

Abstract
It has been reported that mechanical strain activates extracellular signal-regulated protein kinases (ERK) without the involvement of angiotensin II (Ang II) in cardiomyocytes. We examined the effects of mechanical strain on ERK phosphorylation levels in the absence of Ang II using rat mesangial cells. The ratio of phosphorylated ERK (p-ERK) to total ERK expression was increased by cyclic mechanical strain in a time- and elongation strength-dependent manner. With olmesartan [Ang II type 1 receptor (AT1R) antagonist] pretreatment, p-ERK plateau levels decreased in a dose-dependent manner (EC50 = 1.3 × 10−8 M, maximal inhibition 50.6 ± 11.0% at 10−5 M); a similar effect was observed with RNA interference against Ang II type 1A receptor (AT1AR) and Tempol, a superoxide dismutase mimetic. In addition to the inhibition of p-ERK levels, olmesartan blocked the increase in cell surface and phosphorylated p47phox induced by mechanical strain and also lowered the mRNA expression levels of NADPH oxidase subunits. These results demonstrate that mechanical strain stimulates AT1R to phosphorylate ERK in mesangial cells in the absence of Ang II. This mechanotransduction mechanism is involved in the oxidative stress caused by NADPH oxidase and is blocked by olmesartan. The inverse agonistic activity of this AT1R blocker may be useful for the prevention of mesangial proliferation and renal damage caused by mechanical strain/oxidative stress regardless of circulating or tissue Ang II levels.
Keywords: hypertension
hypertension is a dominant pathogenetic factor in target organ damage such as ischemic heart disease, stroke, and renal dysfunction. Emerging evidence indicates that renal sclerosis is a major cause of renal dysfunction in hypertension, and mesangial proliferation is known to play a pivotal role in the pathophysiology of renal sclerosis. Since mesangial cells are located between the vascular and urinary space in glomeruli, they can be influenced by many hormonal/humoral substances, including angiotensin II (Ang II). Ang II stimulates cell differentiation and proliferation of glomerular mesangial cells, leading to glomerulosclerosis (1). Extracellular signal-regulated kinases (ERK), among the mitogen-activated protein kinases (MAPK), are serine/threonine kinases acting as transducers of signals from the cell membrane to the nucleus in response to various cellular stresses, such as cytokines or signals from G protein-coupled receptors, including the Ang II type 1 receptor (AT1R) (27). The activation of ERK, via Ang II through AT1R, modulates cell growth and differentiation in mesangial cells, as well as in cardiac myocytes. Presently, AT1R blockers (ARBs) are widely used clinically for the treatment of hypertension and the prevention of heart disease and renal injury. However, Ang II-independent effects of ARBs on AT1R are not well defined.
Recently it was reported that mechanical strain activates the MAPK pathway via the AT1R without the involvement of Ang II in cardiomyocytes (29). We have also demonstrated that selective knockdown of renal AT1R expression with the use of antisense oligodeoxynucleotides markedly reduces urinary protein excretion and glomerular sclerosis in spontaneously hypertensive rats, independent of circulating Ang II levels (28). However, in the kidney, the influence of mechanical strain and the renin-angiotensin system on mesangial cells remains unclear. It is possible that mechanical strain caused by elevated blood pressure that is not primarily attributable to increased activity of the renin-angiotensin system may be aggravated by Ang II-independent activation of AT1R.
It is well recognized that ERKs are activated by mechanical strain through modulation of intracellular calcium ion concentration (10) or through integrins that connect the cytoskeleton to the extracellular matrix (10, 15). It is also known that NADPH oxidase is activated by Ang II via AT1R, which leads to phosphorylation of ERKs (3, 4, 7). We tested the hypothesis that NADPH oxidase activity can be modulated by mechanical strain through AT1R and activation of ERK in the absence of Ang II. In the present study, we examined ERK phosphorylation caused by mechanical strain in the absence of Ang II using primary cultures of rat mesangial cells (RMCs) and also examined the role of NADPH oxidase in this phenomenon.
MATERIALS AND METHODS
Isolation and cultivation of RMCs.
RMCs were isolated from 5–7-wk-old male Sprague-Dawley rats with the use of a conventional sieving method as reported previously (22). Isolated RMCs (passages 4–6) were cultured in DMEM (Gibco-Invitrogen, Carlsbad, CA) containing 20% fetal bovine serum and penicillin-streptomycin at 37°C in a humidified 5% CO2 water-jacketed incubator. All procedures were approved by the Fukushima Medical University School of Medicine Animal Committee.
Test compounds.
Ang II was supplied from Peptide Institute (Osaka, Japan). Olmesartan was kindly supplied by Daiichi-Sankyo Pharmaceutical (Tokyo, Japan), and losartan potassium was purchased from Wako Pure Chemical Industries (Osaka, Japan). Tempol, BAPTA-TM, and cytochalasin D were purchased from Sigma-Aldrich Japan (Tokyo, Japan).
Mechanical strain application.
Mesangial cells were seeded into six-well silicon elastomer-base culture plates with collagen type I coating (Flex I plates; Flexcell International, Hillsborough, NC) or standard six-well cell-culture plates at a density of 12,000 cells/cm2. The cells achieved confluence after 5–7 days. After serum starvation for 12 h, the cells were preincubated with the test compounds or their corresponding vehicle as control for designated periods. Thereafter, cells were subjected to stretch/relaxation cycles using the Flexercell Tension Plus system, FX-4000T (Flexcell International). Vacuum was cyclically applied (60 cycles/min) to the rubber-based plates via the base plate, which was placed in a water-jacketed incubator with 5% CO2 at 37°C (9). Cells were exposed to elongation stretch strengths of 5, 10, 15, or 20% for periods ranging from 2.5 min to 12 h.
Silencing AT1R by miR RNAi.
BLOCK-iT Pol II miR RNAi expression vector (Invitrogen) with the target sequence of TGTCATCCACCGAAATGTATA was used for silencing of Ang II type 1A receptor (AT1AR) in Rattus norvegicus. Purified oligodeoxynucleotides were designed and synthesized by Invitrogen primer team (Tokyo, Japan). AT1AR micro RNA (miRNA) insert caused a 85.48 ± 4.71% decrease in AT1AR transcript, as verified by real-time RT-PCR. Transfection was performed using Lipofectamine LTX and Plus Reagent (Invitrogen). Briefly, trypsinized and suspended cells in normal growth medium were seeded at a concentration of 1 × 105 cells/well in six-well plates containing miR vector (500 ng) in transfection reagent mixture. After incubation at 37°C for 12 h, the culture medium containing transfection reagent was replaced with fresh medium with Blastcidin for selection. Transfection efficiency was determined by the fluorescent intensity of the Emerald-green fluorescent protein tag.
Membrane protein preparation and immunoprecipitation.
Protein sample preparation and immunoblotting analysis were performed as previously described (28). Whole cell protein samples were extracted with single detergent lysis buffer containing 50 mM Tris·HCl, 150 mM NaCl, 0.02% sodium azide, 1% NP-40, and Complete (a protease inhibitor cocktail; Roche Diagnostics, Mannheim, Germany). Phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO) were also added to the lysis buffer to prevent dephosphorylation. Membrane protein samples were separated using a cell-surface biotinylation method. In brief, cells were incubated in ice-cold PBS supplemented with cell-impermeant and noncleavable sulfo-NHS-SS-biotin (500 μg/ml) with gentle agitation on a rocking platform. After quenching excess biotin with HEPES-buffered saline, cells were lysed with single detergent lysis buffer and cell debris removed with low-speed centrifugation. Biotinylated membranes in lysis buffer were captured by Immobilized NeutrAvidin gel and then eluted in LDS sample buffer with heating at 75°C. Immunoreactive p47phox protein sample was separated and purified by immunoprecipitation. The whole cell protein sample (500 μg protein/ml) was incubated with anti-p47phox antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 2 μl/ml for 2 h, followed by incubation with protein-G agarose at 4°C overnight. The immunoprecipitates were pelleted and washed four times with lysis buffer containing the protease inhibitor cocktail. The pellets were suspended in sample buffer (Invitrogen), heated at 75°C for 10 min, and subjected to immunoblotting with the anti-p47phox and anti-phosphoserine antibodies.
Immunoblotting.
Each sample was electrophoretically size separated under denaturing conditions in 10% bis-Tris-polyacrylamide gels, followed by the transfer of proteins onto polyvinylidene difluoride membranes. The blots were soaked overnight at 4°C in commercially available blocking agent (BlockAce; Dainihon Pharmacy, Osaka, Japan). The membranes were then probed for 1 h with polyclonal rabbit anti p42/p44 MAP kinase (ERK), anti-phospho-p42/p44 MAP kinase (phospho-ERK; Cell Signaling, Beverly, MA), anti-p47phox, anti-AT1R, anti-phosphoserine, or anti-β-actin antibodies (Santa Cruz Biotechnology) in Tris-buffered saline containing 0.1% Tween 20. The membranes were subsequently washed and incubated with peroxidase-conjugated goat anti-rabbit or goat anti-mouse secondary antibody. When necessary, probed membranes were stripped in Tris buffer solution containing 2% SDS and 100 mM β-mercaptoethanol at 65°C. Quantitative assessment of band densities was performed by scanning densitometry.
Quantitative real-time RT-PCR.
Total RNA was prepared from RMCs growing on elastic-bottomed plates using TRIzol (Invitrogen) and column-purified after DNase treatment with RNase-free DNase set (Qiagen, Valencia, CA). Subsequently, 1 μg of total RNA was reverse transcribed into cDNA using oligo (dT) method (iScript cDNA synthesis kit; Bio-Rad, Hercules, CA) in 20 μl reaction volume. cDNA in 1 μl of reaction mix was used for real-time quantitative PCR using Light Cycler and FastStart DNA Master SYBR Green I (Roche Diagnostics). Quantification of mRNA was based on Ct value, normalized to β-actin, and expressed as the magnitude of change under mechanical strain application relative to the appropriate control. Primers for rat angiotensinogen, p22phox, p47phox, p67phox, gp91phox, Nox-1, Nox-4, and β-actin were used to replicate cDNAs reverse transcribed from the experimental, positive, and negative control RNA samples, as described previously (12).
Determination of Ang II concentration in culture medium.
Supernatant was collected from the culture medium after the cells were subjected to cyclic mechanical strain. The concentration of Ang II was measured by an enzyme-linked immunosorbent assay using an Ang II enzyme immunoassay (EIA) Kit (Cayman Chemical, Ann Arbor, MI).
Statistical analysis.
Differences among groups were analyzed by one-way ANOVA with Bonferroni correction. Statistical significance was assumed at P < 0.05. Data are given as means ± SE.
RESULTS
Mechanical strain-induced ERK phosphorylation is decreased by olmesartan in the absence of Ang II.
In primary cultures of rat renal mesangial cells, cyclic mechanical strain alone significantly increased the ratio of phospho-ERK (p-ERK) to total ERK expression in a time-dependent manner. ERK phosphorylation peaked after 5 min of strain (Fig. 1A). With continued strain, ERK phosphorylation decreased and reached a nadir at 60 min and plateaued at a higher level than baseline thereafter. The increase in p-ERK at the peak phase was also elongation strength dependent, and maximal response was achieved with 20% elongation (Fig. 1B). Ang II-induced activation of ERK was confirmed; Ang II administration induced the phosphorylation of ERK in a time- and concentration-dependent manner (Fig. 1, C and D, respectively).
Fig. 1.

Mechanical strain and angiotensin II (Ang II) induce the activation of ERK in a time-dependent and elongation strength (dose)-dependent manner. Phosphorylation (p) levels of ERK were expressed as % change from the control. A: increase in p-ERK by stretch was time dependent; maximal response was obtained at 5 min after the initiation of stretch. B: increase in p-ERK by stretch was elongation strength dependent; maximal response was obtained with 20% elongation. The increase in p-ERK by Ang II was time dependent (C) and concentration dependent (D). Ang II concentration of 10−10 M was used for the time-course study, and an incubation time of 10 min was used for the dose-response study. The maximal response was obtained with 10−10 M of Ang II at an incubation time of 10 min. Means ± SE (n = 4) are given. Ctl, control.
In cardiomyocytes, it has been recently reported that AT1R can be activated by mechanical strain in the absence of its specific agonist, Ang II (29). Since AT1R stimulation leads to ERK phosphorylation, we hypothesized that the stretch-induced ERK phosphorylation is mediated by AT1R activation in mesangial cells. To determine whether mechanical strain can activate ERK through AT1R in mesangial cells, the effect of olmesartan, an AT1R blocker, on mechanical strain-induced ERK activation in the absence of Ang II was examined. Pretreatment with olmesartan for 60 min before the application of mechanical strain significantly inhibited ERK phosphorylation at the plateau phase, which occurred at 60 min after the initiation of stretch stress. As shown in Fig. 2A, p-ERK levels at the plateau phase were reduced in a concentration-dependent manner (□, EC50 = 1.3 × 10−8 M, maximal inhibition, 50.6 ± 11.0% at 10−5 M) with olmesartan treatment. In contrast, olmesartan did not have a significant inhibitory effect on the activation of ERK at 5 min of mechanical stress when maximal p-ERK activation was obtained by mechanical strain (Fig. 2A, •). Because ERK was only minimally active at basal conditions without added Ang II in RMCs, olmesartan did not significantly affect the basal phosphorylation levels of ERK (Fig. 2B). To compare the effects of two different ARBs, the cells were preincubated with losartan or olmesartan. In contrast to olmesartan, losartan did not show a significant inhibitory effect on the activation of ERK induced by mechanical strain at the plateau phase. Losartan did not change the basal phosphorylation levels of ERK, similar to the result obtained with olmesartan (Fig. 2B).
Fig. 2.
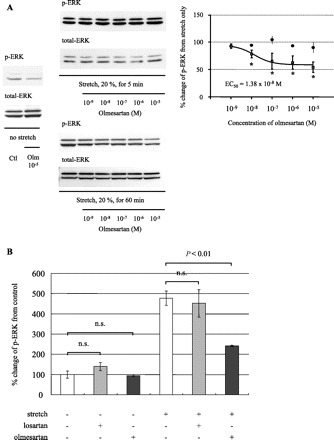
Effect of mechanical strain and olmesartan on the phosphorylation levels of ERK. A: concentration-dependent effect of olmesartan (Olm) on mechanical strain-induced activation of ERK. Phosphorylated ERK levels were expressed as % change from the control. *P < 0.01 vs. control. Means ± SE (n = 5) are given. p-ERK levels at the plateau phase (□) were reduced in a concentration-dependent manner (EC50 = 1.3 × 10−8 M, maximal inhibition 50.6 ± 11.0% at 10−5 M) with olmesartan treatment. In contrast, p-ERK levels at early phase (•) were not attenuated by pretreatment with olmesartan. B: olmesartan as well as losartan (10−5 M) did not change basal p-ERK levels but attenuated the elevated levels of p-ERK induced by cyclic mechanical strain at plateau phase. In contrast to olmesartan, losartan did not have any inhibitory effect on the stretch-induced ERK activation at the plateau phase of 60 min. Means ± SE (n = 5) are given. n.s., not significant.
The effect of AT1R blockade by olmesartan was observed only at plateau phase. For this reason, in subsequent studies, the levels of ERK phosphorylation were studied at 60 min of cyclic stretch at 20% elongation. Because several mechanisms have been described as pathways of mechanotransduction in mechanosensitive tissues including the heart, vasculature, and kidney, the rapid activation of ERK not blocked by olmesartan may be mediated by an AT1R-independent pathway. Integrins, interacting with the connecting matrix/environment, mediate increases in intracellular Ca2+ levels and activate MAPK cascades to cause ERK phosphorylation. Also, previous reports have implicated a stretch-activated ion channel in mediating mechanotransduction (10).
To determine the autocrine/paracrine effects of Ang II, activation of the renin-angiotensin system was assessed. There were no changes in angiotensinogen mRNA levels following 60 min of cyclic mechanical strain (control: 1.51 ± 0.04; olmesartan only: 1.50 ± 0.05; stretch only: 1.52 ± 0.03; stretch after olmesartan pretreatment: 1.51 ± 0.03, no significant differences among groups; data are expressed as ΔCt value normalized to β-actin ± SE, n = 6). The concentration of Ang II in the culture medium was below the detection limit using EIA (3 pg/ml) before and 10, 20, 30, and 60 min after application of the mechanical strain. The stimulatory effect of Ang II on ERK peaked at 10 min and disappeared after 60 min (Fig. 1C). This result supports our contention that the effects of 60 min of mechanical strain on ERK phosphorylation were not attributable to an increase in Ang II that was below the detection limit of our method.
Effect of AT1R miRNA on mechanical strain-induced ERK activation.
It is possible that the inhibition of stretch-induced p-ERK elevation by olmesartan is independent of AT1R blockade. To further demonstrate that the activation via mechanical strain was transduced through the AT1R even in the absence of Ang II, Rattus norvegicus AT1AR gene expression was inhibited using RNA interference (RNAi) technique. miRNA against rat AT1AR, which was produced by BLOCK-iT Pol II miRNA RNAi expression vector, decreased protein expression levels of AT1AR by 76.8 ± 6.8% relative to the mock vector-treated control (Fig. 3A). AT1AR miRNA treatment attenuated the p-ERK levels at the plateau phase (60 min) (53.5 ± 9.7%, Fig. 3B) but did not affect p-ERK levels at the initial peak (5 min) (Fig. 3C), corroborating the results obtained with olmesartan.
Fig. 3.

Effect of micro RNA (miRNA, miR) against Ang II type 1 receptor (AT1R) on ERK phosphorylation induced by mechanical strain. A: transfection with miRNA significantly decreased the basal expression levels of AT1R (76.8 ± 6.8% reduction). B: miRNA treatment significantly attenuated the stretch-induced elevation of phosphorylation levels of ERK at plateau phase (53.5 ± 9.7% reduction). Means ± SE (n = 3) are given. $P < 0.01 vs. mock control, *P < 0.01 vs. mock + stretch. C: AT1R silencing did not have any inhibitory effect on ERK phosphorylation at initial peak 5 min after stretch application (1,304.38 ± 93.8% increasing of p-ERK in mock group vs. 1,284.98 ± 111.3% in miR group, n = 4). AT1AR, Ang II type 1A receptor.
Involvement of oxidative stress caused by mechanical strain through AT1R.
Oxidative stress is known to be involved in the activation of ERK by AT1R. In this study, Tempol, a superoxide dismutase mimetic, also lowered the plateau phase p-ERK levels induced by mechanical strain in a dose-dependent manner (Fig. 4; maximal inhibition of ∼50% was obtained at 10−5 M). NADPH oxidase is activated by Ang II, and NADPH oxidase-dependent O2− and H2O2 production transactivate the MAPK cascade via receptor tyrosine kinases (8, 26). Hydrogen peroxide time and concentration dependently stimulated ERK phosphorylation in RMCs (maximal response of 521.6 ± 47.5% increase in ERK phosphorylation from control was obtained with 0.001% H2O2 at 60 min; means ± SE, n = 4). Thus, to determine the relationship between mechanical strain-induced activation of ERK through AT1R and NADPH oxidase activity, the translocation of p47phox, a cytosolic component of NADPH oxidase, to the cell membrane was examined by Western blotting. The abundance of p47phox in the cell membrane fraction and phosphorylated p47phox in whole cell was significantly increased by mechanical strain (Fig. 5). Olmesartan was able to block the stretch-induced translocation and the serine phosphorylation of p47phox.
Fig. 4.

The effect of Tempol on plateau-phase ERK phosphorylation induced by mechanical strain (Str). Phosphorylation levels of ERK were expressed as % of the control. Similar to the result obtained with olmesartan pretreatment, Tempol dose dependently lowered ERK activity at plateau phase induced by mechanical strain. Maximal inhibition was observed with 10−5 M of Tempol (Tem). Means ± SE (n = 3) are given. $P < 0.01 from control; *P < 0.01 vs. control.
Fig. 5.

Olmesartan attenuates the membrane translocation and phosphorylation of p47phox by stretch (Str). Abundance of p47phox on cell surface membranes (A) and phosphorylation levels of p47phox (B) are shown. Olmesartan (10−5 M) significantly decreased membrane translocation and phosphorylation of p47phox. Means ± SE (n = 4) are given. *P < 0.05 vs. control; $P < 0.05 vs. stretch group. IP, immunoprecipitate.
We also examined the effect of olmesartan on the mRNA expression levels of NADPH oxidase subunits p22phox, p47phox, p67phox, gp91phox, Nox-1, and Nox-4 using real-time RT-PCR. The basal expression levels of p22phox and p67phox were significantly decreased after olmesartan treatment (Fig. 6). Cyclic mechanical strain increased the expressions of p47phox, p22phox, and p67phox, and olmesartan attenuated the elevated expression levels. In contrast, cyclic mechanical strain did not have any significant effect on the expression levels of gp91phox, Nox-1, and Nox-4.
Fig. 6.

Changes in the mRNA expression levels of NADPH oxidase subunits p47phox, p22phox, and p67phox. Quantification of mRNA was determined on the basis of the Ct value, normalized to β-actin, and expressed as the magnitude of change under mechanical strain application relative to the corresponding control. Means ± SE (n = 3) are given. &P < 0.01 vs. control; *P < 0.01 vs. stretch group.
The effect of BAPTA and cytochalasin D on stretch-induced ERK phosphorylation.
Finally, an additional experiment was performed to explore the reason why neither olmesartan nor AT1AR gene silencing had any inhibitory effect on ERK phosphorylation at the rapid phase (5 min) after the initiation of mechanical strain. BAPTA-TM, a potent chelator of calcium ion, and cytochalasin D, an inhibitor of actin polymeration, were used to study the AT1R-induced, oxidative stress-independent activation of ERK (Fig. 7). The early-phase activation of ERK induced by cyclic mechanical strain was abolished almost completely by preincubation with either 1 μM BAPTA-TM or 1 μM cytochalasin D. In contrast, the phosphorylation of ERK at the plateau phase by mechanical strain was still observed with BAPTA-TM or cytochalasin D.
Fig. 7.

Effect of BAPTA and cytochalasin D on stretch-induced ERK phosphorylation. Phosphorylation levels of ERK were expressed as % change from control. The early-phase (5 min) activation of ERK induced by cyclic mechanical strain was abolished almost completely by preincubation with either 1 μM of BAPTA-TM or 1 μM of cytochalasin D. In contrast, the phosphorylation of ERK at the plateau phase (60 min) by mechanical strain was still observed with BAPTA-TM or cytochalasin D (BAPTA-TM; 273.7 ± 20.5%, cytochalasin D; 208.5 ± 19.8% compared with no stretch control). Means ± SE (n = 4) are given. *P < 0.01 vs. control.
DISCUSSION
This study demonstrates for the first time that mechanical strain increases the phosphorylation levels of ERK in RMCs via AT1R even in the absence of Ang II. An ARB, olmesartan, attenuates ERK activation after 60 min of mechanical strain. This is not due to inhibition of locally de novo synthesized Ang II (13, 20, 21) because the concentrations of secreted Ang II and the expression levels of angiotensinogen are unchanged by stretch. Indeed, it is widely known that the intrarenal renin-angiotensin system is highly active, especially in the renal tubules (2). However, in mesangial cells, there are only a few reports on the activation of local renin-angiotensin system, and the existence of mechanical strain-induced local Ang II formation has not been reported. In the present study, evidence of a local activation of the renin-angiotensin system is not observed, but the effect seems to be mediated by AT1R because the knockdown of AT1AR using miRNA has the same effect as olmesartan. These observations indicate that cyclic mechanical strain stimulates AT1R independent of Ang II.
A recent study reported AT1R activation by mechanical strain in the absence of Ang II in cardiomyocytes, leading to the activation of JNK, p38 MAPK, and ERK (29). Endogenous activation of renin-angiotensin system is also not detected in strain-exposed cardiomyocytes since the cells were obtained from angiotensinogen-null mice. Our present report demonstrates that a similar signal transduction system may exist in renal mesangial cells. However, the findings more closely resemble the physiological situation because in the present study the cells were isolated from normal Sprague-Dawley rats with an intact renin-angiotensin system. There are also differences between the reported studies in cardiac myocytes and our studies in renal mesangial cells. In cardiac myocytes, the maximal inhibition of ERK phosphorylation with AT1R blockade is observed at the initial peak after 8 min in cardiomyocytes, as opposed to the plateau phase after 60 min of stretch stress in mesangial cells. There are several cell signaling molecules that activate ERK, such as integrins (10, 15) and intracellular calcium ion accumulation (10). In additional studies, early-phase activation of ERK induced by cyclic mechanical strain is abolished by preincubation with either cytochalasin D, a potent inhibitor of actin polymerization, or BAPTA-TM, a calcium ion chelator. However, these treatments minimally affect the phosphorylation levels of ERK at the plateau phase (Fig. 7). It is possible that the initial peak of ERK phosphorylation with mechanical strain is caused mainly by the early signal transduction systems (e.g., calcium influx or cytoskeleton modification), such that the effect of AT1R may be masked by these short-term signals but becomes apparent only at the plateau phase. Since stretch stress attributable to high blood pressure is a chronic stimulus, the effects of AT1R activation and antagonism at the plateau phase may be more relevant in the pathogenesis of long-term organ damage.
We found that Tempol, a potent superoxide dismutase mimetic, decreases the phosphorylation levels of ERK at the plateau phase. To evaluate further the mechanisms of ERK activation caused by strain through AT1R at the plateau phase, the role of NADPH oxidase activation, one of the major sources of O2− and H2O2 in oxidative stress, was evaluated. Previous studies have shown that Ang II activates NADPH oxidase (5, 11, 14), and the resulting oxidative stress in turn activates the MAPK cascade (3, 7). In the present study, we show that mechanical strain increases both the translocation of p47phox to the cell membrane and phosphorylated levels of p47phox, which may reflect NADPH oxidase activation (25). As stated earlier, calcium ion influx leads to rapid activation of ERK, whereas the activation of ERK through H2O2 production via NADPH oxidase activation may take longer (26). This is indeed the case; H2O2 increases phospho-ERK gradually with the highest value observed at 60 min. The activation of NADPH oxidase and production of reactive oxygen species may explain the delayed effect of AT1R blockade on ERK activation induced by mechanical strain. However, there have been reports that ARBs, including olmesartan, are potent antioxidants (11). It is possible that part of the attenuation of mechanical strain-induced ERK phosphorylation may be exerted through the free-radical scavenging of olmesartan in combination with the Ang II-independent AT1R inactivation.
Olmesartan has a greater blood pressure-lowering and organ-protective effects than other ARBs (23, 24). Some investigators speculate that these enhanced effects may be related to the inverse agonistic activity of ARBs. Some receptor antagonists have inhibitory effects in spite of the absence of specific agonists; these drugs are called inverse agonists (17, 29). Olmesartan has strong inverse agonist activity against constitutively active AT1-F77A or N111G mutant AT1R, relative to losartan (18, 19). Indeed, losartan has been reported to prevent Ang II-induced, but not stretch-induced, VEGF protein secretion in human mesangial cells (8). In the present study, olmesartan but not losartan significantly attenuates stretch-induced, agonist-independent ERK activation in mesangial cells. This observation can be explained by the strong inverse-agonist effect of olmesartan (18, 19). Olmesartan, unlike losartan, has α-carboxyl groups, and of the currently available ARBs olmesartan has the largest number of estimated binding domains that can interact with AT1R. The greater number of interacting domains and affinity to AT1R possibly affect the potency of inverse agonistic activity of ARBs. Olmesartan also significantly decreases basal and stretch-induced NADPH oxidase subunit mRNA expression. Our results show that there may be a low level of constitutive AT1R activity in cultured RMCs, which can be blocked by a potent inverse agonist of AT1R such as olmesartan. Another inverse agonist of AT1R, candesartan, was also able to attenuate the agonist-independent, stretch-induced AT1R activation in cardiomyocytes (29). In that report, a competitive inhibitor for Ang II, (Sar1, Ile8)-Ang II, was unable to replicate the effect of candesartan. Further investigation will be required to determine whether other ARBs with inverse-agonist effect can also block Ang II-independent, stretch-induced AT1R activation.
We have recently demonstrated that selective knockdown of renal AT1R expression using antisense oligodeoxynucleotides markedly reduces urinary protein excretion and glomerular sclerosis in spontaneously hypertensive rats (28). The reduction in urinary protein excretion and glomerular sclerosis occurs despite elevated circulating Ang II and aldosterone levels and sustained high blood pressure. This may be due to the fact that both Ang II-dependent and Ang II-independent mechanical stretch-induced AT1R activation are blocked by the knockdown of the AT1R gene. Although a high salt intake decreases plasma renin activity and aldosterone in hypertensive patients (6, 16), renal damage is not prevented. It is possible that the hypertensive renal damage and mesangial proliferation in these patients may partly be caused by agonist-independent, stretch stress-induced AT1R activation.
In conclusion, cyclic mechanical strain increases the phosphorylation levels of ERK and activates NADPH oxidase in mesangial cells in the absence of local Ang II production. These effects are blocked by olmesartan, an ARB with inverse agonistic activity. The ARBs with inverse agonistic activity such as olmesartan could be beneficial in the prevention of renal mesangial proliferation and hypertensive organ damage regardless of circulating or tissue Ang II levels.
GRANTS
This work was supported in part by the Fukushima Society for the Promotion of Medicine (No. 18) and Salt Science Foundation (No. 0835).
Acknowledgments
The authors thank Hiroko Ohashi for technical assistance. Olmesartan was kindly supplied by Daiichi-Sankyo Pharmaceutical Co., Ltd. (Tokyo, Japan).
REFERENCES
- 1.Ardaillou R, Chansel D, Chatziantoniou C, Dussaule JC. Mesangial AT1 receptors: expression, signaling, and regulation. J Am Soc Nephrol 10, Suppl 11: S40–S46, 1999 [PubMed] [Google Scholar]
- 2.Bader M, Peters J, Baltatu O, Muller DN, Luft FC, Ganten D. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med 79: 76–102, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res 65: 16–27, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 24: 471–478, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol 285: R117–R124, 2003 [DOI] [PubMed] [Google Scholar]
- 6.de la Sierra A, Lluch MM, Coca A, Aguilera MT, Giner V, Bragulat E, Urbano-Marquez A. Fluid, ionic and hormonal changes induced by high salt intake in salt-sensitive and salt-resistant hypertensive patients. Clin Sci (Lond) 91: 155–161, 1996 [DOI] [PubMed] [Google Scholar]
- 7.Gorin Y, Ricono JM, Wagner B, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochem J 381: 231–239, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gruden G, Thomas S, Burt D, Zhou W, Chusney G, Gnudi L, Viberti G. Interaction of angiotensin II and mechanical stretch on vascular endothelial growth factor production by human mesangial cells. J Am Soc Nephrol 10: 730–737, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Hori Y, Katoh T, Hirakata M, Joki N, Kaname S, Fukagawa M, Okuda T, Ohashi H, Fujita T, Miyazono K, Kurokawa K. Anti-latent TGF-beta binding protein-1 antibody or synthetic oligopeptides inhibit extracellular matrix expression induced by stretch in cultured rat mesangial cells. Kidney Int 53: 1616–1625, 1998 [DOI] [PubMed] [Google Scholar]
- 10.Iqbal J, Zaidi M. Molecular regulation of mechanotransduction. Biochem Biophys Res Commun 328: 751–755, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Izuhara Y, Nangaku M, Inagi R, Tominaga N, Aizawa T, Kurokawa K, van Ypersele de Strihou C, Miyata T. Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J Am Soc Nephrol 16: 3631–3641, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol 14: 2775–2782, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Lee MA, Bohm M, Paul M, Ganten D. Tissue renin-angiotensin systems. Their role in cardiovascular disease. Circulation 87: IV7–IV13, 1993 [PubMed] [Google Scholar]
- 14.Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J Biol Chem 278: 12094–12100, 2003 [DOI] [PubMed] [Google Scholar]
- 15.MacKenna DA, Dolfi F, Vuori K, Ruoslahti E. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J Clin Invest 101: 301–310, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKnight JA, Roberts G, Sheridan B, Atkinson AB. The effect of low and high sodium diets on plasma atrial natriuretic factor, the renin-aldosterone system and blood pressure in subjects with essential hypertension. Clin Endocrinol (Oxf) 40: 73–77, 1994 [DOI] [PubMed] [Google Scholar]
- 17.Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol 64: 1271–1276, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Miura S, Kiya Y, Kanazawa T, Imaizumi S, Fujino M, Matsuo Y, Karnik SS, Saku K. Differential bonding interactions of inverse agonists of angiotensin II type 1 receptor in stabilizing the inactive state. Mol Endocrinol 22: 139–146, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miura S, Fujino M, Hanzawa H, Kiya Y, Imaizumi S, Matsuo Y, Tomita S, Uehara Y, Karnik SS, Yanagisawa H, Koike H, Komuro I, Saku K. Molecular mechanism underlying inverse agonist of angiotensin II type 1 receptor. J Biol Chem 281: 19288–19295, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Navar LG, Harrison-Bernard LM, Imig JD, Wang CT, Cervenka L, Mitchell KD. Intrarenal angiotensin II generation and renal effects of AT1 receptor blockade. J Am Soc Nephrol 10, Suppl 12: S266–S272, 1999 [PubMed] [Google Scholar]
- 21.Navar LG, Inscho EW, Majid SA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev 76: 425–536, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Onozaki A, Midorikawa S, Sanada H, Hayashi Y, Baba T, Katoh T, Watanabe T. Rapid change of glucose concentration promotes mesangial cell proliferation via VEGF: inhibitory effects of thiazolidinedione. Biochem Biophys Res Commun 317: 24–29, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Oparil S, Williams D, Chrysant SG, Marbury TC, Neutel J. Comparative efficacy of olmesartan, losartan, valsartan, and irbesartan in the control of essential hypertension. J Clin Hypertens (Greenwich) 3: 283–291; 318, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanada S, Kitakaze M, Node K, Takashima S, Ogai A, Asanuma H, Sakata Y, Asakura M, Ogita H, Liao Y, Fukushima T, Yamada J, Minamino T, Kuzuya T, Hori M. Differential subcellular actions of ACE inhibitors and AT(1) receptor antagonists on cardiac remodeling induced by chronic inhibition of NO synthesis in rats. Hypertension 38: 404–411, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 23: 981–987, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens 22: 1141–1149, 2004 [DOI] [PubMed] [Google Scholar]
- 27.van Biesen T, Luttrell LM, Hawes BE, Lefkowitz RJ. Mitogenic signaling via G protein-coupled receptors. Endocr Rev 17: 698–714, 1996 [DOI] [PubMed] [Google Scholar]
- 28.Yoneda M, Sanada H, Yatabe J, Midorikawa S, Hashimoto S, Sasaki M, Katoh T, Watanabe T, Andrews PM, Jose PA, Felder RA. Differential effects of angiotensin II type-1 receptor antisense oligonucleotides on renal function in spontaneously hypertensive rats. Hypertension 46: 58–65, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6: 499–506, 2004 [DOI] [PubMed] [Google Scholar]