

Abstract
Human HtrA1 (high-temperature requirement protein A1) belongs to a conserved family of serine proteases involved in protein quality control and cell fate. The homotrimeric ubiquitously expressed protease has chymotrypsin-like specificity and primarily targets hydrophobic stretches in selected or misfolded substrate proteins. In addition, the enzyme is capable of exerting autolytic activity by removing the N-terminal insulin-like growth factor binding protein (IGFBP)/Kazal-like tandem motif without affecting the protease activity. In this study, we have addressed the mechanism governing the autolytic activity and find that it depends on the integrity of the disulfide bonds in the N-terminal IGFBP/Kazal-like domain. The specificity of the autolytic cleavage reveals a strong preference for cysteine in the P1 position of HtrA1, explaining the lack of autolysis prior to disulfide reduction. Significantly, the disulfides were reduced by thioredoxin, suggesting that autolysis of HtrA1 in vivo is linked to the endogenous redox balance and that the N-terminal domain acts as a redox-sensing switch.
Human HtrA1 (high-temperature requirement protein A1, Uniprot entry Q92743) is a 51 kDa nonglycosylated serine protease primarily located in the extracellular space. Here, it is involved in remodeling of the extracellular matrix by interactions and proteolysis of components like decorin, fibronectin, aggrecan, and collagen.1−5 The protease also regulates cellular signaling cascades by specific breakdown of mediators like transforming growth factor-β, fibroblast growth factor 8, and epidermal growth factor receptor.6−9 In addition, HtrA1 is colocalized in the cytoplasmic compartment with several intracellular identified substrates, such as tubulin and the X-linked inhibitor of apoptosis (XIAP).10 Dysregulation of HtrA1 expression levels is associated with cerebral small-vessel disease,11 age-related macular degeneration (AMD),12,13 osteoarthritis,14 amyloid neurological disorders,15,16 corneal dystrophy,17 preeclampsia,18,19 and numerous types of cancer.20,21 The specific downregulation of HtrA1 in the latter case promotes the sustained survival of cancer cells and the development of malignant metastatic behavior,20,22,23 most likely correlated with the intracellular HtrA1 associated with microtubules and affecting cell migratory and signaling properties.3,14,24−27 Interestingly, autolytically N-terminally truncated forms of HtrA1 exist in vivo, and such forms have been suggested as prognostic markers in urothelial bladder cancer28 and in preeclamptic pregnancies.18 The exact role and trigger of autolytic maturation is at present unknown.
HtrA1 is composed of three domains; the central core contains the catalytic triad (His220, Asp250, and Ser328) and is responsible for enzymatic activity and most reported functions, such as substrate specificity, proteolysis, trimerization, and liposome and cell-surface interactions.29 The N- and C-terminal domains of HtrA1 comprise an IGFBP-like/Kazal-like tandem domain motif (termed the N-domain) and a PDZ domain, respectively. Both seem largely dispensable for catalytic function,30 and whereas the PDZ domain facilitates protein–protein interactions by tethering of the HtrA1 protease to specific sites in the ECM or in the cell,4,26,31 the role of the N-domain remains enigmatic. The domain contains a total of 16 cysteines, all forming part of an intensive disulfide bond network (Figure 1). It has previously been shown that HtrA1 may undergo autolytic cleavage, and it was therefore suggested that the N-terminal Kazal-like subdomain could play an autoinhibitory function in this context.14 However, recent studies have shown that neither the IGFBP- nor the Kazal-like structures have retained their canonical functions required for IGF interaction or protease inhibition, respectively.30 The lack of autoinhibition by the N-domain is further corroborated by studies showing similar catalytic activities of full-length HtrA1 and ΔN-HtrA1 lacking the N-domain.11,30
Figure 1.
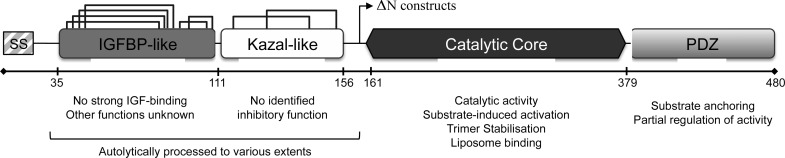
HtrA1 domain organization and disulfide bond network.
The presence of processed HtrA1 forms in the intracellular reducing environment suggests that autolysis could be affected by changes in the redox state of HtrA1, more specifically the integrity of the N-domain disulfide bonds. In this study, we investigate the effect of antioxidant components dithiothreitol (DTT), reduced glutathione (GSH), and thioredoxin (TRX) on the activation of autolytic processing of full-length HtrA1. We show that the TRX redox regulator is a physiologically relevant trigger of autolysis and identify the major autolytic forms, which are in good correlation with previous observations. In addition, the autolytic activity displays a remarkably strong preference for cysteine in the P1 position, which is novel insight into the action and specificity of HtrA1 and explains the sensitivity toward the redox state of the surrounding environment. Thus, we speculate that the autolysis of HtrA1 and the presence of various mature forms in vivo could be correlated to disease-related dysregulations in the endogenous redox balance.
Materials and Methods
Cloning and Expression
HtrA1 cDNA was obtained from Invitrogen and amplified to incorporate a C-terminal His tag separated from the C-terminus of HtrA1 by a FX protease cleavage site. The construct was cloned into the pcDNAFRT/TO vector (Invitrogen) using KpnI and XhoI restriction sites. The resulting mature translated protein (theoretical molecular mass of 50.3 kDa) is termed HtrA1. Production of HtrA1 was achieved in Freestyle 293-F cells (Life technologies) transfected with the FRT-HtrA1-His plasmid at a cell density of 1 × 106 cells/mL using polyethyleneimine (PEI). The culture supernatant was collected 48 h after transfection and dialyzed overnight at 4 °C into 20 mM Tris-HCl and 300 mM NaCl (pH 8.0). The HtrA1 protein was purified by Ni affinity chromatography using a 5 mL Hitrap Chelating column (GE Healthcare Life Sciences). The imidazole-eluted fractions were pooled and dialyzed into TBS {Tris-buffered saline [20 mM Tris-HCl and 150 mM NaCl (pH 7.4)]}. The purity and concentration was estimated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The N-terminal amino acid sequence of the 50 kDa HtrA1 band was determined to be Ser30-Ala-Pro by Edman degradation, in agreement with previous findings.32
HtrA1 Reduction and Autolytic Cleavage
Reaction mixtures for testing HtrA1 disulfide reducibility were set up with 65 μL of TBS containing 1.5 μg of phenylmethanesulfonyl fluoride (PMSF)-treated HtrA1 (2 mM PMSF for 2 h) and increasing concentrations of reducing agents DTT, GSH, and TRX, all from Sigma-Aldrich. Reaction mixtures were incubated at 37 °C for 30 min (DTT) or 1 h (GSH and TRX). The TRX concentration range was selected to reach a 1:1 ratio of the total number of cysteine bridges to TRX molecules at 4 μM TRX. After incubation, samples were then treated for 10 min with 15 mM iodoacetamide (IAA) to block free cysteines and to scavenge the excess reducing agents present in the reaction mixture before SDS–PAGE. For the assessment of autolytic cleavage in the presence of reducing agents, reaction mixtures with catalytically active HtrA1 (1.5 μg) were set up with either 1 mM DTT, 1 mM GSH, or 4 μM TRX and incubated for 20 h at 37 °C. The activity was tested by titrating HtrA1 (full-length or the autolytic products) against a fixed amount of β-casein (3 μg). The proteolytic β-casein products were analyzed after incubation for 30 min at 37 °C by SDS–PAGE.
SDS–PAGE and Edman Degradation
Samples were boiled for 5 min in SDS–PAGE sample buffer in the presence or absence of reducing agents, as specified in the text and figure legends. Proteins were separated in 8 or 10 to 15% (w/v) gradient polyacrylamide gels and run using the discontinuous ammediol/glycine buffer system.33 Gels were stained with Coomassie Brilliant Blue or blotted for 20 min to an Immobilon-P membrane (Millipore) for N-terminal protein sequence analysis using a Procise 494-HT protein sequencer (Applied Biosystems).
Analysis of Autolytic Peptides by Mass Spectrometry
HtrA1 (0.5 μg, 20 μg/mL) was incubated at 37 °C in the presence or absence of 5 mM DTT for 3 or 20 h. The reaction was quenched by the addition of formic acid to a final concentration of 1%. The samples, incubated for 20 h, were pretreated with 10 mM fresh DTT for 15 min to ensure complete reduction of the disulfides before the formic acid quenching step. Then they were micropurified using C18 stage tips (Thermo Fisher Scientific), dried down in a SpeedVac, and dissolved in 0.1% formic acid for liquid chromatography–tandem mass spectrometry analysis. The samples were run three consecutive times on an AB Sciex TripleTOF 5600 instrument. Data analysis was conducted using the Mascot search engine with a peptide fragment mass tolerance of 0.2 Da. Only peptides with a significance score of <0.01 and an ion score cutoff of >30 were considered reliable hits and included in the final list of identified HtrA1 N-terminal peptides. For an autolytic cut site to be valid, the given P1 residue had to be identified in at least two of the three replicates. MS Data Miner34 was used for the extraction of relevant data from the Mascot search file.
Results
Reducing Agents Differentially Affect HtrA1 Disulfide Bridges
Full-length HtrA1 was expressed in the Freestyle 293 human cell line and purified by Ni-NTA chromatography. To explore the stability of the disulfides in the N-domain, we analyzed the susceptibility of PMSF-inhibited HtrA1 to reduction by DTT, GSH, or TRX followed by nonreducing SDS–PAGE (Figure 2). DTT and TRX but not GSH effectively led to a concentration-dependent HtrA1 reduction, which was evident in the disappearance of the oxidized HtrA1 band (Figure 2). The reduction of HtrA1 happened gradually as a smear between the fully oxidized band (Figure 2A lane "0") and the fully reduced control (Figure 2A, lane "R") was clearly visible with increasing concentration of reductant. Higher-molecular mass HtrA1 bands were also observed, most likely because of reoxidation and formation of intermolecular disulfides during the incubation (not shown). This was more pronounced following the incubation with TRX, where the concentration of the oxidant was lower. The experiment showed that TRX was able to reduce the disulfide bridges in the N-domain of HtrA1. This has previously been shown to occur in other proteins by an intramolecular disulfide exchange cascade,35 and a similar mechanism could be utilized during the TRX-mediated reduction of HtrA1.
Figure 2.

Stability of HtrA1 disulfide bonds toward reducing agents. HtrA1 (1.5 μg in each lane) was incubated with freshly prepared reducing agents at the indicated concentrations for either 30 min (DTT) or 1 h (TRX and GSH) at 37 °C. Samples were then alkylated with IAA and analyzed by nonreducing SDS–PAGE using 8% gels. The reduction of HtrA1 by the different agents is reflected by the disappearance of protein from the fully oxidized band. For 4 μM TRX reduction, large cross-linked species were present at the top of the gel, representing reoxidized intermolecular disulfides and thereby explaining the apparent lack of protein at the expected sizes. A reduced control sample (R) was boiled in the presence of 5 mM DTT and subsequently alkylated with IAA before gel electrophoresis. The asterisk indicates a lower-molecular mass form of HtrA1 that is released upon reduction, representing a portion of HtrA1 that is nicked at loop residue Arg-103 or Arg-104 during cellular production and purification. The reduction of the disulfide bridges in HtrA1 is effectively achieved by DTT and TRX but not by GSH.
Autolysis Correlates with the Reduction of Disulfide Bonds
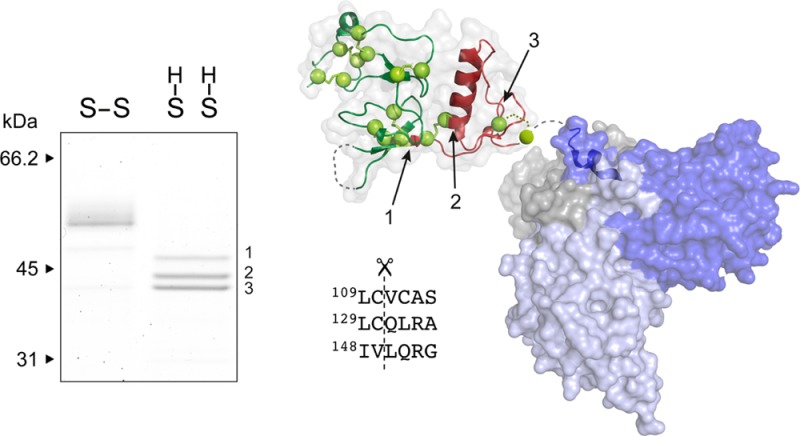
The consequence of disulfide bridge reduction was investigated by incubating active HtrA1 for 20 h in the presence of 1 mM DTT, 1 mM GSH, or 4 μM TRX and analyzing the reaction products by SDS–PAGE (Figure 3). The analysis revealed three major bands with lower molecular masses when the incubation was conducted in the presence of DTT or TRX. GSH did not display noticeable differences with respect to the control; however, if the concentration was increased to 10 mM GSH, a small fraction of HtrA1 was also found to be processed to the same three major bands (data not shown). The proteolytic activity of the truncated forms was similar to that of intact HtrA1 as judged by the ability to hydrolyze β-casein (Figure 4). This was in support of a neglectable effect of the N-domain on protease activity. The three autolytic forms were then subjected to Edman degradation to determine the new N-termini resulting from the major autolytic cuts. These were found to be Val-111 (top band, cleavage site LC↓VC), Gln-131 (middle band, cleavage site LC↓QL), and Leu-150 (lower band, cleavage site IV↓LQ), resulting in theoretical molecular masses of 41.5, 39.5, and 37.2 kDa, respectively. While the 37.2 kDa form contained Cys-155, the two larger forms contained several cysteines. The presence of three bands indicated that autolysis was incomplete, and we speculated that reoxidation of cysteine residues during the 20 h incubation was hampering autolysis. This was investigated by incubating HtrA1 for 72 h in the presence of DTT (5 mM) with the addition of fresh DTT (5 mM) after 24 and 48 h. The results showed that HtrA1 autolysis under these conditions proceeded to the 37.2 kDa band, confirming the need for reduced cysteines in the process and explaining the lack of autolysis prior to disulfide reduction (Figure 5).
Figure 3.

Disulfide bond reduction triggers HTRA1 autolysis. To active HtrA1 (1.5 μg) was added a reducing agent (1 mM DTT, 1 mM GSH, or 4 μM TRX) and the mixture was left for 20 h at 37 °C and subsequently analyzed by nonreducing SDS–PAGE using a 5 to 15% gradient gel. Controls were included for the effect of HtrA1 activity (lane 1, PMSF-treated) and for the effect of the reducing agent (lane 5, unreduced HtrA1). TRX alone was included for comparison (lane 6). The reduction of disulfide bridges led to effective HtrA1 autolysis when proteolytic activity was not inhibited, resulting in three major autolytic products for DTT- and TRX-treated samples. The presence of a smear in lane 1 and a faint high-molecular mass band around 100 kDa (dimeric species) was caused by reoxidation of disulfide bridges by a lack of reducing power as a consequence of DTT oxidation over time.
Figure 4.

Autolytic forms display activity similar to that of full-length HtrA1. The activity of full-length HtrA1 and its autolytic products was tested by incubating β-casein (3 μg) with increasing amounts of HtrA1 (preincubated for 20 h with or without DTT). After 30 min, degradation products of β-casein were easily identified, and no significant difference in the intensity or sizes of the products was observed between full-length HtrA1 and the autolytically processed versions. Assays were also performed after incubation of β-casein with HtrA1 for 45 min or 1 h, giving similar results (data not shown).
Figure 5.

Continued reduction and incubation provides full HtrA1 autolysis. HtrA1 was incubated with 5 mM DTT, and 1.5 μg aliquots were removed at indicated time points, treated with 15 mM IAA for 15 min, and then boiled in SDS–PAGE sample buffer to block further autolysis. Additional 5 mM DTT was added after 24 and 48 h, providing fresh reducing power. Unreduced HtrA1 was included for comparison after incubation for 72 h. The continued HtrA1 autolysis in the presence of sufficient reducing power led to more complete trimming to the smallest of the three major autolytic forms.
Autolytic Cleavage Sites Reveal P1 Specificity for Cysteine
The identification of the major autolytic cut sites indicated that HtrA1 cleaves efficiently with a cysteine in the P1 position. This specificity had not previously been described for HtrA1, and to gain further insight into HtrA1 autolytic P1 preferences, we used mass spectrometry to analyze the N-domain (residues 30–157) fragment peptide space generated after incubation with DTT for 3 or 12 h. The weighted frequency of the P1 occurrences from Ser-30 to Gln-157 is presented in Table 1. Indeed, cysteine was found to be one of the preferred cleavage sites with cleavage at 7 of 16 positions in the N-domain. The other identified P1 cleavage sites aligned well with previous reports on HtrA1 specificity.29 Evidently, many autolytic peptides had already been generated after incubation for 3 h, suggesting rapid activation of autolysis upon disulfide bridge reduction. The central role of cysteines in this process probed us to investigate if general reduction of disulfide bridges in other cysteine-rich proteins facilitated proteolysis by HtrA1; however, no significant cleavage of reduced bovine pancreatic trypsin inhibitor (8 cysteines) or reduced human serum albumin (34 cysteines) was seen after incubation at 37 °C for 3 h (data not shown). This could indicate that autolysis by design is an inherent feature of HtrA1 controlled by its cysteine specificity.
Table 1. Unique Autolytic Cuts in the N-Terminal Domain Observed by Mass Spectrometrya.
| no.
of unique cuts |
weighted
frequency |
||||
|---|---|---|---|---|---|
| type (P1) | total no. in the N-domain | incubation for 3 h at 37 °C with 5 mM DTT | incubation for 20 h at 37 °C with 5 mM DTT | incubation for 3 h at 37 °C with 5 mM DTT | incubation for 20 h at 37 °C with 5 mM DTT |
| C | 16 | 6 | 7 | 0.38 | 0.44 |
| A | 19 | 6 | 4 | 0.32 | 0.21 |
| L | 8 | 4 | 2 | 0.50 | 0.25 |
| Q | 7 | 2 | 2 | 0.28 | 0.29 |
| R | 13 | 2 | 2 | 0.15 | 0.15 |
| T | 2 | 1 | 1 | 0.50 | 0.50 |
| V | 9 | 1 | 1 | 0.11 | 0.11 |
The position of each autolytic cut was identified by analysis of the generated peptides, and the unique cuts refer to the P1 residue type, looking exclusively at the N-domain (residues 30–157). The weighted frequency = (the number of unique cuts)/(total number in the N-domain).
Discussion
Autolysis of HtrA family members has been observed previously, but the functional consequences and triggers of autolysis have been described only to a very limited extent. This study shows that HtrA1 autolysis is linked to the integrity of the disulfide bonds in the N-domain. Following reducing conditions, three major bands were observed via SDS–PAGE, corresponding to fragments starting at Val-111 (top band), Gln-131 (middle band), and Leu-150 (lower band) (Figure 3), in agreement with the forms observed in vivo.26,36 The cleavage site at Val-111 is located directly at the border of the IGFBP-like and Kazal-like domain (Figure 6) and is represented mostly at shorter incubation times. Further proteolysis produces the Gln-131 and Leu-150 N-termini, suggesting that initial proteolytic events are required for effective exposure to continued autolysis. The Leu-150 form (lower band) contains Cys-155 and thus differs from commonly produced ΔN-HtrA1 that lacks this residue. The loss of disulfide bridge integrity is likely to compromise the structural stability of the N-domain, possibly leading to partial unfolding. However, the sequential autolysis toward smaller autolytic forms suggests that the process is guided rather than being a random process facilitated by unfolding. Thus, our findings indicate that the N-domain is proteolytically processed to various degrees, correlated to the exposure time in an effective reductive environment that exposes suitable cysteine cleavage sites.
Figure 6.
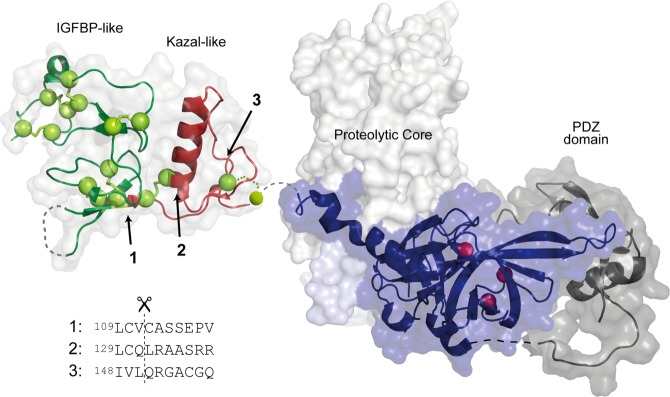
Structural representation of HtrA1 with the indicated locations of the three primary N-termini generated by autolysis. The HtrA1 domain architecture is visualized by representation of the structures for the N-domain [Protein Data Bank (PDB) entry 3TJQ(30)], the trimeric proteolytic core (PDB entry 3NWU(29)), and the PDZ domain (PDB entry 2YTW(48)). Disulfide bridges are shown as yellow spheres, and the catalytic triad is shown as red spheres. The cut sites resulting in the three major autolytic forms are numbered from 1 to 3, and their position in the folded N-domain is indicated with arrows. The new N-termini were identified by Edman degradation, and the surrounding sequence for each cut site is shown below the structure. Cut sites 1 and 2 are the result of cleavages with cysteine at position P1.
Intracellular HtrA1 is exposed to the ubiquitous cellular redox regulators TRX, GSH, and glutaredoxin. Of particular interest in this study is the finding that TRX potentiates N-domain degradation, supporting a correlation among intracellular HtrA1, redox balance, and autolysis. Furthermore, it suggests that the presence of HtrA1 autolytic forms is indicative of high intracellular (or extracellular) endogenous TRX levels. Most studies, in which HtrA1 autolysis has been observed, have been conducted in cancer cell lines, primarily to investigate the pro-apoptotic behavior of HtrA1 and the consequences of HtrA1 knock-down or forced expression.8,26,36,37 It is important to note that increased intracellular levels of TRX and TRX reductase have been associated with several cancer cell types, providing an explanation for the observed autolysis of HtrA1 in such settings.38−42 Treatment with the chemotherapeutic agents cisplatin and paclitaxel in ovarian cancer cell lines leads to upregulation of endogenous intracellular HtrA1 (and HtrA2), and this stimulates apoptosis through the targeted breakdown of XIAP.37 Interestingly, a potential role for autolytic products of HtrA1 in the induction of apoptosis has been suggested because the expression of ΔN-HtrA1 in SKOV3 cells led to an increased level of apoptosis compared to that of WT HtrA1.17,36 Recently, reduced levels of the autolytic ∼38 kDa product of HtrA1 were found to correlate with neoplastic bladder tissue undergoing cancerous transformation, which indeed highlights the importance of understanding the functional maturation of HtrA1 and the potential prognostic value of the autolytic forms.28
Several physiological and pathological conditions may result in increased amounts of local or systemic intra- and extracellular TRX, at levels that directly can impact HtrA1 autolysis. This is likely to be the case in human placentas complicated by preeclampsia where the presence of an autolytic 31 kDa form of HtrA1 correlates with the severity of the condition.18 Preeclamptic placentas display alterations in the global redox balance, and the levels of antioxidant molecules (TRX and glutaredoxin) are elevated in response to increased oxidative insults,43,44 thereby providing a possible explanation for the enhanced HtrA1 autolysis. In general, plasma levels of all HtrA family members are regulated during gestation,45 and an autolytic form of HtrA3 (contains an N-domain similar to HtrA1) can be observed in plasma, suggesting that autolysis goes beyond the intracellular environment.46,47 For HtrA1, it still remains to be established whether direct correlations exist between autolysis and intra- or extracellular location and how functional differences of the forms play a role.
On the basis of the findings in this study, we suggest that the observed autolysis of HtrA1 correlates with the redox balance of the system, in particular the levels of TRX. Our data show that HtrA1 specifically cleaves residues in its N-terminal domain after reduction of cysteine residues. Whether this cysteine cleavage property is used in other cellular settings is yet to be explored. Future studies should aim to resolve the functional aspect of the N-domain of HtrA1 but also how this secreted protein ends up in the intracellular compartment and whether such an event is linked to autolysis in any way.
Glossary
Abbreviations
- DTT
dithiothreitol
- GSH
reduced glutathione
- HtrA1
high-temperature requirement A1
- IAA
iodoacetamide
- IGFBP
insulin-like growth factor binding protein
- TBS
Tris-buffered saline
- TRX
thioredoxin
- WT
wild type
- XIAP
X-linked inhibitor of apoptosis.
The authors declare no competing financial interest.
This work was supported by grants from the Danish National Research Foundation, the Danish Research Council for Strategic Research/Natural Sciences, and the National Eye Institute (R01 EY012712).
Funding Statement
National Institutes of Health, United States
References
- Canfield A. E.; Hadfield K. D.; Rock C. F.; Wylie E. C.; Wilkinson F. L. (2007) HtrA1: A novel regulator of physiological and pathological matrix mineralization?. Biochem. Soc. Trans. 35, 669–671. [DOI] [PubMed] [Google Scholar]
- Campioni M.; Severino A.; Manente L.; De Luca A.; La Porta R.; Vitiello A.; Fiore P.; Toldo S.; Spugnini E. P.; Paggi M. G.; Baldi A. (2011) Identification of protein-protein interactions of human HtrA1. Front. Biosci. 3, 1493–1499. [DOI] [PubMed] [Google Scholar]
- De Luca A.; De Falco M.; Fedele V.; Cobellis L.; Mastrogiacomo A.; Laforgia V.; Tuduce I. L.; Campioni M.; Giraldi D.; Paggi M. G.; Baldi A. (2004) The serine protease HtrA1 is upregulated in the human placenta during pregnancy. J. Histochem. Cytochem. 52, 885–892. [DOI] [PubMed] [Google Scholar]
- Hadfield K. D.; Rock C. F.; Inkson C. A.; Dallas S. L.; Sudre L.; Wallis G. A.; Boot-Handford R. P.; Canfield A. E. (2008) HtrA1 inhibits mineral deposition by osteoblasts: Requirement for the protease and PDZ domains. J. Biol. Chem. 283, 5928–5938. [DOI] [PubMed] [Google Scholar]
- Chamberland A.; Wang E.; Jones A. R.; Collins-Racie L. A.; LaVallie E. R.; Huang Y.; Liu L.; Morris E. A.; Flannery C. R.; Yang Z. (2009) Identification of a novel HtrA1-susceptible cleavage site in human aggrecan: Evidence for the involvement of HtrA1 in aggrecan proteolysis in vivo. J. Biol. Chem. 284, 27352–27359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay S.; Maubert E.; Lebeurrier N.; Tennstaedt A.; Campioni M.; Docagne F.; Gabriel C.; Dauphinot L.; Potier M. C.; Ehrmann M.; Baldi A.; Vivien D. (2008) HtrA1-dependent proteolysis of TGF-β controls both neuronal maturation and developmental survival. Cell Death Differ. 15, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Oka C.; Tsujimoto R.; Kajikawa M.; Koshiba-Takeuchi K.; Ina J.; Yano M.; Tsuchiya A.; Ueta Y.; Soma A.; Kanda H.; Matsumoto M.; Kawaichi M. (2004) HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development 131, 1041–1053. [DOI] [PubMed] [Google Scholar]
- He X.; Ota T.; Liu P.; Su C.; Chien J.; Shridhar V. (2010) Downregulation of HtrA1 promotes resistance to anoikis and peritoneal dissemination of ovarian cancer cells. Cancer Res. 70, 3109–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.-Y.; Kim H.-Y.; Kim H.-T.; Moon J.-M.; Kim C.-H.; Kang S.; Rhim H. (2012) HtrA1 Is a Novel Antagonist Controlling Fibroblast Growth Factor (FGF) Signaling via Cleavage of FGF8. Mol. Cell. Biol. 32, 4482–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.; Khurana A.; Maguire J. L.; Chien J.; Shridhar V. (2012) HtrA1 sensitizes ovarian cancer cells to cisplatin-induced cytotoxicity by targeting XIAP for degradation. Int. J. Cancer 130, 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K.; Shiga A.; Fukutake T.; Nozaki H.; Miyashita A.; Yokoseki A.; Kawata H.; Koyama A.; Arima K.; Takahashi T.; Ikeda M.; Shiota H.; Tamura M.; Shimoe Y.; Hirayama M.; Arisato T.; Yanagawa S.; Tanaka A.; Nakano I.; Ikeda S.-i.; Yoshida Y.; Yamamoto T.; Ikeuchi T.; Kuwano R.; Nishizawa M.; Tsuji S.; Onodera O. (2009) Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 360, 1729–1739. [DOI] [PubMed] [Google Scholar]
- Jones A.; Kumar S.; Zhang N.; Tong Z.; Yang J.-H.; Watt C.; Anderson J.; Amrita; Fillerup H.; McCloskey M.; Luo L.; Yang Z.; Ambati B.; Marc R.; Oka C.; Zhang K.; Fu Y. (2011) Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 14578–14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deangelis M. M.; Ji F.; Adams S.; Morrison M. A.; Harring A. J.; Sweeney M. O.; Capone A.; Miller J. W.; Dryja T. P.; Ott J.; Kim I. K. (2008) Alleles in the HtrA serine peptidase 1 gene alter the risk of neovascular age-related macular degeneration. Ophthalmology 115, 1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S. I.; Carozza M.; Klein M.; Nantermet P.; Luk D.; Crowl R. M. (1998) Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J. Biol. Chem. 273, 34406–34412. [DOI] [PubMed] [Google Scholar]
- Grau S.; Baldi A.; Bussani R.; Tian X.; Stefanescu R.; Przybylski M.; Richards P.; Jones S. A.; Shridhar V.; Clausen T.; Ehrmann M. (2005) Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc. Natl. Acad. Sci. U.S.A. 102, 6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennstaedt A.; Poepsel S.; Truebestein L.; Hauske P.; Brockmann A.; Schmidt N.; Irle I.; Sacca B.; Niemeyer C. M.; Brandt R.; Ksiezak-Reding H.; Tirniceriu A. L.; Egensperger R.; Baldi A.; Dehmelt L.; Kaiser M.; Huber R.; Clausen T.; Ehrmann M. (2012) Human high temperature requirement serine protease A1 (HTRA1) degrades tau protein aggregates. J. Biol. Chem. 287, 20931–20941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karring H.; Runager K.; Thøgersen I. B.; Klintworth G. K.; Højrup P.; Enghild J. J. (2012) Composition and proteolytic processing of corneal deposits associated with mutations in the TGFBI gene. Exp. Eye Res. 96, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi T.; Marzioni D.; Giannubilo S.; Quaranta A.; Crescimanno C.; De Luca A.; Baldi A.; Todros T.; Tranquilli A. L.; Castellucci M. (2009) Expression patterns of two serine protease HtrA1 forms in human placentas complicated by preeclampsia with and without intrauterine growth restriction. Placenta 30, 35–40. [DOI] [PubMed] [Google Scholar]
- Zong L.; Gou W.; Shao W.; Huang P.; Li C. (2012) Changes in the Level of Serum High-Temperature Requirement A1 (HtrA1) during Pregnancy and Its Relationship to Preeclampsia. Hypertens. Pregnancy 31, 389–397. [DOI] [PubMed] [Google Scholar]
- Baldi A.; De Luca A.; Morini M.; Battista T.; Felsani A.; Baldi F.; Catricalà C.; Amantea A.; Noonan D. M.; Albini A.; Natali P. G.; Lombardi D.; Paggi M. G. (2002) The HtrA1 serine protease is down-regulated during human melanoma progression and represses growth of metastatic melanoma cells. Oncogene 21, 6684–6688. [DOI] [PubMed] [Google Scholar]
- Chien J.; Campioni M.; Shridhar V.; Baldi A. (2009) HtrA serine proteases as potential therapeutic targets in cancer. Curr. Cancer Drug Targets 9, 451–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden M. A.; Di Nezza-Cossens L. A.; Jobling T.; Salamonsen L. A.; Nie G. (2006) Serine proteases HTRA1 and HTRA3 are down-regulated with increasing grades of human endometrial cancer. Gynecol. Oncol. 103, 253–260. [DOI] [PubMed] [Google Scholar]
- Esposito V.; Campioni M.; De Luca A.; Spugnini E. P.; Baldi F.; Cassandro R.; Mancini A.; Vincenzi B.; Groeger A.; Caputi M.; Baldi A. (2006) Analysis of HtrA1 serine protease expression in human lung cancer. Anticancer Res. 26, 3455–3459. [PubMed] [Google Scholar]
- Chien J.; He X.; Shridhar V. (2009) Identification of tubulins as substrates of serine protease HtrA1 by mixture-based oriented peptide library screening. J. Cell. Biochem. 107, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clawson G. A.; Bui V.; Xin P.; Wang N.; Pan W. (2008) Intracellular localization of the tumor suppressor HtrA1/Prss11 and its association with HPV16 E6 and E7 proteins. J. Cell. Biochem. 105, 81–88. [DOI] [PubMed] [Google Scholar]
- Chien J.; Ota T.; Aletti G.; Shridhar R.; Boccellino M.; Quagliuolo L.; Baldi A.; Shridhar V. (2009) Serine Protease HtrA1 Associates with Microtubules and Inhibits Cell Migration. Mol. Cell. Biol. 29, 4177–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni M.; Severino A.; Manente L.; Tuduce I. L.; Toldo S.; Caraglia M.; Crispi S.; Ehrmann M.; He X.; Maguire J.; De Falco M.; De Luca A.; Shridhar V.; Baldi A. (2010) The serine protease HtrA1 specifically interacts and degrades the tuberous sclerosis complex 2 protein. Mol. Cancer Res. 8, 1248–1260. [DOI] [PubMed] [Google Scholar]
- Lorenzi T.; Lorenzi M.; Altobelli E.; Marzioni D.; Mensa E.; Quaranta A.; Paolinelli F.; Morroni M.; Mazzucchelli R.; De Luca A.; Procopio A. D.; Baldi A.; Muzzonigro G.; Montironi R.; Castellucci M. (2013) HtrA1 in human urothelial bladder cancer: A secreted protein and a potential novel biomarker. Int. J. Cancer 133, 2650–2661. [DOI] [PubMed] [Google Scholar]
- Truebestein L.; Tennstaedt A.; Mönig T.; Krojer T.; Canellas F.; Kaiser M.; Clausen T.; Ehrmann M. (2011) Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat. Struct. Mol. Biol. 18, 386–388. [DOI] [PubMed] [Google Scholar]
- Eigenbrot C.; Ultsch M.; Lipari M. T.; Moran P.; Lin S. J.; Ganesan R.; Quan C.; Tom J.; Sandoval W.; van Lookeren Campagne M.; Kirchhofer D. (2012) Structural and Functional Analysis of HtrA1 and Its Subdomains. Structure 1–11. [DOI] [PubMed] [Google Scholar]
- Murwantoko; Yano M.; Ueta Y.; Murasaki A.; Kanda H.; Oka C.; Kawaichi M. (2004) Binding of proteins to the PDZ domain regulates proteolytic activity of HtrA1 serine protease. Biochem. J. 381, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J.; Clemmons D. R.; Smeekens S. (2005) Expression and characterization of a serine protease that preferentially cleaves insulin-like growth factor binding protein-5. J. Cell. Biochem. 94, 470–484. [DOI] [PubMed] [Google Scholar]
- Bury A. F. (1981) Analysis of Protein and Peptide Mixtures: Evaluation of 3 Sodium Dodecyl Sulfate-Polyacrylamide Gel-Electrophoresis Buffer Systems. J. Chromatogr. 213, 491–500. [Google Scholar]
- Dyrlund T. F.; Poulsen E. T.; Scavenius C.; Sanggaard K. W.; Enghild J. J. (2012) MS Data Miner: A web-based software tool to analyze, compare, and share mass spectrometry protein identifications. Proteomics 12, 2792–2796. [DOI] [PubMed] [Google Scholar]
- Messens J.; Van Molle I.; Vanhaesebrouck P.; Limbourg M.; Van Belle K.; Wahni K.; Martins J. C.; Loris R.; Wyns L. (2004) How thioredoxin can reduce a buried disulphide bond. J. Mol. Biol. 339, 527–537. [DOI] [PubMed] [Google Scholar]
- Chien J. (2006) Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity. J. Clin. Invest. 116, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien J.; Aletti G.; Baldi A.; Catalano V.; Muretto P.; Keeney G. L.; Kalli K. R.; Staub J.; Ehrmann M.; Cliby W. A.; Lee Y. K.; Bible K. C.; Hartmann L. C.; Kaufmann S. H.; Shridhar V. (2006) Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity. J. Clin. Invest. 116, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z.-H.; Cao H.-Q.; Hu Y.-B.; Wen J.-F.; Zhou J.-H. (2011) TRX is up-regulated by fibroblast growth factor-2 in lung carcinoma. APMIS 119, 57–65. [DOI] [PubMed] [Google Scholar]
- Iwasawa S.; Yamano Y.; Takiguchi Y.; Tanzawa H.; Tatsumi K.; Uzawa K. (2011) Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncol. Rep. 25, 637–644. [DOI] [PubMed] [Google Scholar]
- Kakolyris S.; Giatromanolaki A.; Koukourakis M.; Powis G.; Souglakos J.; Sivridis E.; Georgoulias V.; Gatter K. C.; Harris A. L. (2001) Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin. Cancer Res. 7, 3087–3091. [PubMed] [Google Scholar]
- Lincoln D. T.; Al-Yatama F.; Mohammed F. M. A.; Al-Banaw A. G.; Al-Bader M.; Burge M.; Sinowatz F.; Singal P. K. (2010) Thioredoxin and thioredoxin reductase expression in thyroid cancer depends on tumour aggressiveness. Anticancer Res. 30, 767–775. [PubMed] [Google Scholar]
- Yamada M.; Tomida A.; Yoshikawa H.; Taketani Y.; Tsuruo T. (1996) Increased expression of thioredoxin/adult T-cell leukemia-derived factor in cisplatin-resistant human cancer cell lines. Clin. Cancer Res. 2, 427–432. [PubMed] [Google Scholar]
- Takagi Y.; Nikaido T.; Toki T.; Kita N.; Kanai M.; Ashida T.; Ohira S.; Konishi I. (2004) Levels of oxidative stress and redox-related molecules in the placenta in preeclampsia and fetal growth restriction. Virchows Arch. 444, 49–55. [DOI] [PubMed] [Google Scholar]
- Shibata E.; Ejima K.; Nanri H.; Toki N.; Koyama C.; Ikeda M.; Kashimura M. (2001) Enhanced protein levels of protein thiol/disulphide oxidoreductases in placentae from pre-eclamptic subjects. Placenta 22, 566–572. [DOI] [PubMed] [Google Scholar]
- Marzioni D.; Lorenzi T.; Altobelli E.; Giannubilo S. R.; Paolinelli F.; Tersigni C.; Crescimanno C.; Monsurrò V.; Tranquilli A. L.; Di Simone N.; Castellucci M. (2012) Alterations of maternal plasma HTRA1 level in preeclampsia complicated by IUGR. Placenta 33, 1036–1038. [DOI] [PubMed] [Google Scholar]
- Li Y.; Puryer M.; Lin E.; Hale K.; Salamonsen L. A.; Manuelpillai U.; Tong S.; Chan W.; Wallace E. M.; Nie G. (2011) Placental HtrA3 is regulated by oxygen tension and serum levels are altered during early pregnancy in women destined to develop preeclampsia. J. Clin. Endocrinol. Metab. 96, 403–411. [DOI] [PubMed] [Google Scholar]
- Nie G.; Li Y.; Hale K.; Okada H.; Manuelpillai U.; Wallace E. M.; Salamonsen L. A. (2006) Serine peptidase HTRA3 is closely associated with human placental development and is elevated in pregnancy serum. Biol. Reprod. 74, 366–374. [DOI] [PubMed] [Google Scholar]
- Runyon S. T.; Zhang Y.; Appleton B. A.; Sazinsky S. L.; Wu P.; Pan B.; Wiesmann C.; Skelton N. J.; Sidhu S. S. (2007) Structural and functional analysis of the PDZ domains of human HtrA1 and HtrA3. Protein Sci. 16, 2454–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]