

Abstract
Proteomics techniques have revealed that lysine acetylation is abundant in mitochondrial proteins. This study was undertaken (1) to determine the relationship between mitochondrial protein acetylation and insulin sensitivity in human skeletal muscle, identifying key acetylated proteins, and (2) to use molecular modeling techniques to understand the functional consequences of acetylation of adenine nucleotide translocase 1 (ANT1), which we found to be abundantly acetylated. Eight lean and eight obese nondiabetic subjects had euglycemic clamps and muscle biopsies for isolation of mitochondrial proteins and proteomics analysis. A number of acetylated mitochondrial proteins were identified in muscle biopsies. Overall, acetylation of mitochondrial proteins was correlated with insulin action (r = 0.60; P < 0.05). Of the acetylated proteins, ANT1, which catalyzes ADP–ATP exchange across the inner mitochondrial membrane, was acetylated at lysines 10, 23, and 92. The extent of acetylation of lysine 23 decreased following exercise, depending on insulin sensitivity. Molecular dynamics modeling and ensemble docking simulations predicted the ADP binding site of ANT1 to be a pocket of positively charged residues, including lysine 23. Calculated ADP–ANT1 binding affinities were physiologically relevant and predicted substantial reductions in affinity upon acetylation of lysine 23. Insertion of these derived binding affinities as parameters into a complete mathematical description of ANT1 kinetics predicted marked reductions in adenine nucleotide flux resulting from acetylation of lysine 23. Therefore, acetylation of ANT1 could have dramatic physiological effects on ADP–ATP exchange. Dysregulation of acetylation of mitochondrial proteins such as ANT1 therefore could be related to changes in mitochondrial function that are associated with insulin resistance.
Post-translational modifications provide a mechanism for regulating protein structure and function. Among these modifications, acetylation is widespread. Recent evidence shows lysine acetylation regulates the function of many proteins and is conserved evolutionarily.1,2 In prokaryotes, acetylation coordinates metabolic flux,2,3 so it is no surprise that metabolic and mitochondrial proteins are over-represented in the human acetylome.1,4 Targeted studies show that acetylation regulates mitochondrial function in mammals, where the activities of enoyl-CoA hydratase, malate dehydrogenase, and long chain acyl-CoA dehydrogenase all are regulated by acetylation.5
However, less is known regarding the regulation of the human mitochondrial acetylome or the effects of acetylation on the function of mitochondrial proteins. A variety of changes in functional and proteomic aspects of mitochondria are associated with insulin resistance,6−8 so a more detailed analysis of acetylation of mitochondrial proteins would be useful in this context. Therefore, one reason for undertaking this study was to identify acetylation sites in proteins from mitochondria isolated from skeletal muscle biopsies taken from healthy, nondiabetic volunteers with a range of insulin sensitivity, to test the hypothesis that acetylation of mitochondrial proteins is associated with insulin sensitivity. To accomplish this, we combined euglycemic, hyperinsulinemic clamps and muscle biopsies together with mass spectrometry approaches to show that the mitochondrial acetylome is positively correlated with insulin sensitivity in human muscle. Among the proteins found to be acetylated extensively was adenine nucleotide translocase 1 (ANT1), which was consistently acetylated at lysines 10, 23, and 92. We used molecular modeling simulations to show that acetylation of lysine 23 (Lys23) was sufficient to lower the affinity of ANT1 for ADP; acetylation at other sites had no effect. In turn, these ADP binding affinities were used to adjust the parameter values of a computational model of ANT1 kinetics9 to explore the potential functional consequences of the acetylation of Lys23 on adenine nucleotide flux through ANT1.
Experimental Procedures
Study A
A total of 16 (eight lean and eight obese) normoglycemic volunteers took part in study A (relationship between acetylation of mitochondrial proteins and insulin action), which was approved by the Institutional Review Board of Arizona State University (ASU). All 16 of the subjects had euglycemic clamp studies with basal muscle biopsies (characteristics of the subjects in study A are listed in Table 1). Studies were conducted at the Clinical Research Unit at ASU. Informed consent was obtained from all subjects. The subjects were sedentary, did not engage in regular exercise, and reported no change in body weight for at least 6 months. Subjects were instructed not to exercise for 48 h before studies and to maintain their usual diet. A medical history, physical examination, 12-lead electrocardiogram, and complete chemistry panel were obtained. A 75 g oral glucose tolerance test was performed to ensure normoglycemia (American Diabetes Association criteria). No one was taking any medication affecting glucose metabolism. Some in vivo data for the subjects have been reported.10
Table 1. Characteristics of Subjects in Study Aa.
| lean | obese | |
|---|---|---|
| gender (female/male) | 4/4 | 4/4 |
| age (years) | 39.8 ± 4.8 | 41.0 ± 4.1 |
| BMI (kg/m2) | 23.5 ± 1.4 | 36.6 ± 1.5b |
| insulin-stimulated glucose disposal [mg (kg of FFM)−1 min–1] | 13.4 ± 1.1 | 6.15 ± 0.58 |
| Hba1c (%) | 5.26 ± 0.10 | 5.74 ± 0.10b |
| total cholesterol (mg/dL) | 160 ± 13 | 187 ± 15 |
| plasma triglycerides (mg/dL) | 63 ± 5 | 120 ± 29 |
| fasting plasma glucose (mg/dL) | 82 ± 2 | 94 ± 3b |
| fasting plasma insulin (microunits/mL) | 3.3 ± 0.2 | 13.7 ± 3.0b |
Data are given as means ± SEM.
P < 0.01.
Study B
A total of three lean and two overweight/obese normoglycemic subjects took part in study B (changes in protein acetylation after exercise), which was approved by the Institutional Review Board of ASU. As in study A, all of these subjects had euglycemic clamp studies with basal muscle biopsies (characteristics of the subjects in study B are listed in Table 2). Studies were conducted at the Clinical Research Unit at ASU. Subjects were prepared for study as described for study A. All subjects received a hyperglycemic, euglycemic clamp to assess insulin sensitivity, as described below. The exercise bout was conducted on a separate day after determination of VO2peak and at least 1 week before or after the euglycemic clamp. The design of the exercise bout was previously described.6 Subjects reported to the Clinical Research Unit at 7 a.m. after fasting overnight and exercised on a stationary bicycle, alternating between 70% (8 min) and 90% (2 min) of heart rate corresponding to that at VO2peak, for a total of four sets of exercise, with a 2 min rest between sets.6 After completing exercise, the subject was discharged and returned the following morning after an overnight fast for a biopsy of the vastus lateralis muscle, 24 h after the end of exercise. Peak aerobic capacity (VO2peak) was determined on a separate day in these six subjects as described previously.11
Table 2. Characteristics of Subjects in Study B.
| gender (female/male) | 2/3 |
| age (years) | 33 ± 5 |
| BMI (kg/m2) | 26.2 ± 1.3 |
| insulin-stimulated glucose disposal [mg (kg of FFM)−1 min–1] | 10.1 ± 1.3 |
| total cholesterol (mg/dL) | 177 ± 12 |
| plasma triglycerides (mg/dL) | 118 ± 35 |
| fasting plasma glucose (mg/dL) | 89 ± 2 |
| fasting plasma insulin (microunits/mL) | 6.5 ± 1.3 |
| VO2peak [mL (kg of FFM)−1 min–1] | 44.5 ± 4.8 |
| heart rate max exercise (BPM, predicted, actual) | 187 ± 5, 187 ± 4 |
| heart rate 70% max exercise (BPM, predicted, actual) | 131 ± 3, 139 ± 6 |
| heart rate 90% max exercise (BPM, predicted, actual) | 168 ± 3, 166 ± 5 |
Muscle Biopsy and Hyperinsulinemic, Euglycemic Clamps
Euglycemic clamps and muscle biopsies were performed in all subjects as described previously.12 After an overnight fast, at 7–8 a.m., a percutaneous biopsy of the vastus lateralis muscle was obtained with a Bergstrom cannula under local anesthesia 1 h before starting the insulin infusion. Infusion of [6,6-2H]glucose was used to trace glucose metabolism, and the insulin infusion rate was 80 milliunits m–2 min–1. Enrichment of deuterated glucose was determined using gas chromatography and mass spectrometry as described previously,8 and steady state equations were used to calculate rates of glucose disposal.
Mass Spectrometry
Whole muscle lysates were processed as described previously.7 Mitochondria were isolated from freshly obtained muscle biopsies as described previously.8 Muscle lysate or mitochondrial proteins were resolved by one-dimensional SDS–PAGE. No deacetylase inhibitors were added, so the reported acetylation sites can be taken as a minimum estimate. Because the deacetylases for these sites have not been identified, use of specific inhibitors could introduce biases. For analysis of ANT1, the band corresponding to the molecular weight of ANT1 was excised, prepared, and analyzed using a Thermo HPLC-nanospray LTQ-FTICR hybrid mass spectrometer, as described previously.7
Data Analysis and Bioinformatics
Tandem mass spectra were assigned charge states and searched against the IPI_HUMAN_v3.59 database (http://www.ebi.ac.uk/IPI/).7 Search parameters were as described previously7 with the addition of acetylation as a modification and allowing for three miscleavages. The false discovery rate was determined using a decoy randomized database. The following search parameters were used: mass tolerance of 10 ppm for precursor ion mass and 0.5 Da for product ion mass, digestion with trypsin, maximum of three missed tryptic cleavages, fixed modification of carboamidomethylation, and variable modifications of oxidation of methionine, acetylation of lysine, and phosphorylation of serine, threonine, and tyrosine. Probability assessment of peptide assignments and protein identifications were done using Scaffold (version Scaffold_2_00_06, Proteome Software Inc., Portland, OR). Only peptides with ≥95% probability were considered. All acetylation sites were identified in a data-dependent manner and verified visually using tandem mass spectrometry spectra.7,8 Site-specific acetylation of lysines 10, 23, and 92 was quantified using normalized spectral abundance factors,13−15 and ANT1 abundance was used for normalization. Spectral abundance factor quantification is based on the notion that the abundance of a protein is proportional to the number of spectra observed for peptides derived from that protein, normalized to the length of the protein and the total number of spectra in the complex digest (like a loading control). These methods have previously been described more completely.7
Molecular Modeling
Because the crystal structure of human ANT1 is not known, a human ANT1 homology model was created on the basis of the crystal structure of bovine ANT1 [Protein Data Bank (PDB) entry 1OKC],16 using Modeler17 and VMD18 with the multiseq plugin19 of VMD and the STAMP algorithm.20 The sequence of bovine ANT1 is highly similar with that of human ANT1.
Molecular Dynamics Simulations
All-atom simulations were performed with the NAMD21simulation package (version 2.8). The VMD psfgen plugin was used to create an Xplor protein structure file (psf) of the human ANT1 structure using the CHARMM2222 topology for proteins and lipids with CMAP correction. Bulk water molecules were then added with the Solvate program using a minimal water shell thickness of 6 Å and eight gaussians to define the solvent boundary.
To insert the protein into a lipid bilayer, water molecules that were within a 30 Å slice centered on the transmembrane axis of the protein were trimmed away. A 90 Å × 90 Å palmitoyloleoylphosphatidylcholine (POPC) bilayer was constructed with the VMD membrane builder plugin. The protein was then centered in the membrane by translating its center of mass, and lipid molecules within 3 Å of the protein were removed. Finally, the prepared membrane/protein system was fully solvated in a 90 Å × 90 Å × 90 Å water box with 18 chloride counterions to neutralize the net protein charge.
The solvated ANT1/membrane system was equilibrated via a four-stage process. (1) The membrane was “melted” by performing a 0.5 ns simulation in which only the lipid tails were permitted to move. (2) With the protein constrained with a harmonic energy function, the entire system was subjected to 1000 steps of minimization followed by equilibration for 0.5 ns at 310 K. (3) Equilibration for an additional 0.5 ns was performed without protein constraints to further equilibrate the entire system. (4) Finally, equilibration for an additional 25 ns was performed while holding the area of the lipid patch constant. Equilibration of the system over this lengthy trajectory was validated by plotting the root-mean-square deviation of the system and observing a convergence.
We refer to the end point of this wild-type equilibration as the “equilibrated reference system”, as it served as a common input structure to all of our computational experiments. Using the acetyllysine patch on this system (see below), we prepared a series of three acetylated systems at lysines 10, 23, and 92. In each of these acetylated systems, the furthest chloride ion from the channel was deleted to reneutralize the net charge. The wild type (WT) and three modified systems (Lys10, Lys23, and Lys92) were then subjected to reminimization, independent equilibration for 10 ns, and finally full MD simulation for 30 ns in the isothermal–isobaric (NPT) ensemble. These four systems lacked the presence of ADP and are thus hereafter termed “apo-ANT1” simulations.
Electrostatics Analysis
The VMD PMEPOT23 plugin was used to compute the electrostatic potentials of the assembled ANT1 systems and simulation results. To compare the functional effects of lysine acetylation, potentials were computed on the static protein after the application of an acetyllysine patch to the Lys10, Lys23, and Lys92 residues. Isosurfaces of the potential were visualized in VMD. To produce two-dimensional (2D) slices and line-profile plots of the potential, we used the Python GridDataFormats library to import the OpenDX files exported from PMEPOT. Profiles were calculated by interpolating along a line drawn through the lumen of the channel. The origin of the system was taken as the geometric center of the α-carbon atoms belonging to the three proline hinge residues (Pro28, Pro133, and Pro230) that define the gate of the transporter. 2D slices were rendered with the matplotlib Python library.
Ensemble Docking
Autodock424 was used to perform docking of ADP to an ensemble of protein conformations derived from 1500 equidistant snapshots from each of our four apo-ANT1 systems of MD simulations. A custom TCL script was written to convert the NAMD dcd trajectory files. For each simulation snapshot, the protein was aligned with the equilibrated reference system and the protein structure was written to a separate file.
Instead of assigning partial charges via the Gasteiger method implemented in Autodock tools, we preserved the partial charge assignments previously defined from the CHARMM22 topology files used for all-atom simulations. To export these partial charges explicitly, PQR files were exported from the NAMD trajectories. The prepare\_receptor4.py script from Autodock Tools (ADT) was then used to convert the PQR files to the native Autodock PDBQT format. We manually validated that protein snapshots from wild-type and acetylated systems had net charges of +18 and +17, respectively.
Autogrid4 was invoked with a default grid spacing of 0.375 Å. The bounding box was created for the lower pocket of the channel, positioned at the center of mass of the protein. An XYZ grid size of 40 × 40 × 40 points was used. Docking was then performed on each ANT1MD snapshot conformation with a Lamarckian genetic algorithm. A population of 256 was used, with 2500000 maximal energy evaluations and 27000 maximal generations. We adjusted these parameters extensively but arrived at similar distributions for each experiment. The ADP ligand was exported from VMD into PQR format and prepared manually with Autodock Tools. Nonpolar hydrogens were merged, and nine active torsions were defined by default. The net charge of ADP was reported to be −3. After docking, binding energies and estimated binding constants were parsed from results files with the Unix programs egrep, awk, and sort.
In addition to ensemble docking, we also docked ADP to the static equilibrated wild-type system. The docked pose was similar to the best poses found in docking of the ensemble to the apo-ANT1 ensembles. The docked ligand was subsequently incorporated into the ANT1 structure, and two additional MD simulations were performed on both the wild-type (WT) and acetylated Lys23 systems. These bound “ANT1/ADP” simulations were subsequently analyzed and subjected to independent rounds of ensemble docking.
Finally, the physiological relevance of acetylation was assessed by converting these binding energies to ADP dissociation constants (Kd): Kb = e–(ΔG)/(RT) and Kd = 1/Kb, where Kb is the equilibrium binding constant.
Computational Model of ANT Flux
The velocity of ATP–ADP exchange by ANT1 was modeled using the complete kinetic description developed by Metelkin et al.9 Because the exchange of ATP4– for ADP3– by ANT involves the translocation of one charge, ATP–ADP antiport is energetically driven by the sum of electrical and chemical potentials:
where F is the Faraday constant, ΔΨ is the electrical potential across the inner membrane, R is the gas constant, T is absolute temperature, and o denotes the cytosolic and i the matrix spaces. Metelkin et al.9 have developed a bi-bi random kinetic model of ANT, which takes into account these electrochemical driving forces and their impact on kinetic binding and rate constants. For ANT flux simulations, we used the data of Glancy et al.25 obtained from isolated skeletal muscle mitochondria respiring at five experimentally maintained levels of ATP free energy (see Table 5). These data included steady state values of ATP and ADP concentrations in the extramitochondrial (cytosolic) space, ΔΨ, oxygen consumption rates, and ATP production rates. To estimate the matrix ATP and ADP concentrations at each of the five steady states, we used the ANT rate equation.26 Briefly, because the matrix total adenine nucleotide pool is constant (we used a value of 10 mM), the matrix ATP and ADP concentrations can be calculated when other values in the rate equation (ATP flux, ΔΨ, and cytosolic ATP and ADP concentrations) are known. Because Glancy et al. observed a KmADP for respiration of 43 μM,25 the baseline value for the ANT1 dissociation constant in the rate equation was equal to this value. All other parameter values were taken from ref (26).
Table 5. Estimated Binding Energies of ADP with Unacetylated and Lys23-Acetylated ANT1a.
| unacetylated | Lys23 | |
|---|---|---|
| binding energy (mean) (kcal/mol) | –7.7 | –5.816 |
| Kd (of mean energy) (μM) | 3.66 | 79.4 |
| binding energy (SD) (kcal/mol) | 0.78 | 1.28 |
| binding energy range (±1 SD) (kcal/mol) | –8.49 to −6.93 | –7.09 to −4.53 |
| corresponding Kd range (μM) | 1.02–13.04 | 9.94–634 |
Means of binding energies were obtained through fitting the normal distribution to histograms of binding energies from ensemble docking experiments. The estimated standard deviations were used to consider a range of binding energies of 1σ to either side of the mean. Dissociation constants (Kd) were calculated from these estimated mean binding energies.
Results
Acetylated Proteins Are Abundant in Human Skeletal Muscle and Mitochondria
To assess the extent and sites of lysine acetylation in skeletal muscle proteins, portions of whole muscle biopsies from three of the lean control subjects were lysed. Proteins were resolved by one-dimensional SDS–PAGE. Samples were processed, and mass spectrometry analysis was performed as described previously.7,8 Acetylated proteins and specific sites of acetylation in whole muscle are listed in Table 3. To obtain better proteomic coverage of mitochondrial proteins, mitochondria were isolated from freshly obtained skeletal muscle biopsies from the same three lean volunteers, and lysates of these mitochondria were processed in the same way as whole muscle lysates. Acetylated mitochondrial proteins are listed in Table 4. Pathways represented included shuttle activity (aspartate aminotransferase), fatty acid metabolism (hydroxyacyl-coenzyme A dehydrogenase), citric acid cycle (malate dehydrogenase), NADP-dependent isocitrate dehydrogenase, ATP synthesis (ATP synthase subunits b, d, and O), reactive oxygen species metabolism (Mn-superoxide dismutase), and adenine nucleotide translocase activity (ANT1).
Table 3. Acetylation Sites in Whole Muscle Homogenatesa.
| protein | acetylated amino acid sequence |
|---|---|
| myosin-2 | 773AGLLGLLEEM(ox)RDDK(ac)786 |
| myosin-7 | 146K(ac)RSEAPPHIFSISDNAYQYM(ox)LTDR169 |
| myosin light chain 3 | 124NK(ac)DTGTYEDFVEGLR138 |
| γ-enolase | 202DATNVGDEGGFAPNILENSEALELVK(ac)227 |
| histone H3.2 | 19KQLATK(ac)AAR27 |
| voltage-dependent anion-selective channel protein 1 | 35SENGLEFTSSGSANTETTK(ac)VTGSLETK61 |
| transitional endoplasmic reticulum ATPase | 2ASGADSK(ac)GDDLSTAILK18 |
| cytochrome b–c1 complex subunit 2, mitochondrial | 353AAYNQVK(ac)TIAQGNLSNTDVQAAK375 |
| tubulin β-2C chain | 319GRMSMK(ac)EVDEQMLNVQNK336 |
| myoglobin | 65HGATVLTALGGILK(ac)K79 |
| 33LFK(ac)GHPETLEK43 | |
| isoform 1 of myomesin-1 | 372YK(ac)GEFDETRFHAGASTM(ac)PLSFGVTPYGYASR402 |
| isoform 2 of l-lactate dehydrogenase A chain | 2ATLK(ac)DQLIYNLLK14 |
| trifunctional enzyme subunit α, mitochondrial | 494K(ac)MGLVDQLVEPLGPGLK516 |
| 213VIGM(ox)HYFSPVDK(ac)M(ox)QLLEIITTEK230 |
Acetylation sites are denoted K(ac); methionine oxidation is denoted M(ox).
Table 4. Acetylation Sites in Isolated Human Muscle Mitochondria.
| mitochondrial protein | acetylated amino acid sequence |
|---|---|
| ADP/ATP translocase 1 | 2GDHAWSFLK(ac)DFLAGGVAAAVSK23 |
| 8DFLAGGVAAAVSK(ac)TAVAPIER31 | |
| 81YFPTQALNFAFK(ac)DK94 | |
| trifunctional enzyme subunit β | 292LK(ac)PAFIKPYGTVTAANSSFLTDGASAM(ox)LIM(ox)AEEK325 |
| trifunctional enzyme subunit α | 214K(ac)M(ox)GLVDQLVEPLGPGLKPPEER235 |
| 494VIGM(ox)HYFSPVDK(ac)M(ox)QLLEIITTEK516 | |
| aspartate aminotransferase | 82K(ac)AEAQIAAK90 |
| 356TQLVSNLK(ac)K364 | |
| 394LIK(ac)EFSIYM(ox)TK404 | |
| 397EFSIYM(ox)TK(ac)DGR407 | |
| 410VAGVTSSNVGYLAHAIHQVTK(ac)430 | |
| hydroxyacyl-CoA dehydrogenase | 166FAGLHFFNPVPVM(ox)K(ac)LVEVIK179 |
| isoform 1 | 254GDASK(ac)EDIDTAM(ox)K266 |
| malate dehydrogenase | 53LTLYDIAHTPGVAADLSHIEK(ac)AAVK78 |
| 177ANTFVAELK(ac)GLDPAR191 | |
| 298GIEK(ac)NLGIGK307 | |
| 302NLGIGK(ac)VSSFEEK314 | |
| 329K(ac)GEDFVK355 | |
| Mn-superoxide dismutase | 54HHAAYVNNLNVTEEK(ac)YQEALAK75 |
| 115GELLEAIK(ac)R123 | |
| ATP synthase subunit d | 59ANVAK(ac)AGLVDDFEKK73 |
| 112IVEYEK(ac)EM(ox)EK121 | |
| isocitrate dehydrogenase (NADP) | 46VAK(ac)PVVEM(ox)DGDEM(ox)TR60 |
| mitochondrial | 70LILPHVDIQLK(ac)YFDLGLPNR89 |
| 81YFDLGLPNRDQTDDQVTIDSALATQK(ac)YSVAVK112 | |
| 173HAHGDQYK(ac)ATDFVADR188 | |
| 383GK(ac)LDGNQDLIR393 | |
| ATP synthase subunit b | 155SQQALVQK(ac)R163 |
| ATP synthase subunit O | 58QNKLEQVEK(ac)ELLR64 |
| 159TVLK(ac)SFLSQGQVLK172 | |
| 163SFLSQGQVLKLEAK(ac)176 | |
| 189IGEK(ac)YVDMSVK199 |
Acetylation of Mitochondrial Proteins Is Related to Insulin Sensitivity (study A)
To determine whether the extent of acetylation of mitochondrial proteins overall is associated with insulin sensitivity, normalized spectral abundance factor (NSAF) quantification was performed using a total of 16 nondiabetic, healthy volunteers (eight lean and eight obese), summing results for all sites that were found in all mitochondrial proteins listed in Table 3. The overall acetylation state of mitochondrial proteins was positively correlated with insulin-stimulated glucose disposal [r = 0.60; P < 0.01 (Figure 1)]. Total acetylation also was correlated positively with BMI (r = 0.55; P < 0.05) but not with fasting plasma insulin (r = 0.43; P = NS). Analyzed as groups, the levels of total mitochondrial protein acetylation were higher in lean [(3.23 ± 0.32) × 10–6 NSAF unit] than obese volunteers [(2.24 ± 0.31) × 10–6 NSAF unit] (P < 0.05).
Figure 1.

Acetylation of mitochondrial proteins is correlated with insulin action. The number of total spectra assigned to all acetylated peptides derived from mitochondrial proteins was normalized to the total number of spectra for mitochondrial proteins (NSAF), using mitochondria isolated from subjects described in Table 1. Empty circles represent data for individuals with a BMI of <25.0 and filled circles data for individuals with a BMI of >30.0. N = 16; *P < 0.05.
Adenine Nucleotide Translocase 1 (ANT1) Contains Multiple Acetylation Sites
Among the mitochondrial proteins we identified, ANT1 was observed to be present at a high level (based on spectral abundance) and acetylated in all individuals tested (based on the presence of spectra for acetylated peptides) in the initial screen of three subjects at lysines 10, 23, and 92. Because ANT1 has significant control of mitochondrial respiration under physiological conditions, we performed more detailed studies of regulation of acetylation of this protein.27 The amino acid sequence of human ANT1, with verified acetylation sites highlighted, is shown in Figure 2A. The yeast sequence also is shown to illustrate the homology of acetylation sites. The crystal structure of ANT1, in the “c” conformation (open to the intermembrane space), complexed with the inhibitor carboxyatractyloside, is shown in panels B and C of Figure 2. The human ANT1 homology model was prepared and inserted into a POPC membrane for simulation (Figure 2D). NSAF analysis showed that ANT1 abundance, expressed relative to total mitochondrial proteins, did not differ between lean [(7.15 ± 0.92) × 10–7 NSAF unit] and obese subjects [(7.27 ± 0.63) × 10–7 NSAF unit] (n = 16). Moreover, ANT1 acetylation at Lys23 was unaltered by obesity (7.15257 × 10–7 ± 9.17958 × 10–8 NSAF unit vs 6.33032 × 10–7 ± 6.27261 × 10–8 NSAF unit, normalized to ANT1 abundance). ANT1 acetylation, overall or Lys23, was not correlated with insulin-stimulated glucose disposal, BMI, or fasting plasma insulin levels.
Figure 2.

(A) Sequence alignment of human ANT1 and yeast AAC2. For the human sequence, yellow and green highlighting represent mass spectrometry coverage and observed lysine acetylations, respectively. For the yeast sequence, red highlighting indicates residues required for ADP transport. Arrows indicate residues that bind ADP phosphates in our modeling experiments. This sequence alignment was drawn with ESPript.43 (B) Top-down and (C) side cut-away views of bovine ANT1 complexed with the inhibitor carboxyatractyloside. Acetylated lysines (10, 23, and 92) are colored blue. (D) Human ANT1 homology model inserted into a POPC membrane for modeling experiments.
Acetylation of Lys23 of ANT1 Changes after Exercise, Depending on Insulin Sensitivity (study B)
We reasoned that if acetylation of mitochondrial proteins were regulated in a physiological manner, exercise and its attendant increases in ATP production rates and changes in mitochondrial redox state would be a prime candidate to cause changes. To assess this, an acute exercise protocol that we had previously used to show that insulin resistant muscle is also exercise resistant was employed.6 Biopsies taken in the resting state and 24 h after exercise were subjected to analysis of ANT1 Lys23 acetylation. Overall, there was a significant reduction in Lys23 acetylation that was highly dependent on insulin sensitivity (Figure 3; r = 0.90; P < 0.05).
Figure 3.
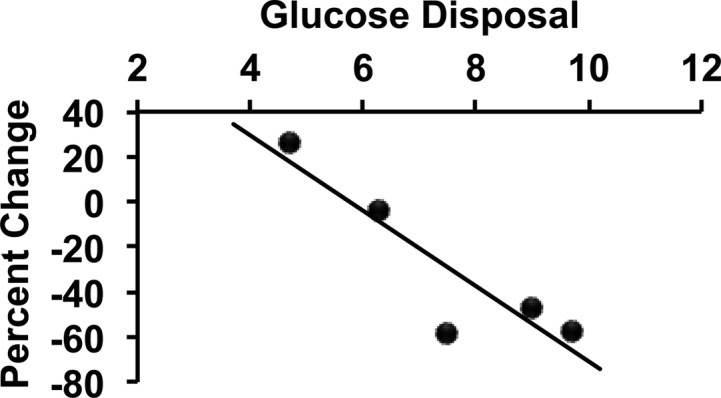
Change in acetylation of ANT1 at lysine 23 that is inversely correlated with insulin sensitivity. Subjects with a range of insulin sensitivities, as measured by glucose disposal (milligrams per kilogram of FFM per minute) during a euglycemic clamp had a single exercise bout with resting and 24 h post-exercise muscle biopsies. Lysine 23 acetylation was assessed as described in Experimental Procedures and expressed here as a percent change from basal (negative percent change indicating a decrease in acetylation). r = 0.90; P < 0.05.
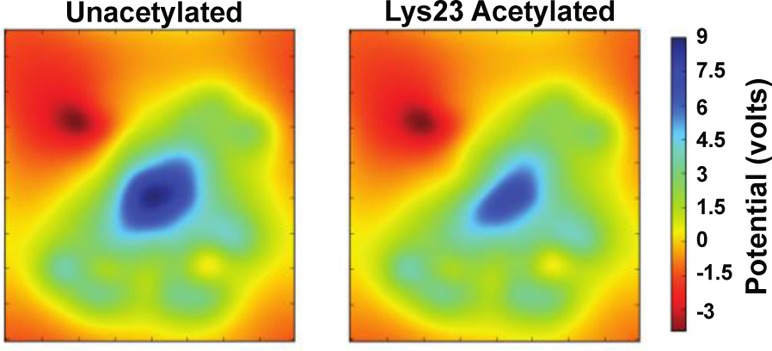
Acetylation Affects the Electrostatic Properties of ANT1
To assess the functional significance of lysine acetylation that results in the loss of positive charge, we analyzed the electrostatic properties of unacetylated and acetylated (Lys10, Lys23, and Lys92) ANT1. We produced 2D slices (Figure 4A) of the electrostatic potential using transverse sections through the region of ANT1 with the highest charge, where ANT1 exhibited a strong electrostatic potential at the bottom of the cavity, probably directing the binding of negatively charged ADP. The potential was highest near the side chain of Lys23 (Figure 4B). To visualize the changes to the positions and shapes of the isopotential after acetylation, we interpolated the potentials along a line drawn through the axis of the channel (Figure 4B). The electrostatic potential of these plots near the binding pocket experienced a rapid decline with distance “upward” through the channel opening, toward the intermembrane space or cytosol (Figure 4C). The potential at the binding pocket was reduced with acetylation of only Lys23, suggesting that acetylation specifically at this site could affect ADP binding affinity.
Figure 4.

ANT1 has a +18 charge and acts as a funnel to attract ADP. (A) 2D slices of the electrostatic potential. (B) To visualize the electrostatic profile along a hypothetical reaction coordinate in which ADP would translocate, the potential was interpolated along a 30 Å line (orange) drawn in the vertical axis. The origin was defined as the geometric center of the proline hinge α-carbon atoms. The three mitochondrial carrier motif gate helices are colored purple. The electrostatic potential is greatest at a site near the side chain of Lys23 (red sphere). In the profile plots (C), this potential maximum is clearly visible and substantially reduced in the Lys23 acetylated system.
Modeling of Apo-ANT1
All-atom molecular dynamics (MD) simulations were performed for 30 ns using unacetylated and acetylated (Lys10, Lys23, and Lys92) ANT1 systems. Simulations were performed without ADP (apo-ANT1 simulations) to test whether lysine acetylation affects the structure or dynamics of apo-ANT1. Fluctuation values [root-mean-square fluctuation (rmsf)] for the protein were plotted versus sequence position. A broad increase was seen near hydrophobic residues 76–87 (midpoint of the second transmembrane helix) in the acetylated Lys23 simulation (Figure 4A), representing the side chain of acetylated Lys23 drifting away from the binding pocket. Rather than strong electrostatic interactions of Lys23 with Glu30 in the salt bridge network observed in simulations of unacetylated ANT1, the hydrophobic part of the side chain of acetylated Lys23 interacted with residues Tyr81 and Asn77, pulling the second transmembrane helix inward toward the channel lumen. In the three other simulations, deacetylated Lys23 remained localized to the binding pocket due to electrostatic interactions with Glu30 (refer to Figure 6A for positions of residues).
Figure 6.

(A) Overlaid binding pocket regions of both unacetylated and acetylated Lys23 systems before ADP is docked. The acetyl group atoms in the acetylated system are colored red (oxygen) and teal (carbon). (B) The overlaid binding pockets for both systems after ADP is docked at the systems are simulated for 30 ns. From the overlay, we see that all charged residues have similar ending conformations except acetylated Lys23, which drifts away from ADP. Only the phosphates of ADP are shown for the sake of clarity. Dark blue side chains interact the strongest with ADP and are homologues of critical yeast mutants (Figure 1A). Lightly colored residues form the underlying salt bridge network. (C) Location of the ADP binding pocket in ANT1. ADP is shown bound after docking and MD simulation for 30 ns. (D) We measured the radial distance of basic side chains in the binding pocket in the apo and bound simulations. The dramatic stabilization after ADP insertion demonstrates an induced fit. (E) Distribution of binding energies obtained from ensemble docking experiments for the unacetylated/ADP and Lys23/ADP simulations.
To investigate the consequences of acetylation on ADP binding, we used an ensemble docking approach. Docking of ADP was focused to the binding pocket of the channel. Docking of ADP to the acetylated Lys10 and Lys92 systems replicated the unacetylated system. ADP adopted similar docking poses in all three acetylated systems. The two phosphate groups were arranged in a vertical orientation near the center of the pocket, preferentially interacting with four basic residues: Arg80, Arg280, Arg236, and Lys23. Despite no differences in the conformation of bound ADP, there were differences in binding affinity. The distributions of the estimated binding energies were similar among the unacetylated, Lys10-acetylated, and Lys92-acetylated systems. However, acetylating Lys23 resulted in a large shift in the total binding energy of ADP from −3 to approximately −1 kcal/mol (Figure 5B).
Figure 5.
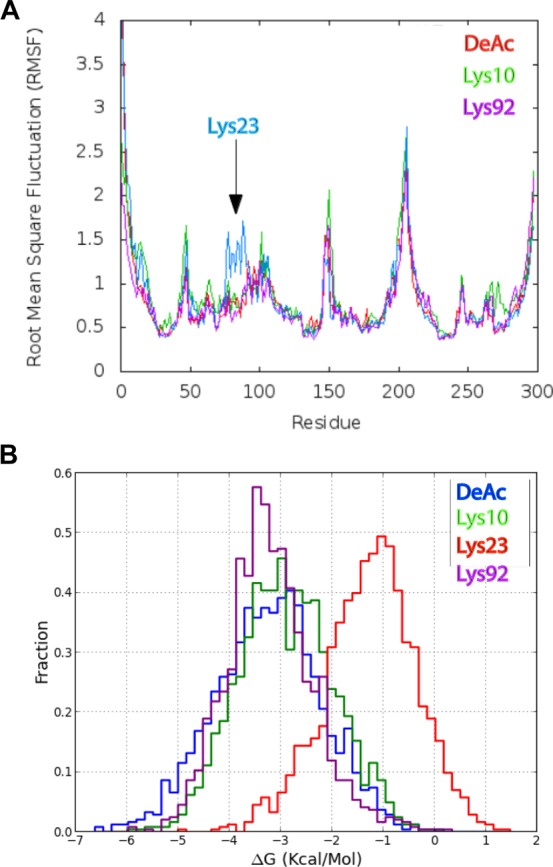
(A) rmsf profiles of the four apo-ANT1 simulations. A broad perturbation can be seen between residues 76 and 87 in the acetylated Lys23 simulation. (B) Acetylated Lys23 is observed departing the binding pocket (bottom left) and participating in hydrophobic interactions with the sidewall of the channel. These interactions pull the second transmembrane helix slightly inward over the course of a 30 ns simulation. (C) Binding energy distributions obtained from ensemble docking of the four simulated systems.
Modeling of ANT1 with ADP Bound
In the apo-ANT1 system, docking experiments might yield weak binding affinities, because the binding pocket is unable to rearrange to more favorably accommodate the absent ligand. To allow such rearrangements, we conducted molecular dynamics experiments with ADP docked to the binding pocket. We performed docking of ADP to the static equilibrated unacetylated structure and used this system as an input for two further simulations. Wild-type-bound (unacetylated/ADP) and acetylated Lys23-bound (acetylated Lys23/ADP) systems were prepared and simulated as before for 30 ns. The charged residues within the binding pocket rapidly reorganized. Using unacetylated ANT1, Arg80, Arg280, Arg236, and Lys23 rearranged to form tight hydrogen bonds with the negatively charged ADP phosphate groups. Figure 6B shows superimposed structures of the unacetylated and Lys23-acetylated simulations with ADP bound. Both systems had the same initial configuration, in which critical residues of the binding pocket interacted with charged residues at the periphery. After simulation for 30 ns, the side chains of these residues rearranged to a more centralized conformation to bind the ADP phosphates (Figure 6B). In both systems, the final side chain conformations were similar for each charged residue, with the exception of acetylated Lys23, which drifted away from the binding pocket (Figure 6B). The location of the docked ADP within the unacetylated ANT1 after simulation for 30 ns is illustrated in Figure 6C. The terminal phosphates of ADP interacted primarily with four basic residues, one of which is Lys23, the others being arginines 80, 236, and 280.
Next, we analyzed the radial positions of the conserved critical residues in the binding pocket (Lys23, Arg80, Arg280, Lys33, Arg138, Arg235, and Arg236) as a function of time. From the unacetylated system simulation, we computed the average position of the terminal phosphorus atom of ADP. Side chain distances were calculated from this position to the NZ atoms of Lys23 and Lys33 and the CZ atoms of Arg80, Arg138, Arg235, Arg236, and Arg280 for the 25 ns unbound (unacetylated) equilibration and the 30 ns bound simulation (unacetylated/ADP). Time evolution of these distances, which indirectly measures the interaction of ADP with these conserved residues critical for binding, showed a substantial increase in binding pocket stability at the transition of the equilibration and bound simulations (Figure 6D).
We hypothesized that this induced fit strengthens ADP binding and re-evaluated binding affinities of ADP for unacetylated and acetylated Lys23 ANT using ensemble docking. We conducted an independent set of docking experiments on snapshots from the two bound ANT1/ADP simulations and fit normal distributions to the binding energy histograms obtained from these experiments (Figure 6E). Binding energies increased dramatically compared to those of the apo-ANT1 docking experiments (Figure 5C). The ADP binding affinity of the unacetylated ANT1 increased from −3 kcal/mol in the apo-ANT1 ensemble to −7.7 kcal/mol for the ADP-bound unacetylated structure (unacetylated/ADP). For the acetylated Lys23 system, ADP bound with an estimated energy of −5.8 kcal/mol to the bound (Lys23/ADP) ensemble, confirming and emphasizing how Lys23 acetylation reduces the affinity of ANT1 for ADP.
We used the range of binding energies, from one standard deviation to the left and right of the estimated means, to calculate KdADP values (Table 5). These calculations clearly show the dramatic impact of Lys23 acetylation on the binding affinity of ANT1 for ADP. Mean binding energies gave KdADP values of 3.66 and 79.4 μM for unacetylated and acetylated Lys23, respectively (Table 5). Thus, MD modeling predicts that Lys23 acetylation causes large increases in KdADP, at values that reflect the physiological range of ADP concentrations in vertebrate skeletal muscle.
Modeling of ANT1 Function with Changes in KdADP
To explore potential consequences of such KdADP changes on ANT1 function, ANT1 flux (JANT in nanomoles of ATP per minute per milligram of mitochondrial protein) was simulated using the kinetic model of Metelkin et al.9 at three values of KdADP: 43.0, 79.4, and 3.66 μM. The “baseline” condition, KdADP of 43.0 μM, was based on the data of Glancy et al. and was used to calculate matrix ATP/ADP. The other two values represent modeling-derived estimates for ANT1 with Lys23 acetylated (79.4 μM) or unacetylated (3.66 μM). As reported in Table 6 and illustrated in Figure 7, model output predicts that increasing the KdADP from 43.0 to 79.4 μM due to ANT1 Lys23 acetylation would result in a dramatic decrease in mitochondrial ATP export. Moreover, this decline in ANT flux would occur across the entire aerobic scope of the mitochondrion. A KdADP of 3.66 μM obviously implies near saturation of ANT at ADP concentrations in the range often observed in resting skeletal muscle. In this case, the model of Metelkin et al.26 not surprisingly predicted near-maximal flux at very low ADP concentrations.
Table 6. ANT Fluxes (JANT) Simulateda with the Kinetic Model of Metelkin et al.9.
| matrix | cyto | matrix |
cyto |
JANT (nmol min–1 mg–1) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| JO2 (nmol min–1 mg–1) | JATP (nmol min–1 mg–1) | ΔΨ (mV) | ATP/ADP | [ADP] (mM) | [ATP] (mM) | [ADP] (μM) | [ATP] (mM) | KdADP = 43.0 μM | KdADP = 79.4 μM | KdADP = 3.66 μM | |
| 123 | 664 | 201 | 0.39 | 299 | 7.22 | 2.78 | 16.7 | 4.98 | 665 | 469 | 1932 |
| 151 | 815 | 200 | 0.31 | 224 | 7.65 | 2.35 | 22.2 | 4.98 | 815 | 586 | 1941 |
| 191 | 1031 | 198 | 0.21 | 149 | 8.27 | 1.74 | 33.3 | 4.97 | 1031 | 768 | 1740 |
| 225 | 1215 | 197 | 0.17 | 112 | 8.58 | 1.43 | 44.4 | 4.96 | 1214 | 929 | 1608 |
| 261 | 1409 | 195 | 0.11 | 74 | 8.98 | 1.02 | 66.7 | 4.93 | 1410 | 1134 | 1284 |
The rate of export of mitochondrial ATP by ANT (JANT) was simulated with the kinetic model of Metelkin et al.10 All model kinetic parameters were taken from ref (26), except for ADP affinity. The data of Glancy et al.25 from isolated mitochondria respiring at five steady state levels of ATP free energy provided values for O2 consumption rate (JO2), ATP production rate (JATP), membrane potential (ΔΨ), and the cytosolic (i.e., extramitochondrial) concentrations of ADP and ATP, at an observed KMADP for respiration of 43.0 μM. This ADP affinity was inserted into the model to calculate the apparent matrix ADP and ATP concentrations.26 This procedure yielded excellent matching of model output JANT to the experimentally observed JATP at reasonable matrix ATP/ADP ratios. Next, the functional consequences of ANT1 Lys23 acetylation were explored by running model simulations with a step increase in KdADP to 79.4 μM. This resulted in dramatic reductions in ANT flux. Finally, deacetylation, and the corresponding decrease in KdADP to 3.66 μM, resulted in very high JANT values even at very low cytosolic ADP concentrations.
Figure 7.

Data reported in Table 5 are graphically illustrated here. The rate of export of mitochondrial ATP by ANT (JANT) is simulated with the kinetic model of Metelkin et al.10 at KdADP values of 3.66, 43.0, and 79.4 μM. These KdADP values correspond to deacetylated ANT1, the observed data of Glancy et al.25 in isolated mitochondria, and ANT1 acetylated at Lys23, respectively. See the text for further discussion.
Discussion
This study was undertaken in part to identify and quantify acetylation sites in proteins from skeletal muscle from humans with a wide range of insulin sensitivity, focusing on mitochondrial and other proteins involved in metabolism. To accomplish this, we used euglycemic clamps to quantify insulin sensitivity on groups of lean and obese individuals, and muscle biopsies together with mitochondrial fractionation and a proteomics approach to identify lysine acetylation sites in mitochondrial and other proteins in skeletal muscle. Because we did not specifically enrich acetylated peptides, our acetylome likely represents the most with the highest levels of acetylation. Many of the acetylated proteins we observed are involved in metabolism.4 Employing isolated mitochondria to enrich the preparation identified a number of additional sites in mitochondrial proteins. Our analyses showed that overall, when total acetylation was measured, it was found to be positively associated with insulin sensitivity in resting muscle. That is, total protein acetylation was higher in individuals whose skeletal muscle was the most sensitive to insulin, as assessed by the euglycemic clamp technique, suggesting that a balance between acetylation and deacetylation favoring acetylation characterizes insulin sensitive skeletal muscle, specifically under basal, resting conditions. However, because alterations in mitochondrial function have been associated with insulin resistance,6−8 in addition to the overall protein acetylation state, acetylation of individual mitochondrial proteins also should be assessed within the context of insulin sensitivity and the potential affects of acetylation on the function of specific proteins.
Among the acetylated mitochondrial proteins we identified, the adenine nucleotide translocase ANT1 (SLC25A4) stood out because of its multiple acetylation sites, its importance in physiologic control of respiration,28 and its major role in proton leak in skeletal muscle mitochondria.29 Lysine residues contribute to the net positive charge of ANT1 that is critical for transport of ADP/ATP, and acetylation of these residues could affect the function of the protein by altering charge-based interactions.30 We identified three lysine acetylation sites in ANT1, namely, lysines 10, 23, and 92. Moreover, we also showed that lysine 23 acetylation of ANT1 is regulated by an acute bout of exercise in an insulin-dependent manner.
Given the evidence that ANT1 is a central point of control for oxidative metabolism and that mutations in ANT1 in yeast31 and humans32 can cause profound dysfunction, we focused on characterizing the biophysical mechanisms by which ANT1 function might be altered by acetylation of these charged residues. Acetylation of critical positively charged residues of ANT1 could affect the electrostatic properties and inhibit the initial capture or binding of ADP or could perturb its structure or dynamics, possibly changing the shape of its binding pocket or altering molecular dynamics that facilitate ligand recognition or channel opening. To capture and transport ADP, ANT1 relies upon a substantial +18 charge to attract negatively charged ADP molecules. Wang et al.33 showed that this charge is high in comparison to those of most mitochondrial proteins, so it is critical to understand the effects of acetylation on the electrostatic properties of ANT1. Of the lysine residues we identified to be acetylated in our study, Lys23 is uniquely positioned at the binding pocket where the electrostatic potential is highest.
After initial ADP capture and binding, ANT1 undergoes a conformational change leading to channel opening and passage of ADP into the mitochondrial matrix. ANT1 is characterized by three mitochondrial carrier motifs, PX(D/E)XX(K/R). The motifs colocalize in the structure of bovine ANT1 to form a triple-helix gate at the matrix side of the channel. The proline residues form kinks in the α-helices, allowing a hinge motion to occur during opening and closing of the channel. The two charged D/E and K/R residues of each helix form salt bridges that bind the neighboring helices together in a closed conformation. Channel opening may require disruption of this salt bridge network, presumably by the binding of ADP to the three basic residues located on these helices. ANT1 may undergo a conformational switch between a cytosol-facing c state and a matrix-facing m state, in which either ADP or ATP is bound and translocated in alternating order. We postulated that a perturbation of these dynamics by lysine acetylation could influence the function of the channel. A large structural difference in the channel occurred when Lys23 was acetylated, manifested by the second transmembrane helix (residues 76–87) being perturbed due to interactions with acetylated Lys23. The results suggest that channel narrowing may be a structural consequence of Lys23 acetylation that might affect ADP transport. Parenthetically, although phosphorylated tyrosine residues, particularly Tyr191 and Tyr195, may participate in the passage of ADP through ANT1,34 this model did not test their involvement with acetylation at Lys23. In some cases, we did observe phosphorylation at these residues in human ANT1.
With regard to ADP capture and binding, two previous studies simulated the early capture and translocation events of ADP using all-atom molecular dynamics techniques.33,35 Both studies focused on the strong electrostatic properties of ANT1 to act as “funnel” that captures ADP molecules from the intermembrane space and transports them to the binding pocket. In these simulations, ADP became trapped in various locations and orientations for extended durations that we felt precluded the use of all-atom molecular dynamics to simulate capture events for determining the effects of lysine acetylation. Therefore, we focused on docking of ADP directly to the binding pocket where Lys23 is located. Autodock has been employed successfully to perform a screen of ATP binding sites on the matrix-facing surface of ANT1,36 so we adopted similar methodologies for ADP binding in the cytosol-facing pocket. These experiments resulted in highly reproducible bound conformations of ADP agreeing with all-atom molecular dynamics results33 and yielded estimates of binding energies that indicate a single acetylation event at a crucial site can have significant effects.
Molecular dynamics data provide strong evidence that acetylation of Lys23 profoundly reduces the affinity of ADP for ANT1. The ADP dissociation constant (Kd; the concentration of ADP required for half-occupancy of ANT1), calculated from the binding energies from ensemble docking experiments, was 3.66 μM in unacetylated ANT1 but increased to 79.4 μM with the acetylation of Lys23. Human skeletal muscle rests at a free cytosolic ADP concentration of roughly 10–20 μM, with an apparent KMADP for respiration of 30–50 μM.37,38 These values fall between our estimates of the binding affinities of ADP for the unacetylated and Lys23-acetylated systems. In human skeletal muscle contracting in the mild-to-moderate aerobic metabolic range, ANT1 is the major locus of aerobic flux control27 and 31P NMR measurement of energy phosphate concentrations has shown that simple Michaelis–Menten kinetics provide a reasonable fit of the [ADP]:VO2 relationship,38 although higher-order models also have been demonstrated.39 To gain insight into the functional consequences that could result from Lys23 acetylation, ANT function was simulated using a computational model.9 Metelkin et al. assigned a value of 39 μM to the kinetic parameter corresponding to ANT–ADP binding on the outside of de-energized mitochondria, which is similar to the observed KMADP for respiration of intact muscle (above) and isolated mitochondria, as reported by Glancy et al.25 Simulations run with the data of Glancy et al., including their observed KmADP of 43 μM,25 allowed the model to match observed ATP production rates by isolated mitochondria across five levels of ATP free energy. In contrast, simulating acetylated Lys23 by increasing the model KdADP to 79.4 μM, with all other model input variables held constant, dramatically reduced the mitochondrial ATP output. This analysis does not necessarily imply that mitochondria with ANT1 Lys23 acetylation would be catalytically incompetent to match cellular ATP demand, but it does strongly suggest that acetylation would require compensatory changes in other variables, and these changes ultimately would be associated with a reduction in cytosolic energy status. Important caveats obviously attend such hypothetical scenarios, but the analysis nevertheless affords useful insight into the role of ANT1 in the control of respiration and the profound impact of ANT1 acetylation. The observation that the apparent in vivoKMADP for respiration is intermediate between the Kd values of deacetylated and acetylated ANT1 modeled here may reflect a mechanism that adjusts mitochondrial sensitivity to the ADP respiratory signal geared to metabolic demand. These computational simulations effectively are tests of the immediate effect on ANT1 flux of a change in the acetylation status of Lys23. In a single ANT1 molecule, acetylation of Lys23 would abruptly increase KdADP from 43 to ∼79.4 μM, and the ATP export/ADP import rate would fall, as shown in Figure 7. Presumably, a period of metabolic adjustment would follow, in which matrix ATP concentration, cytosolic ADP concentration, and ΔΨ would all increase to attempt to meet the rate of ATP demand. However, the crossover at ANT implies that such a return toward the initial turnover rate would necessarily require a new steady state with a lower cytosolic ATP energy status (“downstream of ANT”) and higher mitochondrial driving forces (protonmotive force and matrix redox, which are “upstream of ANT”). The former, downstream, adjustment would tend to inhibit cytosolic ATP-utilizing sites, while the latter, upstream, adjustment would tend to impede fuel oxidation by substrate dehydrogenases and the citric acid cycle. Thus, without compensatory activation from an external effector, ATP turnover might not return to its initial rate, as formalized in the connectivity property of metabolic control analysis.40 Moreover, the higher mitochondrial “pressures” would tend to promote energy-dissipating processes, such as the proton leak across the inner membrane41 and the electron leak along the respiratory chain, that is, superoxide or ROS production.42
Studies in yeast, where the sequence of the AAC2 transporter is highly similar to that of ANT1, have provided functional information about the molecular mechanism of ADP/ATP transport by ANT1, showing the importance of charged residues.30 Mutants of AAC2 that affected ADP/ATP transport were screened, and the results showed that one lysine and six arginines were essential for growth on oxidative substrates. Of the six arginines identified, three are part of the RRRMMM motif, characteristic of all ADP/ATP transporters.31 Another two mutant arginines in yeast correspond to Arg80 and Arg280 in the human transporter, which bind ADP and flank Lys23 in the bovine crystal structure. Finally, the single most critical lysine in yeast, Lys38, is in a region with a sequence almost identical to that of the regions surrounding Lys23 in human ANT1. When Lys38 in yeast AAC2 was mutated to alanine, the mutant yeast were incapable of growth on nonfermentable carbon sources, indicating that mitochondria from these yeast were incapable of producing ATP.30 Biochemical assays and all-atom molecular dynamics simulations also have been used to characterize pathological mutants of ANT1 observed in humans.32 Such mutations are linked to fatigue, weakness, ptosis, and exercise intolerance,32 confirming that ANT1 is a point of flux control for proper metabolism in muscle. Taken together, the data in yeast and humans indicate the importance of charged residues in general, and Lys23 in particular, with respect to the ability of ANT1 to translocate adenine nucleotides. Although our modeling studies are consistent with those results in yeast, and we showed other acetylation sites (Lys10 and Lys92) to have little impact on modeled parameters, one caveat is that these modeling results were derived from the crystal structure of ANT1 in the so-called c state (open to the intermembrane space) and might be different with ANT1 modeled in the as yet unsolved m conformational state.
In summary, acetylation of mitochondrial proteins, and in particular the adenine nucleotide translocase ANT1, is abundant in human skeletal muscle mitochondria and correlated with insulin sensitivity. The use of simulations and docking experiments revealed that Lys23 is a critical residue for controlling the affinity of ADP for ANT1, with the acetylated system having a much lower binding affinity for ADP. Taking the modeling, computational, and physiologic results as a whole, we conclude that the positive charge at lysine 23 of ANT1 is critical for regulating the KmADP for oxidative phosphorylation. The ability of insulin sensitive individuals to reduce acetylation at this site by exercise, hence decreasing the KmADP, would relieve the need for higher mitochondrial energetic driving forces, and hence ROS production, to meet cellular ATP demand. We found no difference between insulin sensitive and resistant sedentary individuals for lysine 23 acetylation, when they were studied at rest, at least 48 h after having done any voluntary exercise. However, 24 h after an exercise bout, a decrease in lysine 23 acetylation was evident. This suggests that this effect is transitory, like many of the effects of exercise, and the deacetylation effect likely was even more pronounced in the immediate post-exercise period. Ascertainment of the mechanisms underlying this deacetylation event likely will require the use of different models.
Acknowledgments
We thank the participants who volunteered for this study and gratefully acknowledge the editorial assistance of Irene Beauvais from the Mayo Clinic. We also thank Dianne DeNardo, RN, from Arizona State University for expert nursing assistance and Mark Mattern, MD, from Arizona State University for medical oversight of the euglycemic clamp experiments.
Glossary
Abbreviations
- AAC2
ADP/ATP carrier
- ANT1
adenine nucleotide translocase 1
- BPM
beats per minute
- LTQ-FTICR
linear triple-quadrupole Fourier transform ion cyclotron resonance
- ROS
reactive oxygen species
- SD
standard deviation
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SEM
standard error of the mean.
The authors declare no competing financial interest.
This study was supported by National Institutes of Health Grants R01 DK047936 (L.J.M.) and DK066483 (L.J.M.) and an allocation of supercomputer time from the Arizona State University Advanced Computing Center.
Funding Statement
National Institutes of Health, United States
References
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Yang C.; Xiong H.; Lin Y.; Yao J.; Li H.; Xie L.; Zhao W.; Yao Y.; Ning Z. B.; Zeng R.; Xiong Y.; Guan K. L.; Zhao S.; Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Sprung R.; Pei J.; Tan X.; Kim S.; Zhu H.; Liu C. F.; Grishin N. V.; Zhao Y. (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Xu W.; Jiang W.; Yu W.; Lin Y.; Zhang T.; Yao J.; Zhou L.; Zeng Y.; Li H.; Li Y.; Shi J.; An W.; Hancock S. M.; He F.; Qin L.; Chin J.; Yang P.; Chen X.; Lei Q.; Xiong Y.; Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey M. D.; Shimazu T.; Goetzman E.; Jing E.; Schwer B.; Lombard D. B.; Grueter C. A.; Harris C.; Biddinger S.; Ilkayeva O. R.; Stevens R. D.; Li Y.; Saha A. K.; Ruderman N. B.; Bain J. R.; Newgard C. B.; Farese R. V. Jr.; Alt F. W.; Kahn C. R.; Verdin E. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis E.; Alvarez G.; Berria R.; Cusi K.; Everman S.; Meyer C.; Mandarino L. J. (2008) Insulin-resistant muscle is exercise resistant: Evidence for reduced response of nuclear-encoded mitochondrial genes to exercise. Am. J. Physiol. 294, E607–E614. [DOI] [PubMed] [Google Scholar]
- Hwang H.; Bowen B. P.; Lefort N.; Flynn C. R.; De Filippis E. A.; Roberts C.; Smoke C. C.; Meyer C.; Hojlund K.; Yi Z.; Mandarino L. J. (2010) Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 59, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort N.; Glancy B.; Bowen B.; Willis W. T.; Bailowitz Z.; De Filippis E. A.; Brophy C.; Meyer C.; Hojlund K.; Yi Z.; Mandarino L. J. (2010) Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 59, 2444–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metelkin E.; Goryanin I.; Demin O. (2006) Mathematical modeling of mitochondrial adenine nucleotide translocase. Biophys. J. 90, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort N.; Yi Z.; Bowen B.; Glancy B.; De Filippis E. A.; Mapes R.; Hwang H.; Flynn C. R.; Willis W. T.; Civitarese A.; Hojlund K.; Mandarino L. J. (2009) Proteome profile of functional mitochondria from human skeletal muscle using one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. J. Proteomics 72, 1046–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ-Roberts C. Y.; Mandarino L. J. (2004) Glycogen synthase: Key effect of exercise on insulin action. Exercise Sport Sci. Rev. 32, 90–94. [DOI] [PubMed] [Google Scholar]
- Cusi K.; Maezono K.; Osman A.; Pendergrass M.; Patti M. E.; Pratipanawatr T.; DeFronzo R. A.; Kahn C. R.; Mandarino L. J. (2000) Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Invest. 105, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais P.; Yi Z.; Finlayson J.; Luo M.; Mapes R.; De Filippis E.; Meyer C.; Plummer E.; Tongchinsub P.; Mattern M.; Mandarino L. J. (2011) Global IRS-1 phosphorylation analysis in insulin resistance. Diabetologia 54, 2878–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais P.; Mandarino L. J.; Yi Z. (2010) Label-free relative quantification of co-eluting isobaric phosphopeptides of insulin receptor substrate-1 by HPLC-ESI-MS/MS. J. Am. Soc. Mass Spectrom. 21, 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z.; Luo M.; Mandarino L. J.; Reyna S. M.; Carroll C. A.; Weintraub S. T. (2006) Quantification of phosphorylation of insulin receptor substrate-1 by HPLC-ESI-MS/MS. J. Am. Soc. Mass Spectrom. 17, 562–567. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Dahout-Gonzalez C.; Kahn R.; Trezeguet V.; Lauquin G. J.; Brandolin G. (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44. [DOI] [PubMed] [Google Scholar]
- Pieper U.; Webb B. M.; Barkan D. T.; Schneidman-Duhovny D.; Schlessinger A.; Braberg H.; Yang Z.; Meng E. C.; Pettersen E. F.; Huang C. C.; Datta R. S.; Sampathkumar P.; Madhusudhan M. S.; Sjolander K.; Ferrin T. E.; Burley S. K.; Sali A. (2011) ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 39, D465–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 27–38. [DOI] [PubMed] [Google Scholar]
- Roberts E.; Eargle J.; Wright D.; Luthey-Schulten Z. (2006) MultiSeq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinf. 7, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. B.; Barton G. J. (1992) Multiple protein sequence alignment from tertiary structure comparison: Assignment of global and residue confidence levels. Proteins 14, 309–323. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Banavali N.; Foloppe N. (2000) Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56, 257–265. [DOI] [PubMed] [Google Scholar]
- Aksimentiev A.; Schulten K. (2005) Imaging α-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 88, 3745–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B.; Willis W. T.; Chess D. J.; Balaban R. S. (2013) Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metelkin E.; Demin O.; Kovacs Z.; Chinopoulos C. (2009) Modeling of ATP-ADP steady-state exchange rate mediated by the adenine nucleotide translocase in isolated mitochondria. FEBS J. 276, 6942–6955. [DOI] [PubMed] [Google Scholar]
- Jeneson J. A.; Schmitz J. P.; van den Broek N. M.; van Riel N. A.; Hilbers P. A.; Nicolay K.; Prompers J. J. (2009) Magnitude and control of mitochondrial sensitivity to ADP. Am. J. Physiol. 297, E774–E784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko B.; Zilinskiene V.; Borutaite V.; Ivanoviene L.; Toleikis A.; Praskevicius A. (1987) The role of adenine nucleotide translocators in regulation of oxidative phosphorylation in heart mitochondria. FEBS Lett. 223, 247–250. [DOI] [PubMed] [Google Scholar]
- Brand M. D.; Pakay J. L.; Ocloo A.; Kokoszka J.; Wallace D. C.; Brookes P. S.; Cornwall E. J. (2005) The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D. R.; Lawson J. E.; Klingenberg M.; Douglas M. G. (1993) Site-directed mutagenesis of the yeast mitochondrial ADP/ATP translocator. Six arginines and one lysine are essential. J. Mol. Biol. 230, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Clemencon B.; Rey M.; Trezeguet V.; Forest E.; Pelosi L. (2011) Yeast ADP/ATP carrier isoform 2: Conformational dynamics and role of the RRRMMM signature sequence methionines. J. Biol. Chem. 286, 36119–36131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravaud S.; Bidon-Chanal A.; Blesneac I.; Machillot P.; Juillan-Binard C.; Dehez F.; Chipot C.; Pebay-Peyroula E. (2012) Impaired transport of nucleotides in a mitochondrial carrier explains severe human genetic diseases. ACS Chem. Biol. 7, 1164–1169. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Tajkhorshid E. (2008) Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc. Natl. Acad. Sci. U.S.A. 105, 9598–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Lucchinetti E.; Enkavi G.; Wang Y.; Gehrig P.; Roschitzki B.; Schaub M. C.; Tajkhorshid E.; Zaugg K.; Zaugg M. (2010) Tyrosine phosphorylation by Src within the cavity of the adenine nucleotide translocase 1 regulates ADP/ATP exchange in mitochondria. Am. J. Physiol. 298, C740–C748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehez F.; Pebay-Peyroula E.; Chipot C. (2008) Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J. Am. Chem. Soc. 130, 12725–12733. [DOI] [PubMed] [Google Scholar]
- Di Marino D.; Oteri F.; della Rocca B. M.; D’Annessa I.; Falconi M. (2012) Mapping multiple potential ATP binding sites on the matrix side of the bovine ADP/ATP carrier by the combined use of MD simulation and docking. J. Mol. Model. 18, 2377–2386. [DOI] [PubMed] [Google Scholar]
- Jeneson J. A.; Westerhoff H. V.; Brown T. R.; Van Echteld C. J.; Berger R. (1995) Quasi-linear relationship between Gibbs free energy of ATP hydrolysis and power output in human forearm muscle. Am. J. Physiol. 268, C1474–C1484. [DOI] [PubMed] [Google Scholar]
- Chance B.; Leigh J. S. Jr.; Clark B. J.; Maris J.; Kent J.; Nioka S.; Smith D. (1985) Control of oxidative metabolism and oxygen delivery in human skeletal muscle: A steady-state analysis of the work/energy cost transfer function. Proc. Natl. Acad. Sci. U.S.A. 82, 8384–8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeneson J. A.; Wiseman R. W.; Westerhoff H. V.; Kushmerick M. J. (1996) The signal transduction function for oxidative phosphorylation is at least second order in ADP. J. Biol. Chem. 271, 27995–27998. [DOI] [PubMed] [Google Scholar]
- Brand M. D. (1996) Top down metabolic control analysis. J. Theor. Biol. 182, 351–360. [DOI] [PubMed] [Google Scholar]
- Brand M. D. (1990) The proton leak across the mitochondrial inner membrane. Biochim. Biophys. Acta 1018, 128–133. [DOI] [PubMed] [Google Scholar]
- Korshunov S. S.; Skulachev V. P.; Starkov A. A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18. [DOI] [PubMed] [Google Scholar]
- Gouet P.; Courcelle E.; Stuart D. I.; Metoz F. (1999) ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308. [DOI] [PubMed] [Google Scholar]