants or single nucleotide polymorphisms (SNPs) travel together in blocks of linkage disequilibrium.24 For example, 5 variants may commonly be found together, as indicated by 1-5 in figure 1.25, 26 Therefore, one variant (#3 in the figure) that correlates highly with other variants in the linkage disequilibrium block, can serve as a marker or tag of the large area (tag variant) and can predict the other variants that are found in the region without actually genotyping every variant in the linkage disequilibrium block. It is a tag variant that is found in the genotyping arrays used for GWASs. These arrays incorporate a subset, the tag variants, of the 11 million common variants found in the human genome (frequency of greater than 1%). These arrays then allow an investigator to examine the entire complement of common variation in the population in cases compared to controls without a preconceived hypothesis. Based on the large number of variants on these arrays, as many as 1 million on some, the results need to be corrected for the number of independent tests, i.e. every independent variant on the arrays in association with the disorder of interest. The typical p value required to suggest an association must be lower than 1×10-8. The variants on an array can also be associated with quantitative traits, which are a measure of a phenotype parameter that is a continuous variable.
Figure 1.
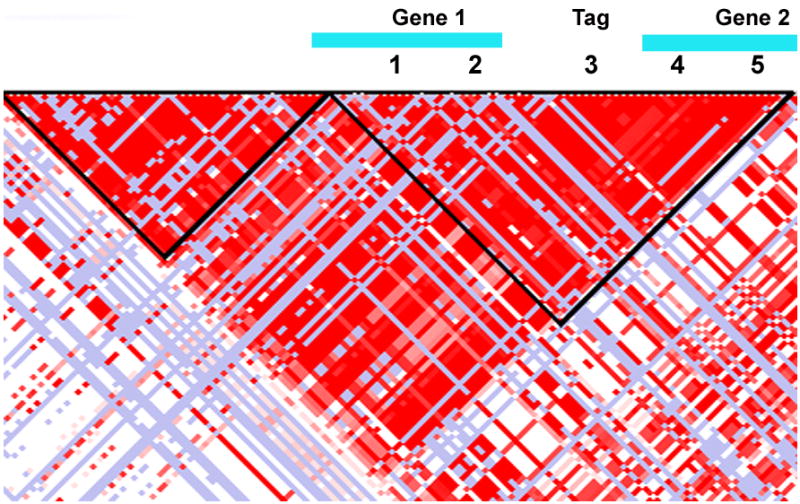
Simplified representation of a region in the genome identified in an association study by a tag variant (#3). The black triangle surrounding the red pixels marks a region of linkage disequilibrium, in which there is little genetic recombination and the variants 1-5 tend to travel together. Although #3 is the variant that was associated with disease, further mapping demonstrated that variant #5 had a stronger association with disease and was found in a coding region for gene 2. Therefore, variant 5 is the causal variant in the study. Functional studies can now be performed to determine how variant 5 disrupts gene function and causes disease.
The largest proportion of common DNA variants fall in intronic regions or non-coding regions of the genome, i.e. not in exons which code for the amino acids of the translated proteins. Therefore, their function is very hard to interpret. Further, a GWAS does not identify a gene associated with a disorder. Rather, it identifies a genetic locus of interest because tagged variants are examined. While variants are often reported in a gene or in the region of a gene, the variant may be affecting the nearest gene or a gene that is much farther away. Therefore, the relationship of the associated variant and the disorder remains to be determined after the GWAS is published.
Two genome-wide association studies have now been performed that demonstrate variants associated with PCOS risk in Han Chinese women.27, 28 These studies have enrolled 10,480 cases and 10,489 controls and identified 11 variants associated with PCOS. Some of the variants are in regions with genes that may influence the development of PCOS (Table 1). In the first GWAS, three susceptibility loci for PCOS were identified at 2p16.3, 2p21 and 9q33.3.27 In a second GWAS, 8 additional variants were uncovered (Table 1).28 Based on the results of both studies, the variants were located within introns of genes or near genes implicated in gonadotropin action (LHCGR and FSHR), insulin signaling (INSR) and type 2 diabetes (THADA and HMGA2), organ size control or cell proliferation (YAP1 and SUMO1P1), architectural factors important for chromatin remodeling (TOX3) and in regions associated with type 1 diabetes (region containing RAB5B, SUOX and ERBB3). These studies provide new hypotheses to test regarding the origin and pathophysiology of PCOS.
Table 1.
Gene variants associated with risk for polycystic ovary syndrome (PCOS) in Han Chinese women and replication in European women. The gene closest to the variant is indicated as is the highest odds ratio (OR) and p value available in a replicate European cohort.
| Locus | Variant | Closest Gene | ORa Han Chinese | Overall p value | OR Replication in European cohortb | P value | Reference European Replicate Group |
|---|---|---|---|---|---|---|---|
| 2p16.3 | rs13405728 | LHCGR | 1.41 | 3.77×10-9 | 1.15 | 0.34 | 31 |
| 2p16.3 | rs2268361 | FSHR | 0.87 | 9.89×10-13 | NAd | ||
| 2p16.3 | rs2349415 | FSHR | 1.19 | 2.35×10-12 | 1.17 | 0.14 | 36 |
| 2p21 | rs12468394 | THADA | 1.39 | 1.15 | 0.0002c | 31 | |
| 2p21 | rs13429458 | THADA | 1.49 | 4.17×10-13 | 1.05 | 0.6 | 36 |
| 9q22.32 | rs4385527 | C9orf3 | 0.84 | 5.87×10-9 | NA | ||
| 9q22.32 | rs3802457 | C9orf3 | 0.77 | 5.28×10-14 | NA | ||
| 9q33.3 | rs10986105 | DENND1A | 1.47 | 5.14×10-10 | 1.68 | 0.0003 | 31, 32 |
| 9q33.3 | rs2479106 | DENND1A | 1.34 | 5.14×10-10 | 1.05 | 0.34 | 31 |
| 11q22.1 | rs1894116 | YAP1 | 1.27 | 1.08×10-22 | 1.37 | 0.002 | 32 |
| 12q13.2 | rs705702 | RAB5B,SUOX | 1.27 | 8.64×10-26 | 1.21 | 0.003 | 32 |
| 12q14.3 | rs2272046 | HMGA2 | 0.70 | 1.95×10-21 | NA | ||
| 16q12.1 | rs4784165 | TOX2 | 1.15 | 3.64×10-11 | NA | ||
| 19p13.3 | rs2059807 | INSR | 1.14 | 1.09×10-8 | NA | ||
| 20q13.2 | rs6022786 | SUMO1P1 | 1.13 | 1.83×10-9 | NA |
OR odds ratio
OR and p value from the strongest available replication in a European cohort, as referenced.
Combined p value in two published data sets
NA-Replication data not available at the time of publication
As yet, there have been no published genome-wide associated variants for PCOS in European women. Indeed, these studies will be important because the susceptibility variants may differ in individual ethnic groups as do the phenotypic features of PCOS. Nevertheless, a number of groups using European cohorts have replicated a subset of the findings in Han Chinese women with PCOS. Three of four groups have replicated two of the loci reported in the first GWAS publication, 2p21 and 9q33.3.29,30 These groups replicated two associated variants in introns of DENND1A that are in linkage disequilibrium and one in THADA.29, 31, 32 The DENND1A protein regulates Rab GTPases,33 which are important for calcium regulated exocytosis in pituitary cells and for basal and GnRH-induced gonadotropin release.34 Therefore, it is possible that the variants in DENND1A affect exocytosis of gonadotropins. A variant in THADA has been replicated in European women by one group and trended toward replication in another. As above, variants in THADA have been associated with type 2 diabetes, although these type 2 diabetes risk variants are found within a separate area of linkage disequilibrium in THADA.35 However, a potential relationship between THADA and factors in women with PCOS predisposing to type 2 diabetes is suggested.
The association in region 2p16.3 is complex. The initial variant identified in Han Chinese women fell within introns of both the LHCGR, the LH/hCG receptor, and GTF21L, the general transcription factor IIA, 1-like.27 An independent signal was also identified in two variants that sit within introns of FSHR, the FSH receptor.28 The variant in the LHCGR is rare in European populations and has not been replicated,29, 31, 36 nor was there evidence for an association in other, more common variants in linkage disequilibrium in the Icelandic population.31 An extensive examination of the region demonstrated that a variant in linkage disequilibrium in the Han Chinese population, but not in Europeans, was associated with PCOS,36 although the association was not replicated by others.20 The most highly associated variant near the FSHR in Europeans was slightly upstream.36 The failure to replicate some findings may speak to differences in risk variants in distinct ethnic groups and the power of the replication studies.
Two additional risk variants identified in the second GWAS of Han Chinese women have been replicated in European women.28, 32 The risk variant on chromosome 11 is located in an intron of YAP1, a downstream nuclear effector of the Hippo signaling pathway which is involved in development, growth, repair, and homeostasis. Interestingly, disruption of YAP phosphorylation increased nuclear YAP levels in mice, resulting in increased downstream growth factors that increased follicle growth and decreased apoptotic factor levels.37These studies suggest a role for YAP in the maintenance of the follicle complement and follicle growth. The variant at locus 12q13.2 is located between two genes. RAB5B is a member of the RAS oncogene family and SUOX, sulfite oxidase, is a homodimeric protein enzxyme localized to the intermembrane space of mitochondria, which catalyzes oxidation of sulfite to sulfate, the final reaction in the oxidative degradation of the sulfur amino acids cysteine and methionine. It is not clear which gene, if either, might be affected by the associated variant. Of note, the same locus has been associated with risk for type 1 diabetes, mapping in the ERBB3 gene, which is also in the region.38
The ultimate goal of GWASs is not to identify a variant associated with a disorder, but to find a causal variant that demonstrates a functional effect and a pathophysiology that explains how a variant predisposes a person to develop the disorder.39 Recall that the variants chosen for the chip arrays are representative, tag variants that mark a region of the genome in linkage disequilibrium. Therefore, the region in linkage disequilibrium with the tag variant must be carefully mapped to determine the causal variant for functional studies. The causal variant is defined as the common variant most significantly associated with PCOS risk and/or with a potential biological effect, therefore explaining the risk at that locus.39 Before beginning functional studies, it is critical to focus on the causal variant to target the precise variant altering function and therefore the right candidate gene and/or pathway.40 For example, #3 is the tag variant identified in the GWAS, but the association was driven by the physical and genetic proximity to #5, which is the causal variant sitting in Gene #2 and altering its function (Figure 1).41
Phenotype and Genetic Risk Variants
In addition to further mapping the associated locus to identify a causal variant, the phenotype associated with the variant can provide insight into the pathophysiology conferred by the locus. Of note, in Han Chinese women, there were no phenotypic traits associated with the identified PCOS variants when an additive model was used, i.e. a model in which the presence of each variant is associated with an additive risk for the trait.42 A relationship was only identified when the initial model on which the association was discovered was changed to a recessive model, in which two copies of the variant are necessarily associated with the trait. However, this analysis is not appropriate given the hypothesis that defined the variants and risk for PCOS. Changing to a recessive model, as was done in this study, also results in reduced subject number and increased the potential for false positive associations because the number of subjects who carry two risk variants is very small.
A similar attempt has been made to identify quantitative traits associated with the variants replicated in European populations. The DENND1A variant is associated with hyperandrogenism and irregular menses,31 and as such is a risk variant for PCOS as documented by the NIH criteria. DENND1A is expressed in the theca cells and testes,43 therefore it may be associated with hyperandrogenism by increasing androgen levels. However, there is no relationship between the DENND1A variants and testosterone or androstenedione when using an additive genetic model for phenotypic traits,
Since DENND1A regulates Rab GTPases,33 which are important for calcium regulated exocytosis in pituitary cells and for basal and GnRH-induced gonadotropin release,34 we hypothesized that the risk variants would be associated with gonadotropin levels and LH pulse dynamics. Results revealed that there was no difference in mean LH or FSH levels in the carriers of the PCOS risk variants in DENND1A versus those who did not carry the variants.31 LH pulse secretion, a marker of GnRH pulse secretion, was also examined in a subset of the subject with PCOS (n=47) whom had undergone frequent blood sampling and genotyping. LH pulse secretion parameters were calculated using the modified Santen and Bardin method. Pulse parameters and hormone levels were log normalized and the relationship between these parameters and genotype was examined using linear regression with an additive genetic model. LH pulse amplitude (7.2±0.8 vs. 8.9±2.6 IU/L) and LH pulse frequency (15.5±0.9 vs. 19.4±2.6 pulses/24 hours) were not different in carriers of the common rs10986105-T or the risk G allele (all p>0.05) or in LH pulse amplitude (8.0±2.6 vs. 7.5±0.8 IU/L) or LH pulse frequency (19.7±2.6 vs. 15.7±0.9 pulses/24 hours) between carriers of the common rs10818854-G or the risk A allele (all p>0.05). Thus, DENND1A PCOS risk variants do not appear to be associated with gonadotropin levels or LH pulse parameters, although larger studies are needed.
Other phenotypic traits have been identified that are associated with PCOS risk loci. Variants found in linkage disequilibrium with the risk FSHR variants were associated with FSH levels and triglycerides.36 Paradoxically, the variant within the THADA gene that conferred risk for PCOS was associated with lower testosterone levels.31
Relationship of PCOS with Type 2 Diabetes Mellitus and Obesity
One of the most interesting aspects of the PCOS GWASs is the relationship between variants that confer risk for type 2 diabetes and those for PCOS.44-46 The most notable is the absence of risk for PCOS from variants in TCF7L2, which have the strongest association with type 2 diabetes.44, 45 In contrast, there is overlap with between PCOS and type 2 diabetes risk in regions that have smaller associated effect size for type 2 diabetes including those in regions encompassing the genes THADA, INSR and HMGA2.35, 47 However, these relationships are complicated. For example, variants in THADA associated with risk for PCOS in a GWAS of Han Chinese women were not in linkage disequilibrium with the type 2 diabetes risk variants. In other words, the regions in the genes that confer risk for the two disorders do exhibit significant overlap. Further, the minor allele of a variant in THADA is associated with decreased risk for type 2 diabetes but nominally associated with increased risk for PCOS.44 The INSR relationship in both PCOS and type 2 diabetes suggests that insulin resistance may be the stronger phenotypic feature in the overlap between the two disorders, because the TCF7L2 variant associated with diabetes has been demonstrated to decrease insulin secretion rather than increase insulin resistance.48
One additional question that may be answered by genetics is whether obesity is a predisposing or causal factor for PCOS. If markers that confer risk for obesity also confer risk for PCOS even when corrected for BMI, it would suggest an underlying risk for development of PCOS by genes predisposing to obesity. To answer this question, the strongest risk variants for BMI, variants in the FTO gene (rs11642841 and rs9939609),49 have been examined in association studies of PCOS using a candidate gene approach.44, 50-52. The majority of studies demonstrate no relationship between the FTO variants and PCOS when controlled for BMI,44, 50 or when only lean women with PCOS were examined.51 However, some studies still demonstrate an association with PCOS when controlled for BMI.51, 52 Importantly, these variants have not reached genome-wide significance in studies to date, suggesting they are not the strongest genetic factors influencing PCOS risk.27, 28
Next Steps in PCOS Genetics
What are the next steps for genetics in PCOS? GWASs identify common variants, typically present in greater than 1% of the population. These variants have a small effect size for disease, as demonstrated by the OR for risk of less than 2 for all variants discovered to date. Therefore, identifying additional common variants of small effect will require large numbers of cases and controls. Large cohorts will come together to meta-analyze data for the identification of additional variants that are associated with PCOS in European cohorts. The small number of variants found to date indicates that larger subject numbers will need to be pooled to determine additional risk variants with small effect.
In addition, the next steps in PCOS genetics studies will take advantage of the decreasing technology costs. The cost of whole exome and whole genome sequencing has decreased enough to make large scale studies in cases and controls plausible. The use of such in depth sequencing will facilitate the discovery of rare variants with large effect size. However, the low frequency of the variants will make it more difficult to achieve statistical significance for association studies. On the other hand, the variants with large effect size will more likely fall in regions that disrupt proteins and may therefore be easier to test for functional effects. We may also come full circle in the populations we study. Whereas linkage analyses started with families to identify rare variations (mutations) with a large effect, then moved to case:control studies with large numbers to find variants with small effects, we now have the ability to examine a large number of variants within families to identify relatively rare variants with large effects. The prospects for improving our understanding of PCOS pathophysiology are exciting.
Acknowledgments
This work was supported by the National Institutes of Health U01 HD 4417 (WFC) and 1R01HD065029 (CKW), ADA 1-10-CT-57 (CKW), 1 UL1 RR025758 Harvard Clinical and Translational Science Center and M01-RR-01066 from the National Center for Research Resources.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
Reference List
- 1.Knochenhauer ES, Key TJ, Kahsar-Miller M, Waggoner W, Boots LR, Azziz R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern United States: a prospective study. J Clin Endocrinol Metab. 1998;83:3078–3082. doi: 10.1210/jcem.83.9.5090. [DOI] [PubMed] [Google Scholar]
- 2.Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab. 1999;84:4006–4011. doi: 10.1210/jcem.84.11.6148. [DOI] [PubMed] [Google Scholar]
- 3.Asuncion M, Calvo RM, San Millan JL, Sancho J, Avila S, Escobar-Morreale HF. A prospective study of the prevalence of the polycystic ovary syndrome in unselected Caucasian women from Spain. J Clin Endocrinol Metab. 2000;85:2434–2438. doi: 10.1210/jcem.85.7.6682. [DOI] [PubMed] [Google Scholar]
- 4.Taylor AE, McCourt B, Martin KA, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2248–2256. doi: 10.1210/jcem.82.7.4105. [DOI] [PubMed] [Google Scholar]
- 5.Rebar R, Judd HL, Yen SSC, Rakoff J, Vandenberg G, Naftolin F. Characterization of inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57:1320–1329. doi: 10.1172/JCI108400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waldstreicher J, Santoro NF, Hall JE, Filicori M, Crowley WF., Jr Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab. 1988;66:165–172. doi: 10.1210/jcem-66-1-165. [DOI] [PubMed] [Google Scholar]
- 7.Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab. 1983;57:356–359. doi: 10.1210/jcem-57-2-356. [DOI] [PubMed] [Google Scholar]
- 8.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165–1174. doi: 10.2337/diab.38.9.1165. [DOI] [PubMed] [Google Scholar]
- 9.Stein IF, Leventhal ML. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181–191. [Google Scholar]
- 10.Adams J, Polson DW, Franks S. Prevalence of polycystic ovaries in women with anovulation and idiopathic hirsutism. Br Med J. 1986;293:355–359. doi: 10.1136/bmj.293.6543.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pigny P, Merlen E, Robert Y, et al. Elevated Serum Level of Anti-Mullerian Hormone in Patients with Polycystic Ovary Syndrome: Relationship to the Ovarian Follicle Excess and to the Follicular Arrest. Journal of Clinical Endocrinology & Metabolism. 2003;88:5957–5962. doi: 10.1210/jc.2003-030727. [DOI] [PubMed] [Google Scholar]
- 12.Adams JM, Taylor AE, Crowley WF, Jr, Hall JE. Polycystic ovarian morphology with regular ovulatory cycles: insights into the pathophysiology of polycystic ovarian syndrome. J Clin Endocrinol Metab. 2004;89:4343–4350. doi: 10.1210/jc.2003-031600. [DOI] [PubMed] [Google Scholar]
- 13.Legro RS, Chiu P, Kunselman AR, Bentley CM, Dodson WC, Dunaif A. Polycystic ovaries are common in women with hyperandrogenic chronic anovulation but do not predict metabolic or reproductive phenotype. J Clin Endocrinol Metab. 2005;90:2571–2579. doi: 10.1210/jc.2004-0219. [DOI] [PubMed] [Google Scholar]
- 14.Welt CK, Arason G, Gudmundsson JA, et al. Defining constant versus variable phenotypic features of women with polycystic ovary syndrome using different ethnic groups and populations. J Clin Endocrinol Metab. 2006;91:4361–4368. doi: 10.1210/jc.2006-1191. [DOI] [PubMed] [Google Scholar]
- 15.Johnson TRB, Kaplan LK, Ouyang P, Rizza RA. Evidence-based Methodology Workshop on Polycystic Ovary Syndrome. prevention nih gov/workshops/2012/pcos/docs/PCOS_Final_Statement pdf. 2013 [Google Scholar]
- 16.Cussons AJ, Stuckey BG, Walsh JP, Burke V, Norman RJ. Polycystic ovarian syndrome: marked differences between endocrinologists and gynaecologists in diagnosis and management. Clin Endocrinol (Oxf) 2005;62:289–295. doi: 10.1111/j.1365-2265.2004.02208.x. [DOI] [PubMed] [Google Scholar]
- 17.Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome (PCOS) in a Dutch twin-family study. J Clin Endocrinol Metab. 2005;91:2100–2104. doi: 10.1210/jc.2005-1494. [DOI] [PubMed] [Google Scholar]
- 18.Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- 19.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 20.Pau C, Saxena R, Welt CK. Evaluating reported candidate gene associations with polycystic ovary syndrome. Fertil Steril. 2013;10 doi: 10.1016/j.fertnstert.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ewens KG, Stewart DR, Ankener W, et al. Family-based analysis of candidate genes for polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95:2306–2315. doi: 10.1210/jc.2009-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbanek M. The genetics of the polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2007;3:103–111. doi: 10.1038/ncpendmet0400. [DOI] [PubMed] [Google Scholar]
- 23.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 24.A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 26.Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet. 2005;6:109–118. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 27.Chen ZJ, Zhao H, He L, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet. 2011;43:55–59. doi: 10.1038/ng.732. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Zhao H, Shi Y, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44:1020–1025. doi: 10.1038/ng.2384. [DOI] [PubMed] [Google Scholar]
- 29.Goodarzi MO, Jones MR, Li X, et al. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet. 2012;49:90–95. doi: 10.1136/jmedgenet-2011-100427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lerchbaum E, Trummer O, Giuliani A, Gruber HJ, Pieber TR, Obermayer-Pietsch B. Susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21, and 9q33.3 in a cohort of Caucasian women. Horm Metab Res. 2011;43:743–747. doi: 10.1055/s-0031-1286279. [DOI] [PubMed] [Google Scholar]
- 31.Welt CK, Styrkarsdottir U, Ehrmann DA, et al. Variants in DENND1A are Associated with Polycystic Ovary Syndrome in Women of European Ancestry. 2012 doi: 10.1210/jc.2011-3478. In Revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louwers YV, Stolk L, Uitterlinden AG. Replication of Chinese PCOS susceptibility loci in patients diagnosed with PCOS from Caucasian descent. 95th Annual Meeting of the Endocrine Society; 2013. [Google Scholar]
- 33.Marat AL, Dokainish H, McPherson PS. DENN domain proteins: regulators of Rab GTPases. J Biol Chem. 2011;286:13791–13800. doi: 10.1074/jbc.R110.217067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tasaka K, Masumoto N, Mizuki J, et al. Rab3B is essential for GnRH-induced gonadotrophin release from anterior pituitary cells. J Endocrinol. 1998;157:267–274. doi: 10.1677/joe.0.1570267. [DOI] [PubMed] [Google Scholar]
- 35.Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mutharasan P, Galdones E, Penalver BB, et al. Evidence for chromosome 2p16.3 polycystic ovary syndrome susceptibility locus in affected women of European ancestry. J Clin Endocrinol Metab. 2013;98:E185–E190. doi: 10.1210/jc.2012-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawamura K, Cheng Y, Suzuki N, et al. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. Proceedings of the National Academy of Sciences. 2013 doi: 10.1073/pnas.1312830110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Jin Y, Reddy MV, et al. Genetically dependent ERBB3 expression modulates antigen presenting cell function and type 1 diabetes risk. PLoS ONE. 2010;5:e11789. doi: 10.1371/journal.pone.0011789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet. 2008;17:R156–R165. doi: 10.1093/hmg/ddn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Todd JA. Statistical false positive or true disease pathway? Nat Genet. 2006;38:731–733. doi: 10.1038/ng0706-731. [DOI] [PubMed] [Google Scholar]
- 41.Tsao H, Florez JC. Introduction to genetic association studies. J Invest Dermatol. 2007;127:2283–2287. doi: 10.1038/sj.jid.5701054. [DOI] [PubMed] [Google Scholar]
- 42.Cui L, Zhao H, Zhang B, et al. Genotype-phenotype correlations of PCOS susceptibility SNPs identified by GWAS in a large cohort of Han Chinese women. Hum Reprod. 2013;28:538–544. doi: 10.1093/humrep/des424. [DOI] [PubMed] [Google Scholar]
- 43.Strauss JF, III, McAllister JM, Urbanek M. Persistence pays off for PCOS gene prospectors. J Clin Endocrinol Metab. 2012;97:2286–2288. doi: 10.1210/jc.2012-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena R, Welt CK. Polycystic ovary syndrome is not associated with genetic variants that mark risk of type 2 diabetes. Acta Diabetol. 2012 doi: 10.1007/s00592-012-0383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ewens KG, Jones MR, Ankener W, et al. Type 2 diabetes susceptibility single-nucleotide polymorphisms are not associated with polycystic ovary syndrome. Fertil Steril. 2011 doi: 10.1016/j.fertnstert.2011.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biyasheva A, Legro RS, Dunaif A, Urbanek M. Evidence for association between polycystic ovary syndrome (PCOS) and TCF7L2 and glucose intolerance in women with PCOS and TCF7L2. J Clin Endocrinol Metab. 2009;94:2617–2625. doi: 10.1210/jc.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyssenko V, Lupi R, Marchetti P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007;117:2155–2163. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ewens KG, Jones MR, Ankener W, et al. FTO and MC4R gene variants are associated with obesity in polycystic ovary syndrome. PLoS ONE. 2011;6:e16390. doi: 10.1371/journal.pone.0016390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barber TM, Bennett AJ, Groves CJ, et al. Association of variants in the fat mass and obesity associated (FTO) gene with polycystic ovary syndrome. Diabetologia. 2008;51:1153–1158. doi: 10.1007/s00125-008-1028-6. [DOI] [PubMed] [Google Scholar]
- 52.Li T, Wu K, You L, et al. Common variant rs9939609 in gene FTO confers risk to polycystic ovary syndrome. PLoS ONE. 2013;8:e66250. doi: 10.1371/journal.pone.0066250. [DOI] [PMC free article] [PubMed] [Google Scholar]