

Abstract
Purpose of review
Recent evidence has linked n-3 polyunsaturated fatty acid (PUFA) supplementation with dramatic alterations of mitochondrial phospholipid membranes and favorable changes in mitochondrial function. In the present review, we examine the novel effects of n-3 PUFA on mitochondria, with an emphasis on cardiac mitochondrial phospholipids.
Recent findings
There is growing evidence that dietary n-3 PUFA, particularly docosahexaenoic acid (DHA), has profound effects on mitochondrial membrane phospholipid composition and mitochondrial function. Supplementation with n-3 PUFA increases membrane phospholipid DHA and depletes arachidonic acid, and can increase cardiolipin, a tetra-acyl phospholipid that is unique to mitochondrial and essential for optimal mitochondrial function. Recent studies show that supplementation with DHA decreases propensity for cardiac mitochondria to undergo permeability transition, a catastrophic event often leading to cell death. This finding provides a potential mechanism for the cardioprotective effect of DHA. Interestingly, other n-3 PUFAs that modify membrane composition to a lesser extent have substantially less of an effect on mitochondria and do not appear to directly protect the heart.
Summary
Current data support a role for n-3 PUFA supplementation, particularly DHA, on mitochondria that are strongly associated with changes in mitochondrial phospholipid composition.
INTRODUCTION
Fatty acids are important regulators of mitochondrial structure and function through their role as oxidative substrates and inhibitors of carbohydrate oxidation, ligands for nuclear receptors that regulate the expression of mitochondrial proteins, and structural components in mitochondria membrane phospholipids. The role of fatty acids as a mitochondrial substrate for ATP production and as an inhibitor of pyruvate dehydrogenase has been investigated since the 1960s, and it is now well described [1]. Fatty acid regulation of gene expression via activation of various nuclear receptors was extensively studied over the past 30 years, and it is now well established that stimulation of peroxisome proliferator-activated receptors up-regulates the expression of genes involved in mitochondrial fatty acid metabolism [2,3]. It has become clear that dietary fatty acids affect the composition of mitochondrial phospholipids, which in turn impacts mitochondrial function. Supplementation with n-3 polyunsaturated fatty acid (PUFA) can increase cardiolipin, a tetra-acyl phospholipid that is unique to mitochondrial and essential for optimal mitochondrial function. Mitochondrial dysfunction plays a causal role in many debilitating medical conditions, such as heart failure, neurodegenerative disorders, and diabetes. Thus there is currently great interest in understanding how dietary long chain fatty acids can be used to prevent or reverse mitochondrial dysfunction in human disease. In this brief review we will provide an update on recent work investigating the impact of dietary n-3 PUFAs on mitochondrial phospholipids and function.
RECENT ADVANCES IN THE UNIQUE ASPECTS OF MITOCHONDRIAL PHOSPHOLIPIDS
Similar to other cell membranes, the primary phospholipids in mitochondrial membranes are phosphatidylethanolamine and phosphatidylcholine. However, unlike other membranes in mammalian cells, mitochondrial membranes contain high levels of cardiolipin, a tetra-acyl phospholipid. Cardiolipin comprises 10–20% of the mass of total mitochondrial phospholipid. Depletion of cardiolipin results in severe mitochondrial dysfunction, as evidenced in Barth syndrome patients, a rare X-linked mutation resulting in the absence of tafazzin, an enzyme that is essential for formation of functional cardiolipin. These patients present with skeletal muscle weakness and cardiomyopathy, consistent with defective mitochondrial ATP formation [4]. Linoleic acid is the main fatty acyl moiety in cardiolipin, with 60–80% of cardiolipin being tetralinoleoyl cardiolipin (L4CL) in cardiac mitochondria in humans, dogs and rats [5,6,7]. A major new tool in the study of the pathophysiology of Barth syndrome recently became available with the creation of a tafazzin knockdown mouse using RNA interference [8]. These mice recapitulated key aspects of human Barth syndrome in terms of depletion of L4CL and long chain tetraacyl cardiolipin from skeletal and cardiac muscle mitochondria, accumulation of immature monolysocardiolipin, mitochondrial proliferation and myofibrillar disarray, and functional myopathy [8]. Future studies utilizing this mouse model will further our understanding of the mechanisms underlying Barth syndrome and cardiolipin remodeling.
It has been proposed that high levels of L4CL are essential for optimal mitochondrial function in the heart [6], though recent evidence runs counter to this concept. Minkler and Hoppel [5] showed that cardiolipin acyl chains vary greatly by species, showing that there are very different fatty acyl compositions between rat liver, mouse heart and dog heart mitochondria. They used a novel high-performance liquid chromatography-mass spectrometry method to separate and characterize cardiolipin molecular species, and showed that cardiolipin in cardiac mitochondria from dogs was 77% tetralinoleoyl cardiolipin (L4CL), which is similar to previously published data from humans and rats [7,9]. The other molecular species in the dog heart comprised 18 : 1, 18 : 0 and 16 : 0. Surprisingly, they found that in the mouse heart L4CL comprises only 22% of the total cardiolipin [5], and that there was high abundance of various species containing docosahexaenoic acid (DHA; 22 : 6n6) (53% of total cardiolipin) [5,7] (Fig. 1).
FIGURE 1.
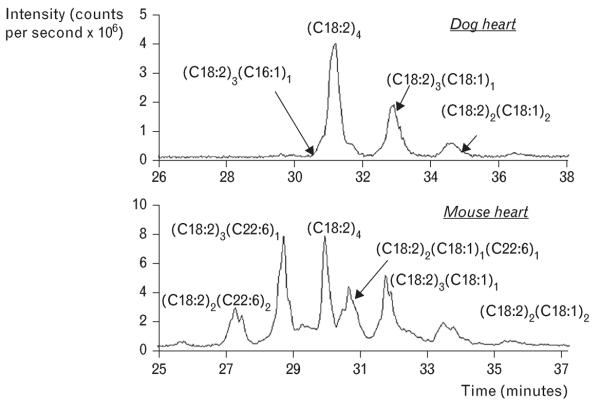
Data from [5] showing profound species differences in the relative amount of the cardiolipin containing four linoleic acid side chains [(C18 : 2)4] in cardiac mitochondria. Dogs had high levels of (C18:2)4 (upper panel) compared to mice (lower panel). Mouse heart mitochondrial had a high abundance of various cardiolipin species containing docosahexaenoic acid (C22 : 6), which were not detected in mitochondrial from dog heart. Reproduced with permission from [5].
These findings are significant because changes in cardiolipin acyl composition may influence the activity of essential inner membrane proteins, as high levels of DHA will increase the fluidity of membrane and alter protein–lipid interaction [6]. Cardiolipin is necessary for formation of contact sites between inner and outer mitochondrial membranes, stabilization of essential inner membrane proteins and respiratory complexes, and in mitochondrial apoptotic signaling pathways [6]. A decrease in the total cardiolipin in cardiac mitochondria and less L4CL has been observed in acquired cardiac pathologies such as hypertension-induced hypertrophy and heart failure in rodents [6]. On the contrary, a decrease in whole tissue content of L4CL was not observed in cardiac biopsies from heart failure patients [7], though it was observed in explanted hearts from heart failure patients compared to nonfailing organ donors [10]. In recent studies it was shown that dogs with established heart failure secondary to irreversible myocardial ischemic damage had severe mitochondrial respiratory dysfunction but normal mitochondrial total cardiolipin content and L4CL levels [11,12]. Taken together, it now appears that the well documented impairment in mitochondrial respiratory function in heart failure is not due to depletion of total cardiolipin or L4CL in humans or dogs.
IMPACT OF DIETARY POLYUNSATURATED FATTY ACIDS ON MITOCHONDRIAL PHOSPHOLIPIDS AND FUNCTION
There is growing evidence that dietary n-3 PUFA, particularly DHA, have profound effects on mitochondrial membrane phospholipids and mitochondrial function. Initial studies showed that high intake of fish oil rich in DHA and eicosapentaenoic acid (EPA; 20:5n-3) prevented the age-related decrease in the sum of n-3 PUFA and increase in n-6 PUFA in cardiac mitochondrial membranes, restored the cardiolipin content, and prevented age-associated increases in phosphatidylcholine [13]. We recently observed that supplementation of rats with DHA + EPA increased L4CL and cardiolipin containing one DHA side chain and three linoleic acid (18:2) side chains in isolated cardiac mitochondria [14]. Thus dietary long-chain n-3 PUFA impacts mitochondrial phospholipid composition.
The cardioprotective effects of n-3 PUFAs have long been recognized [15]. Both epidemiological and clinical evidence indicate that high consumption of fish or fish oil supplementation lowers the incidence of cardiac death and possibly other cardiovascular events [15]. The precise mechanisms underlying the cardioprotective effect of n-3 PUFAs are not well established, but recent evidence suggests that it may be partially due to changes in membrane phospholipids and resultant improvement in the function of cardiac mitochondria.
We previously found that supplementation with DHA + EPA from fish oil attenuated the development of heart failure in rats subjected to chronic aortic hypertension [16–18]. Rats were subjected to abdominal aortic banding and assigned to 12 weeks of treatment with either a standard chow or a chow supplemented with a mixture of DHA + EPA (70:30 ratio) from fish oil at either 0.7, 2.3 or 7% of the total energy intake [17]. We found that supplementation with fish oil resulted in a dose-dependent decrease in cardiomyocyte apoptosis, measured by TdT-mediated dUTP Nick-End Labeling (TUNNEL) staining. Rats fed the highest dose of fish oil had levels of apoptosis below control sham levels. These data were the first to directly demonstrate that supplementation with DHA + EPA prevented apoptosis and to hint at mitochondrial involvement as a mechanism for the beneficial effects of n-3 PUFA.
Dietary supplementation with fish oil improves cardiac contractile recovery with experimental myocardial ischemia followed by reperfusion [19]. This beneficial effect was associated with a reduction in the rate of oxygen consumption by the myocardium at a given work output, which reflects an enhancement in cardiac mechanical efficiency [19]. A similar general phenomenon was observed in exercising health humans, in whom fish oil supplementation reduced whole body oxygen consumption during cycling exercise at a fixed workload [20]. Lower oxygen consumption by mitochondria isolated from n-3 PUFA-supplemented hearts, most notably during uncoupled respiration, suggests that alterations in membrane phospholipid n-3 PUFA content mitochondrial membranes may increase thermodynamic efficiency [13]. Furthermore, phosphorylating respiration, or state 3 respiration, may also be influenced by increased n-3 PUFAs [21], although others could not replicate this finding [14,22,23]. In contrast, cardiac mitochondria from rats fed sardine oil, rich in n-3 PUFA but low in linoleic acid, had decreased mitochondrial respiration; however, this was accompanied by a decrease in linoleic acid in total phospholipids and specifically in cardiolipin [24]. This suggests that the decrease in respiration was due to deficient electron transport chain activity and decreased L4CL. Our group, as well as others, have consistently shown that diets with similar linoleic acid contents, but supplemented in DHA, EPA, or DHA + EPA do not deplete L4CL [14,22], and supplementation with DHA alone can increase it [22].
We have recently investigated the independent effects of DHA and EPA on cardiac mitochondria. Clinical studies have established that the triglyceride-lowering effects of DHA and EPA are equivalent [25]; however, studies show that DHA is readily shortened to form EPA, but EPA is not readily elongated to DHA [26], particularly in the heart [27]. We compared supplementation with DHA to EPA and found that DHA supplementation caused a greater increase in total n-3 PUFA (DHA + EPA) in cardiac mitochondrial phospholipids than did EPA [22,23].
Mitochondria determine cell survival through the opening of the MPTP, which occurs under conditions of cell stress, causing mitochondrial depolarization and triggering of cell death. The MPTP is a large diameter (3 nm), high conductance, voltage-dependent channel that allows passage of water, ions, and molecules up to 1500 Da [28,29]. Ca2+, oxidative stress and numerous reactive chemicals induce MPTP opening. The precise structure and regulation of the MPTP is not well understood [28,29]. As a consequence of MPTP opening, the mitochondria depolarize, uncouple, and swell. The loss of ATP production and release of cytochrome C can result in cell death via either apoptosis or necrosis, thus MPTP opening is generally considered to be a catastrophic event. We recently found that dietary supplementation with a mixture of DHA + EPA (70 : 30 ratio) increased DHA and EPA in cardiac mitochondrial phospholipids and the tolerance of isolated mitochondrial to Ca2+-induced MPTP opening [14]. A recent study confirmed this observation in mice, and showed a decrease in hydrogen peroxide production in cardiac mitochondria with n-3 PUFA supplementation [30].
We subsequently showed that supplementation with only DHA delayed Ca2+-induced MPTP opening (Fig. 2) [22,23]. This effect was associated with an increase in DHA and total n-3 PUFA in cardiac mitochondrial phospholipids, a reduction in the amount of arachidonic acid (ARA) and an increase in L4CL. EPA supplementation, on the contrary, had no effect of MPTP opening in response to Ca2+, despite increasing membrane EPA and moderately reducing ARA. The shorter n-3 PUFA, alpha-linoleic acid (ALA), also had no effect on MPTP opening (Fig. 2). At physiological doses, ALA had little effect on membrane composition, further supporting the hypothesis that the beneficial effects are linked to modulation of mitochondrial phospholipids.
FIGURE 2.
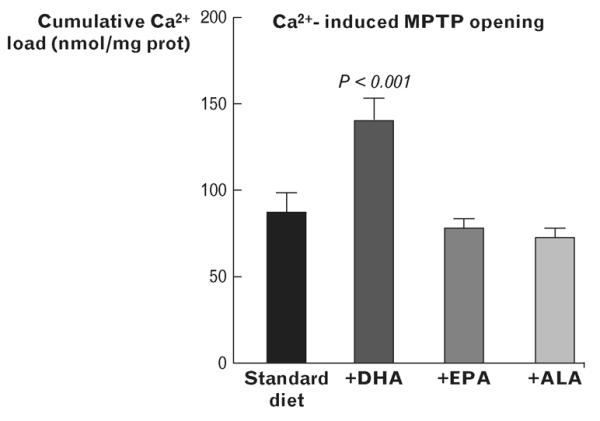
Ca2+-induced opening of the mitochondrial permeability transition pore (MPTP) in cardiac mitochondria from rats fed either a standard low n-3 PUFA diet or supplemented with DHA, EPA or ALA at 2.3% of energy intake. Redrawn from data taken from [22] for the standard diet, DHA and EPA groups, and from unpublished data for the ALA group. ALA, alpha-linoleic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; MPTP, mitochondrial permeability transition pore; PUFA, polyunsaturated fatty acid.
CONCLUSION
In conclusion, it is evident that dietary n-3 PUFAs have profound effects on mitochondrial phospholipid composition and mitochondrial resistance to stress. In particular, cardioprotection observed in pathological states such as heart failure may be due in part to mitochondrial phospholipid remodeling and improved mitochondrial function. In addition, supplementation with a specific n-3 PUFA, DHA, induced greater effects on mitochondrial membrane phospholipid composition and function as compared to other n-3 PUFAs such as EPA. These studies emphasize the importance of delineating the specific roles of dietary fatty acids on mitochondrial membranes to prevent or reverse mitochondrial dysfunction with various human pathologies.
Table 1.
| KEY POINTS |
|---|
|
Acknowledgements
None.
Conflicts of interest
The work was supported by the National Institutes of Health, Grant numbers HL074237, HL101434 and HL072751.
REFERENCES
- 1.Lopaschuk GD, Ussher JR, Folmes CD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 2.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008;44:968–975. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Noy N. Ligand specificity of nuclear hormone receptors: sifting through promiscuity. Biochemistry. 2007;46:13461–13467. doi: 10.1021/bi7018699. [DOI] [PubMed] [Google Scholar]
- 4.Schlame M, Towbin JA, Heerdt PM, et al. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51:634–637. doi: 10.1002/ana.10176. [Context Link] [DOI] [PubMed] [Google Scholar]
- 5.Minkler PE, Hoppel CL. Separation and characterization of cardiolipin molecular species by reverse-phase ion pair high-performance liquid chromatography-mass spectrometry. J Lipid Res. 2010;51:856–865. doi: 10.1194/jlr.D002857. Presents a novel method for measuring cardiolipin, and provide the potentially important observation that mice have a profoundly different cardiolipid fatty acyl side chain composition than seen in humans, dogs and mice.
- 6.Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol. 2009;53:290–301. doi: 10.1097/FJC.0b013e31819b5461. [DOI] [PubMed] [Google Scholar]
- 7.Schlame M, Kelley RI, Feigenbaum A, et al. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Acehan D, Vaz F, Houtkooper RH, et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286:899–908. doi: 10.1074/jbc.M110.171439. Presents a novel mouse model of tafazzin deficiency which should prove very useful for studying cardiolipidin physiology and the pathology of Barth syndrome.
- 9.Sparagna GC, Johnson CA, McCune SA, et al. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J Lipid Res. 2005;46:1196–1204. doi: 10.1194/jlr.M500031-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Sparagna GC, Chicco AJ, Murphy RC, et al. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res. 2007;48:1559–1570. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Rosca M, Minkler P, Hoppel CL. Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim Biophys Acta. 2011;1807:1373–1382. doi: 10.1016/j.bbabio.2011.02.003. Analysis of cardiolipin composition and content of cardiac mitochondria from dogs with heart failure, showing normal cardiolipin content and compositon despite severe mitochondrial respiratory dysfunction.
- 12.Rosca MG, Vazquez EC, Kerner J, et al. Cardiac mitochondria in coronary microembolization-induced heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pepe S, Tsuchiya N, Lakatta EG, Hansford RG. PUFA and aging modulate cardiac mitochondrial membrane lipid composition and Ca2+ activation of PDH. Am J Physiol. 1999;276:H149–H158. doi: 10.1152/ajpheart.1999.276.1.H149. [DOI] [PubMed] [Google Scholar]
- 14.O’Shea KM, Khairallah RJ, Sparagna GC, et al. Dietary omega-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J Mol Cell Cardiol. 2009;47:819–827. doi: 10.1016/j.yjmcc.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duda MK, O’Shea KM, Stanley WC. omega-3 polyunsaturated fatty acid supplementation for the treatment of heart failure: mechanisms and clinical potential. Cardiovasc Res. 2009;84:33–41. doi: 10.1093/cvr/cvp169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duda MK, O’Shea KM, Lei B, et al. Dietary supplementation with omega-3 PUFA increases adiponectin and attenuates ventricular remodeling and dysfunction with pressure overload. Cardiovasc Res. 2007;76:303–310. doi: 10.1016/j.cardiores.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duda MK, O’Shea KM, Tintinu A, et al. Fish oil, but not flaxseed oil, decreases inflammation and prevents pressure overload-induced cardiac dysfunction. Cardiovasc Res. 2009;81:319–327. doi: 10.1093/cvr/cvn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah KB, Duda MK, O’Shea KM, et al. The cardioprotective effects of fish oil during pressure overload are blocked by high fat intake: role of cardiac phospholipid remodeling. Hypertension. 2009;54:605–611. doi: 10.1161/HYPERTENSIONAHA.109.135806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepe S, McLennan PL. Cardiac membrane fatty acid composition modulates myocardial oxygen consumption and postischemic recovery of contractile function. Circulation. 2002;105:2303–2308. doi: 10.1161/01.cir.0000015604.88808.74. [DOI] [PubMed] [Google Scholar]
- 20.Peoples GE, McLennan PL, Howe PR, Groeller H. Fish oil reduces heart rate and oxygen consumption during exercise. J Cardiovasc Pharmacol. 2008;52:540–547. doi: 10.1097/FJC.0b013e3181911913. [DOI] [PubMed] [Google Scholar]
- 21.Demaison L, Sergiel JP, Moreau D, Grynberg A. Influence of the phospholipid n-6/n-3 polyunsaturated fatty acid ratio on the mitochondrial oxidative metabolism before and after myocardial ischemia. Biochim Biophys Acta. 1994;1227:53–59. doi: 10.1016/0925-4439(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 22.Khairallah RJ, Sparagna GC, Khanna N, et al. Dietary supplementation with docosahexaenoic acid, but not eicosapentaenoic acid, dramatically alters cardiac mitochondrial phospholipid fatty acid composition and prevents permeability transition. Biochim Biophys Acta. 2010;1797:1555–1562. doi: 10.1016/j.bbabio.2010.05.007. Demonstrates that dietary supplementation with DHA, but not EPA, profoundly remodels mitochondrial phospholipids and delays Ca2+-induced permeability transition.
- 23.Khairallah RJ, O’Shea KM, Brown BH, et al. Treatment with docosahexaenoic acid, but not eicosapentaenoic acid, delays Ca2+-induced mitochondria permeability transition in normal and hypertrophied myocardium. J Pharmacol Exp Ther. 2010;335:155–162. doi: 10.1124/jpet.110.170605. Extends the previous observation that dietary supplementation with DHA delays Ca2+-induced mitochondrial permeability transition using a model of cardiac hypertrophy caused by pressure overload.
- 24.Yamaoka S, Urade R, Kito M. Mitochondrial function in rats is affected by modification of membrane phospholipids with dietary sardine oil. J Nutr. 1988;118:290–296. doi: 10.1093/jn/118.3.290. [DOI] [PubMed] [Google Scholar]
- 25.Grimsgaard S, Bonaa KH, Hansen JB, Nordoy A. Highly purified eicosapentaenoic acid and docosahexaenoic acid in humans have similar triacylglycerol-lowering effects but divergent effects on serum fatty acids. Am J Clin Nutr. 1997;66:649–659. doi: 10.1093/ajcn/66.3.649. [DOI] [PubMed] [Google Scholar]
- 26.Mori TA, Woodman RJ. The independent effects of eicosapentaenoic acid and docosahexaenoic acid on cardiovascular risk factors in humans. Curr Opin Clin Nutr Metab Care. 2006;9:95–104. doi: 10.1097/01.mco.0000214566.67439.58. [DOI] [PubMed] [Google Scholar]
- 27.Igarashi M, Ma K, Chang L, et al. Rat heart cannot synthesize docosahexaenoic acid from circulating alpha-linolenic acid because it lacks elongase-2. J Lipid Res. 2008;49:1735–1745. doi: 10.1194/jlr.M800093-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricchelli F, Sileikyte J, Bernardi P. Shedding light on the mitochondrial permeability transition. Biochim Biophys Acta. 2011;1807:482–490. doi: 10.1016/j.bbabio.2011.02.012. Timely review on the biochemistry of mitochondrial permeability transition.
- 29.Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38:841–860. doi: 10.1042/BST0380841. Overview of the role of mitochondrial dysfunction in cardiac injury, with an emphasis on mitochondrial permeability transition pore opening.
- 30.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]