

Abstract
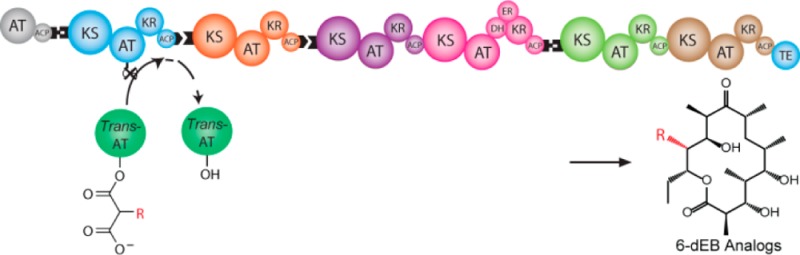
Due to their pivotal role in extender unit selection during polyketide biosynthesis, acyltransferase (AT) domains are important engineering targets. A subset of assembly line polyketide synthases (PKSs) are serviced by discrete, trans-acting ATs. Theoretically, these trans-ATs can complement an inactivated cis-AT, promoting introduction of a noncognate extender unit. This approach requires a better understanding of the substrate specificity and catalytic mechanism of naturally occurring trans-ATs. We kinetically analyzed trans-ATs from the disorazole and kirromycin synthases and compared them to a representative cis-AT from the 6-deoxyerythronolide B synthase (DEBS). During transacylation, the disorazole AT favored malonyl-CoA over methylmalonyl-CoA by >40000-fold, whereas the kirromycin AT favored ethylmalonyl-CoA over methylmalonyl-CoA by 20-fold. Conversely, the disorazole AT had broader specificity than its kirromycin counterpart for acyl carrier protein (ACP) substrates. The presence of the ACP had little effect on the specificity (kcat/KM) of the cis-AT domain for carboxyacyl-CoA substrates but had a marked influence on the corresponding specificity parameters for the trans-ATs, suggesting that these enzymes do not act strictly by a canonical ping-pong mechanism. To investigate the relevance of the kinetic analysis of isolated ATs in the context of intact PKSs, we complemented an in vitro AT-null DEBS assembly line with either trans-AT. Whereas the disorazole AT efficiently complemented the mutant PKS at substoichiometric protein ratios, the kirromycin AT was considerably less effective. Our findings suggest that knowledge of both carboxyacyl-CoA and ACP specificity is critical to the choice of a trans-AT in combination with a mutant PKS to generate novel polyketides.
Polyketides are a large class of medicinally relevant natural products, many of which are produced in an assembly line fashion by multimodular polyketide synthases (PKSs). Each PKS module is composed of several enzymatic domains that together are responsible for one round of polyketide chain elongation and processing. The acyltransferase (AT) domains within each module are responsible for selection of coenzyme A (CoA)-linked extender units for incorporation into the growing polyketide chain.1 Because this specificity influences the structural diversity and biological activity of the natural product, AT domains are often targeted in the engineering of PKSs for the production of novel polyketides.2
The reaction catalyzed by AT domains is shown in Scheme 1. Each AT transfers an acyl group from an α-carboxyacyl-CoA (carboxyacyl-CoA) extender unit to the phosphopantetheine arm of an acyl carrier protein (ACP) domain. Assembly line PKSs are known to harbor two types of AT domains. In systems such as the 6-deoxyerythronolide B synthase (DEBS), each AT domain is paired with its own ACP domain; these ATs are typically referred to as cis-acting AT domains (Scheme 1A). In contrast, trans-acting ATs exist as stand-alone enzymes; they catalyze extender unit transfer onto one or more ACPs of a multimodular PKS assembly line (Scheme 1B,C).3 Examples of PKSs harboring trans-acting AT domains include assembly lines that synthesize disorazole,4 leinamycin,5 and bryostatin.6
Scheme 1. Reactions Catalyzed by Three Acyltransferase (AT) Domains.

(A) The cis-AT domain from DEBS module 3 transfers a methylmalonyl extender unit from the corresponding carboxyacyl-CoA precursor onto the phosphopantetheine arm of the ACP within the same module; (B) the trans-AT from the DSZS loads malonyl extender units onto the phosphopantetheine arms of all the ACP domains of this polyketide synthase; module 1 of DSZS is shown as a representative example; (C) the trans-AT from the kirromycin synthase, KirCII, exclusively transfers an ethylmalonyl extender unit from the corresponding carboxyacyl-CoA precursor onto the phosphopantetheine arm of the ACP in module 5; the remaining ACP domains of the kirromycin polyketide assembly line are poor substrates for this trans-acting AT21.
trans-Acting AT domains have the potential to be useful engineering tools: a cis-AT could in principle be inactivated by site-directed mutagenesis, and the resulting PKS could be complemented with a trans-AT that transfers a different extender unit onto the ACP domain of the module. However, rational implementation of such a strategy requires a firm understanding of the carboxyacyl-CoA and ACP specificities of these AT domains.
Most trans-AT domains are specific for malonyl-CoA. A trans-AT involved in the biosynthesis of kirromycin, however, was shown to preferentially utilize ethylmalonyl-CoA extender units.7 Indeed, this enzyme was able to load its cognate ACP with a range of non-natural extender units.8trans-Acting ATs may also have good tolerance for unnatural ACP substrates.9,10 For example, the AT from the disorazole synthase (DSZS) had higher specificity constants (kcat/KM) for ACP domains from DEBS than did an AT from the DEBS assembly line.10 In this study, we therefore sought to exploit a recently developed fluorometric assay11 to compare the specificity of the DSZS AT, the ethylmalonyl-specific kirromycin AT (KirCII), and a representative AT domain from DEBS for alternative carboxyacyl-CoA and ACP substrates. In turn, these quantitative insights led us to predict the efficacy with which the two trans-AT domains would complement a mutant of the DEBS assembly line in which a single AT domain had been catalytically inactivated. Our predictions were verified in vitro using a recently established LC-MS assay.12
Experimental Procedures
Reagents and Chemicals
Protein Expression and Purification
Isopropyl β-d-1-thiogalactopyranoside (IPTG) was from Gold Biotechnology. Ni-NTA affinity resin was from MCLab, and SDS-PAGE gradient gels were from Bio-Rad. The Hi-Trap Q anion exchange column was from GE Healthcare. The Ni-NTA superflow cartridge was from Qiagen, and centrifugal filter units were from Millipore.
Synthesis of Ethylmalonyl-CoA
All solvents and chemicals were from Fisher Scientific with the following exceptions: diethylethylmalonate was from Santa Cruz Biotechnology, and coenzyme A, thiophenol, dimethylformamide, and dicyclohexylcarbodiimide were from Sigma-Aldrich.
Fluorometric Assays
Methylmalonyl-CoA, malonyl-CoA, β-NAD+, NADH, α-ketoglutarate dehydrogenase (porcine heart) (αKGDH), α-ketoglutarate, thiamine pyrophosphate (TPP), and EDTA were from Sigma-Aldrich. TCEP was from CalBiochem and BSA was from New England Biolabs. The 96-well microtiter plates (black polystyrene, flat bottom, half area, nonbinding surface) were from Corning.
In vitrotrans-AT Complementation of DEBS
NADPH, propionyl-CoA, methylmalonic acid, malonic acid, and magnesium chloride hexahydrate were from Sigma-Aldrich. ATP was from Teknova.
Constructs for Protein Expression
DEBS AT3, ACP3, and ACP6 were expressed from plasmids pAYC47,13 pVYA05,14 and pFW55,10 respectively. The DSZS AT and ACP1 were expressed from plasmids pFW310 and pFW69,15 respectively. Proteins required to reconstitute DEBS, including the loading didomain, module 1, module 2, DEBS2, and DEBS3 were expressed from pBL12, pBL13, pBL36, pFW98, and pFW100, respectively.12 The genes for Streptomyces coelicolor MatB and methylmalonyl-CoA epimerase, both in pET28 expression vectors, were gifts from Professor Michelle Chang’s laboratory at the University of California, Berkeley.
All remaining proteins were expressed from DNA constructs prepared for this study. Genes encoding KirCII and the ACP domains from modules 4 and 5 of the kirromycin synthase were purchased from Genscript as codon-optimized sequences for expression in Escherichia coli. These synthetic sequences can be found in Table S1, Supporting Information. Expression plasmids for KirCII, ACP4, and ACP5 (pKW29, pFW139, and pFW140 respectively) were engineered by inserting the corresponding NdeI–EcoRI fragments into a pET28 vector. The expression plasmid encoding DSZS ACP7.1 was obtained by PCR amplification of the relevant gene as an NdeI–EcoRI fragment from pKOS254–190.4,4 followed by insertion into a pET28 vector to obtain pBD61. The expression plasmid encoding DEBS module 1 harboring a Ser → Ala mutation in the AT active site was constructed using Quikchange site-directed mutagenesis (Agilent) on plasmid pBL13 as the template,12 resulting in plasmid pTR1. The identity of all constructs was confirmed by DNA sequencing.
Protein Expression and Purification
All plasmids were introduced into E. coli BAP116 cells by electroporation. DEBS AT3 and the DSZS AT were expressed and purified as previously described.10,11 KirCII was purified according to a recent reference7 with slight modifications. Cells were grown in LB medium at 37 °C until the culture optical density reached 0.6. The culture was cooled to 16 °C, induced with 0.2 mM IPTG, and grown for an additional 15 h. Cells were harvested by centrifugation (4420g for 15 min). The cell pellet was resuspended in lysis buffer (50 mM Tris-HCl, pH 7.7, 500 mM NaCl, 20% glycerol) and lysed by sonication (5 × 1 min, on ice). After centrifugation at 42700g for 60 min, the supernatant was incubated with Ni-NTA agarose for 1 h. The resin was washed with 10 column volumes of wash buffer (50 mM Tris-HCl, pH 7.7, 500 mM NaCl, 20 mM imidazole), and the bound protein was eluted with four column volumes of elution buffer (50 mM Tris-HCl, pH 7.7, 500 mM NaCl, 400 mM imidazole). The eluent was concentrated using a 30000 MW cutoff centrifugal filter and redissolved in 25 mL of buffer A (50 mM Tris-HCl, pH 7.7, 500 mM NaCl, 20% glycerol, 10 mM imidazole). The solution was applied to a Ni-NTA Superflow cartridge, and protein was eluted with a linear gradient of increasing imidazole concentration (10–400 mM). Fractions containing KirCII were pooled and concentrated in storage buffer (50 mM Tris-HCl, pH 7.7, 500 mM NaCl, 8% glycerol).
To express ACP proteins, plasmids were introduced into E. coli BAP116 cells by electroporation. Holo-DEBS ACP3, DEBS ACP6, DSZS ACP1, DSZS ACP7.1, Kirr ACP4, and Kirr ACP5 were all expressed and purified using a previously described protocol.11 Complete phosphopantetheinylation of ACP proteins was confirmed by MALDI-TOF mass spectrometry.
The DEBS loading didomain, module 1, module 2, DEBS2, DEBS3, and module 1-ATnull proteins were expressed and purified as previously described.12
Acyltransferase Assays
Assays were performed as previously described11 with the following modifications. Assays were run in 96-well microtiter plates, and NADH fluorescence was monitored using a Synergy HT multimode microplate reader (BioTek). Samples were illuminated with a tungsten light source and a 360 nm filter, and fluorescence emission was monitored using a 460 nm filter. For each assay, data was collected for 5 min using the minimum interval between measurements (8 s). All measurements were made in triplicate at a minimum, and appropriate controls lacking enzyme were run in parallel.
Assay components were prepared in three different solutions. For kinetic measurements involving varying concentrations of alternative carboxyacyl-CoA substrates, solution 1 contained the ACP, α-KGDH, NAD+, TPP, and α-ketoglutarate at four times their final concentrations, solution 2 contained the carboxyacyl-CoA substrate prepared at four times its final concentration, and solution 3 contained the AT prepared at twice its final concentration. For kinetic measurements involving varying concentrations of alternative ACP substrates, solution 1 contained the carboxyacyl-CoA substrate, α-KGDH, NAD+, TPP, and α-ketoglutarate at four times their final concentrations, solution 2 contained the ACP prepared at four times its final concentration, and solution 3 contained the AT domain prepared at twice its final concentration. All solutions were prepared in 50 mM sodium phosphate buffer, pH 7.6, 10% glycerol, 5 mM TCEP, and 1 mM EDTA. The AT stock solutions also contained 0.1 mg/mL BSA. Solutions 1 and 2 (25 μL of each) were combined for approximately 1 min before addition of 50 μL of solution 3 to initiate the reaction. Final assay concentrations were 50 mM sodium phosphate, pH 7.6, 10% glycerol, 1 mM TCEP, 1 mM EDTA, 0.4 mU/μL αKGDH, 0.4 mM NAD+, 0.4 mM TPP, 2 mM α-ketoglutarate, and 0.05 mg/mL BSA. AT, ACP, and carboxyacyl-CoA concentrations were varied as described.
To measure rates of carboxyacyl-CoA hydrolysis by AT domains in the absence of an ACP cosubstrate, enzyme concentrations ranged between 0.1 and 1 μM, and carboxyacyl-CoA concentrations ranged between 0 and 200 μM. To measure acyl transfer kinetics in the presence of varying concentrations of carboxyacyl-CoA or ACP substrates, the concentration of DEBS AT3 was 0.2–0.5 μM, whereas KirCII was present at a concentration of 0.5 μM. DSZS AT was present at a concentration of 3–30 nM in assays involving malonyl-CoA, and 0.2 μM for all other acyl-CoA substrates. Individual holo-ACP and carboxyacyl-CoA concentrations ranged between 0 and 200 μM.
Data Analysis
Data was analyzed as previously described11 with several modifications. Relevant competing hydrolysis rates were subtracted from their transacylation counterparts before the calculation of the kinetic parameters describing transacylation. In analyzing data describing the transacylation of DSZS ACP1 and ACP7.1 with malonyl-CoA by the DSZS AT domain, only the linear portion of the curve was considered due to possible rate limitation by the coupling enzyme at the highest ACP concentrations. The velocity versus concentration curves were fit to the Michaelis–Menten equation using the curve-fitting functionality of GraphPad Prism.
Synthesis of Ethylmalonyl-CoA
Ethylmalonyl-CoA was synthesized as previously described17 with several modifications. For a detailed protocol, see the Supporting Information.
In Vitro Complementation of the Mutant DEBS Assembly Line by DSZS AT or KirCII
The fully reconstituted DEBS assembly line (composed of purified proteins) was assayed as previously described,12 with the AT-null version of module 1 replacing its wild-type counterpart. The DSZS AT was titrated from 0 to 1 μM in the presence of equimolar concentrations of malonate and methylmalonate (1 mM each); these diacids were converted in situ into racemic forms of their corresponding CoA monothioesters using the Streptomyces coelicolor MatB and methylmalonyl-CoA epimerase enzymes, as before. Similarly, KirCII was titrated into the AT1-null DEBS assembly line from 0 to 10 μM in the presence of equimolar concentrations of ethylmalonate and methylmalonate (1 mM each). All reactions were quenched after 3 h by the addition of ethyl acetate and analyzed by LC-MS as previously described.12 The presence of 6-deoxyerythronolide B (6-dEB) was verified by comparison of LC-MS data to that of an authentic 6-dEB standard.12 The putative 12-desmethyl-6-dEB analogue, didesmethyl-6-dEB analogues, and 12-desmethyl-12-ethyl-6-dEB analogue (along with other ethyl-6-dEB analogues produced by natural incorporation of ethylmalonate by the DEBS assembly line) were identified by first extracting the [M + Na]+ species (m/z = 395.22–395.26, m/z = 381.20–381.24, and m/z = 423.26–423.28, respectively). The presence of these analogues was then considered confirmed only if ions for at least two of the following derived species were also present: [M + H]+, [M + H – H2O]+, or [M + H – 2H2O]+. For representative extracted ion chromatograms (EIC), full mass spectra, and details on integration areas used for quantification of EIC values, please see Figure S1 (DSZS) and Figure S2 (KirCII), Supporting Information. Although the 12-desmethyl-12-ethyl-6-dEB analogue could not be identified definitively, we did observe a KirCII-dependent change in analogue ratios (Figure S2, Supporting Information).
Native and Mutant DEBS Assembly Line Control Reactions
The fully reconstituted DEBS assembly line (composed of purified proteins) was assayed as previously described,12 in the presence of 1 mM methylmalonate. The mutant DEBS assembly line in the absence of trans-AT was assayed similarly, with the AT-null version of module 1 replacing its wild-type counterpart. This reaction was also run in the presence of 1 mM methylmalonate. Reactions were run for 30 min.
Results
For this study, we chose to compare the properties of three acyltransferase (AT) domains: (i) the ethylmalonyl-CoA specific trans-AT domain from the kirromycin synthase, KirCII, due to its unique substrate specificity;7,8,18 (ii) the malonyl-CoA specific trans-AT domain from the DSZS,4,19 which was previously shown to transacylate DEBS ACP domains;10 (iii) the methylmalonyl-CoA specific cis-AT domain from module 3 of DEBS, which has been characterized both structurally20 and kinetically.11 The reactions catalyzed by each of these AT domains are shown in Scheme 1. The panel of carboxyacyl-CoA substrates included the natural substrates of each of these three enzymes. The acceptor ACP cosubstrates used in this study included: (i) the ACP domain from module 5 of the kirromycin synthase, which is the natural substrate of KirCII;7,18,21 (ii) the ACP from module 4 of the kirromycin synthase, which is not normally transacylated by KirCII;7,21,22 (iii) both ACP1 and the first ACP from module 7 (ACP7.1) of the DSZS, each of which is apparently recognized by the DSZS AT; (iv) both DEBS ACP3 and ACP6, representing preferred and nonpreferred substrates, respectively, of DEBS AT3.10,11,23
Hydrolytic Activity of cis- and trans-AT Domains
Carboxyacyl-CoA hydrolytic activity has been observed for cis-AT domains from several different PKS assembly lines.11,24 Although hydrolytic cleavage of a thioester linkage by a trans-AT-like enzyme has been proposed as a proofreading mechanism in the biosynthesis of pederin,25 the hydrolytic activity of trans-AT domains toward carboxyacyl-CoA substrates has not been examined. We therefore determined the kinetic parameters describing DSZS AT-, KirCII-, and DEBS AT3-mediated hydrolysis for a panel of three carboxyacyl-CoA substrates (Figure 1). Interestingly, the basal hydrolytic activity varies considerably among the AT domains tested. While the rates of hydrolysis (as measured by kcat/KM) of all three carboxyacyl-CoA substrates were below detectable limits for the DSZS AT, the hydrolytic rate constants of KirCII and DEBS AT3 were comparable to their transacylation rate constants for a number of the substrates tested. KirCII hydrolyzed carboxyacyl-CoA substrates relatively indiscriminately, whereas DEBS AT3 showed the highest rate of hydrolysis with its natural substrate, methylmalonyl-CoA. It is therefore unlikely that the substrate specificity of DEBS AT3 is determined by a hydrolytic mechanism, but rather likely that the enhanced hydrolytic rate simply reflects the strong substrate specificity of this cis-AT domain.11 In contrast, KirCII may utilize a hydrolytic correction mechanism in vivo so as to minimize competing incorporation of methylmalonyl or malonyl extender units.
Figure 1.

Kinetic analysis of the hydrolytic reaction catalyzed by cis- and trans-AT domains in the absence of added ACP acceptor. Note that AT-catalyzed hydrolysis involves attack of the carboxyacyl–enzyme intermediate by a solvent nucleophile rather than the phosphopantetheine arm of the ACP cosubstrate: (A) DEBS AT3; (B) KirCII. aPreviously reported data shown for comparison.11
Extender Unit Specificity in AT-Catalyzed Transacylation Reactions
To quantify the transacylation specificity of DEBS AT3, DSZS AT, and KirCII for alternative extender units, varying concentrations of carboxyacyl-CoA substrates were used in the presence of a fixed concentration of the cognate holo-ACP cosubstrate. As seen in Figure 2, all AT domains had at least 20-fold specificity for their cognate substrate. Of the three, DSZS AT had the highest substrate specificity, with a kcat/KM for malonyl-CoA approximately 5 orders of magnitude higher than the kcat/KM for either methylmalonyl-CoA or ethylmalonyl-CoA. In contrast to DEBS AT3, which could not be saturated by its cognate methylmalonyl-CoA substrate, both trans-ATs had KM values for their cognate substrates in the 2–10 μM range.
Figure 2.

Kinetic analysis of the carboxyacyl-CoA substrate dependence of the transacylation reaction catalyzed by cis- and trans-AT domains. MCoA, malonyl-CoA; MMCoA, methylmalonyl-CoA; EMCoA, ethylmalonyl-CoA. (A) DEBS AT3; (B) DSZS AT; (C) KirCII. aPreviously reported data shown for comparison.11
The data in Figure 2 also revealed an unexpected trend with respect to the specificity constants for transacylation compared with those for hydrolysis. AT-catalyzed transacylation of an ACP acceptor is normally thought to occur by a ping-pong bi-bi mechanism,26 in which the first half-reaction involves formation of a carboxyacyl–enzyme intermediate and release of the coenzyme A (CoASH) product, while the second half reaction involves nucleophilic attack on the carboxyacyl–enzyme intermediate by the terminal thiol of the phosphopantetheine arm of the ACP (Scheme 2A). Competing hydrolysis involves attack on the carboxyacyl–enzyme intermediate by a solvent nucleophile rather than the nucleophilic thiol. According to this mechanism, in the absence of high concentrations of free CoASH, the value of kcat/KM should be the same for carboxyacyl-CoA-limited transacylation and for competing hydrolysis, because the first half-reaction is effectively irreversible in either case. The experimentally determined values of kcat/KM for hydrolysis of the carboxyacyl-CoA substrate and for transfer to the ACP are essentially identical for DEBS AT3. This suggests that the ping-pong mechanism is relevant for this enzyme and the carboxyacyl-CoA specificity is primarily determined by the first half-reaction, since the kcat/KM is intrinsically a function of only those microscopic rates constants up to and including, but not following, the first irreversible step.11 Indeed, the invariability of kcat/KM as a function of the concentration (or identity) of the second substrate is a fundamental characteristic of the canonical ping-pong mechanism. Unexpectedly, however, the observed kcat/KM values for the carboxyacyl-CoA substrates of both KirCII and especially DSZS AT vary considerably when comparing the hydrolytic and transacylation reactions for each trans-AT. Both enzymes show a marked increase in the specificity constant kcat/KM for their cognate carboxyacyl-CoA substrates when the holo-ACP cosubstrate is present. Assuming that hydrolysis is a relevant reaction catalyzed by the DSZS AT (despite measured hydrolysis rates being below detectable limits) and that comparison of kcat/KM values is meaningful for this enzyme, the above observation is inconsistent with a classical ping-pong mechanism. Rather, it suggests that interaction with the ACP cosubstrate occurs prior to irreversible release of the CoASH product during transacylation by both trans-ATs. Examination of the double reciprocal plots (Lineweaver–Burk) at varying ACP concentrations could resolve these mechanistic possibilities. The low KM values (below 10 μM) of these trans-ATs for their cognate carboxyacyl-CoA substrates, however, made detection of product difficult using the coupled assay and therefore precluded accurate generation of double reciprocal plots, despite our best efforts.
Scheme 2. Reaction Mechanisms Describing AT-Catalyzed Transacylation.

(A) The traditional ping-pong bi-bi mechanism; (B) possible sequential or random ordered bi-bi mechanisms in which the ACP binds before formation of the carboxyacyl–enzyme intermediate; (C) possible sequential ordered bi-bi mechanism in which the carboxyacyl–enzyme intermediate forms before ACP binding, but the ACP must bind before CoASH release. The trans-AT domains characterized in this study appear to function by transacylation mechanisms such as those shown in parts B and C, in which ACP association occurs before the first irreversible step (•Association; ∼Covalent intermediate; −Covalent product).
ACP Specificity in AT-Catalyzed Transacylation Reactions
In order to complement a mutant PKS assembly line, a trans-AT domain must be tolerant of a range of noncognate ACP substrates. We therefore quantified the ACP specificity of each of the three AT enzymes by varying the ACP concentration in the presence of fixed concentrations of their respective cognate carboxyacyl-CoA substrates (Figure 3). The ACP specificity of KirCII is comparable to that of DEBS AT3, with both displaying approximately 20-fold preference for their cognate ACP partner compared with the next most active ACP acceptor, as measured by the relative values of kcat/KM. In contrast, the DSZS AT is considerably more promiscuous with respect to the ACP substrate, with only a modest 5–7-fold preference for its cognate ACP partners compared with other ACP proteins. This is consistent with a previous estimate of the relative ACP specificity of DSZS AT, determined by a different assay.10 Interestingly, the kcat/KM values for DSZS AT with the two DEBS ACP domains were three times greater than the kcat/KM displayed by DEBS AT3 for its native partner, DEBS ACP3. By contrast, the corresponding kcat/KM values for KirCII-mediated transacylation are substantially lower than those seen with the DSZS AT. Thus, the tolerance of DSZS AT for variations in the acceptor ACP domain clearly makes it a superior candidate for complementation of module-specific AT lesions, notwithstanding its restricted specificity for the carboxyacyl-CoA substrate. The specificity constants for trans-AT catalyzed transacylation with a noncognate assembly line ACP should therefore be considered when designing biosynthetic complementation systems, as described in the following section.
Figure 3.

Kinetic analysis of the ACP substrate dependence of the transacylation reaction catalyzed by cis and trans-AT domains. (A) DEBS AT3, (B) DSZS AT, (C) KirCII. aPreviously reported data shown for comparison.11
In Vitro Complementation of an AT-null DEBS Assembly Line with trans-Acting Acyltransferases
To test the implications of the above kinetic analysis of stand-alone AT domains on the operation of a complete PKS assembly line, we utilized the reconstituted DEBS enzyme system recently developed in our laboratory.12 For these studies, the protein harboring the native DEBS module 1 was replaced with a mutant in which the AT domain had been inactivated by an active site Ser → Ala substitution (Figures 4A and 5A). Based on the relevant kcat/KM parameters (Figures 2 and 3), we predicted that the DSZS AT would be more effective than the KirCII AT in supplying its preferred malonyl-CoA extender unit to the mutated DEBS assembly line. What could not be predicted a priori, however, was the dependence of trans-complementation on the concentration of the trans-AT or the effect of introducing a non-native extender unit in the modified diketide intermediate produced by the complemented DEBS module 1 on the net turnover rate of the entire PKS assembly line.
Figure 4.

In vitro complementation of the fully reconstituted DEBS assembly line harboring an AT1-null mutation with the trans-acting DSZS AT in the presence of equimolar malonate and methylmalonate concentrations: (A) Schematic of the overall enzyme system and the expected product, 12-desmethyl-6-dEB; (B) LC-MS analysis of product distribution ratios at varying DSZS AT concentrations. For details regarding the identification of products and extracted ion chromatogram (EIC) quantification, see Experimental Procedures. IS, internal standard, N-Boc glutamic acid benzyl ester (11.6 μM).
Figure 5.

In vitro complementation of the fully reconstituted DEBS assembly line harboring an AT1-null mutation with the trans-acting KirCII AT in the presence of equimolar ethylmalonate and methylmalonate concentrations: (A) Schematic of the overall enzyme system and the expected product, 12-desmethyl-12-ethyl-6-dEB; (B) LC-MS analysis of product distribution ratios at varying KirCII concentrations. Because DEBS incorporates ethylmalonate naturally,12 EIC quantification of the 12-desmethyl-12-ethyl-6-dEB analogue also includes other ethyl-6-dEB analogues. For details regarding the identification of products and EIC quantification, see the Experimental Procedures. IS, internal standard, N-Boc-glutamic acid benzyl ester (11.6 μM).
When the DSZS AT was titrated up to a final concentration of 1 μM in the presence of nonlimiting concentrations of methylmalonyl-CoA and malonyl-CoA and 2 μM of each DEBS protein, increasing yields of the putative 12-desmethyl-6-dEB product were observed (Figure 4B). Indeed, this 6-dEB analogue was produced at a DSZS AT concentration as low as 10 nM. Unexpectedly, the natural product, 6-dEB, was also observed under all conditions tested, even in the absence of added DSZS AT, implying that DEBS ACP1 can be transacylated at measurable efficiency by one or more of the catalytically active DEBS AT domains of the remaining five modules. The possibility that the Ser → Ala mutant of AT1 has residual activity is ruled out by our previous observation that the analogous mutation in module 2 of DEBS yields a protein with undetectable turnover capacity.27 Further, we have observed that the AT1-null mutant shows no capacity for turnover in the absence of trans-AT, based on an NADPH consumption assay similar to those performed previously12 (data not shown). Also notable is the appearance of a molecular ion corresponding to the mass of a didesmethyl-6-dEB analogue at the highest DSZS AT concentrations, suggesting competitive incorporation of a malonyl extender unit in other modules of the engineered DEBS assembly line harboring wild-type AT domains. Our data thus indicate that the DSZS AT is potent enough to complement an AT lesion in DEBS at substoichiometric concentrations that could readily be achieved in vivo.
In contrast to the effective complementation of the AT1-null form of DEBS by DSZS AT, the available kinetic evidence predicted that analogous trans-complementation by KirCII (Figure 5A) would be much less effective. This experiment is further complicated by the fact that the wild-type DEBS assembly line naturally incorporates ethylmalonyl extender units.12 Nonetheless, as shown in Figure S2, Supporting Information, in the presence of KirCII, we observed a concentration-dependent increase in the yield of one ethylmalonyl-CoA-derived analogue, provisionally assigned as 12-desmethyl-12-ethyl-6-dEB. This analogue remained a minor product, even in the presence of a 5-fold molar excess of KirCII relative to DEBS (Figure 5B), suggesting that KirCII does not compete effectively with other functional DEBS AT domains in the transacylation of ACP1. The kinetic data in Figures 2 and 3 also suggest that, under the assay conditions, the preferred extender unit for KirCII is methylmalonate.
Discussion
While most PKS assembly lines contain dedicated acyltransferase (AT) domains harbored within each module, several systems utilize stand-alone ATs that act in trans. These trans-acting ATs may have considerable potential for regioselective production of modified polyketide antibiotics if they can be used to complement mutant biosynthetic assembly lines in which the natural cis-AT domains of targeted modules have been inactivated. Rational implementation of such a strategy, however, requires a detailed understanding of the catalytic mechanism of the reactions catalyzed by these stand-alone ATs, as well as their specificity for both carboxyacyl-CoA and ACP substrates. We have characterized both the hydrolytic and the transacylation activities of two representative trans-AT enzymes, the malonyl-CoA specific AT from the disorazole synthase, DSZS AT, and the ethylmalonyl-CoA specific AT from the kirromycin synthase, KirCII. We have also analyzed the individual specificity of these two trans-AT enzymes for different carboxyacyl-CoA and ACP substrates and compared these properties with those of a representative cis-AT domain, DEBS AT3. We have also evaluated the implications of the mechanistic analysis of both trans-AT domains by determining their ability to complement the entire DEBS assembly line harboring a module-specific AT lesion.
Although we anticipated at the onset of these studies that all AT enzymes would catalyze acyl transfer by a standard ping-pong mechanism (Scheme 2A), we were surprised to discover that, in contrast to the behavior of the cis-AT3 domain of DEBS, the relative kcat/KM values for transacylation and for hydrolysis of carboxyacyl-CoA substrates are significantly different for each trans-AT enzyme examined. This indicates that the presence, and likely the identity, of the cosubstrate ACP acceptor protein influences the carboxyacyl-CoA specificity parameter for both the DSZS and KirCII ATs. This observation prompts consideration of alternative mechanisms for this critical enzymatic component of assembly line PKSs (e.g., Scheme 2B,C) and suggests that the trans-AT-catalyzed reaction occurs in a coupled, vectorial manner on an intact assembly line.28 Examination of trans-AT catalytic mechanisms will be the topic of further investigations in our laboratory.
The three AT domains analyzed in this study showed significant specificity for their cognate carboxyacyl-CoA substrate (at least 20-fold as measured by kcat/KM), with the DSZS AT exhibiting the highest level of substrate specificity, with a kcat/KM for malonyl-CoA approximately 5 orders of magnitude higher than that for either methylmalonyl-CoA or ethylmalonyl-CoA. This trans-AT domain thus appears to have evolved to maximize both the kcat and kcat/KM for its native substrate. In contrast, KirCII appears to have evolved toward optimization of KM only compared with other AT domains (Figure 2). At saturating substrate concentrations, KirCII actually favors methylmalonyl-CoA over ethylmalonyl-CoA. Given the likely low titers of both methylmalonyl- and ethylmalonyl-CoA extender units in an actinomycete cellular environment,29,30 optimization of KM is presumably an effective specificity-determining strategy. This behavior should be considered in the design of engineered PKS assembly lines, where extender unit concentrations must be carefully controlled to achieve regiospecific incorporation of an ethylmalonyl extender unit by KirCII.
In contrast to its high carboxyacyl-CoA specificity, the DSZS AT had a more modest, ∼10-fold, specificity for its cognate ACP substrates compared with ACP domains from heterologous PKS systems. This combination bodes well for the potential use of the enzyme as a trans-acting AT in conjunction with an engineered PKS assembly line. On the other hand, KirCII had a low kcat/KM value for noncognate ACP domains. This suggests that, notwithstanding its promising carboxyacyl-CoA tolerance8 and the potential to engineer the AT-ACP protein–protein interface for improved interaction,21trans-complementation using KirCII would only be feasible if it could be drastically overexpressed to give high protein concentrations in vivo (Figure 3).
To test these predictions, in vitro complementation studies were performed with the DSZS AT or KirCII added to a fully reconstituted DEBS assembly line in which the AT domain of the first module had been inactivated by site-directed mutagenesis (Figures 4 and 5). Whereas efficient trans-complementation was observed at substoichiometric concentrations of DSZS AT, production of the putative 12-desmethyl-12-ethyl-6-dEB analogue was extremely low even when KirCII was present in 5-fold molar excess over the DEBS proteins.
Two other notable conclusions can be drawn from these trans-complementation experiments. First, as the concentration of the DSZS AT was increased, the overall preference for the desired 12-desmethyl-6-dEB product decreased due to the competing formation of additional putative didesmethyl-6-dEB analogues, most likely due to competitive incorporation of a malonyl extender unit by the DSZS AT at an otherwise fully functional DEBS module. This suggests that the concentration of DSZS AT must be carefully controlled so as to achieve regiospecific incorporation of a non-native extender unit. Second, even at the highest tested concentrations of both DSZS AT and KirCII, incorporation of a non-native extender unit at module 1 of DEBS led to a significant drop in the rate of product formation (Figure 6). This resulting decrease in overall productivity is not surprising given that the DSZS AT or KirCII mediated transacylation must be followed by at least 20 enzyme-catalyzed reactions that must accommodate the resulting structural change and that the overall specificity of a multistep metabolic pathway reflects the discrimination capacity of individual active sites. The lower yield of 6-dEB analogues might also result from hydrolysis of aberrant intermediates produced by the assembly line, or it could be due to a partial decoupling of the normal vectorial enzyme-catalyzed reactions controlled by the PKS.28 These observations warrant further investigation, given their profound implications for polyketide biosynthetic engineering.
Figure 6.
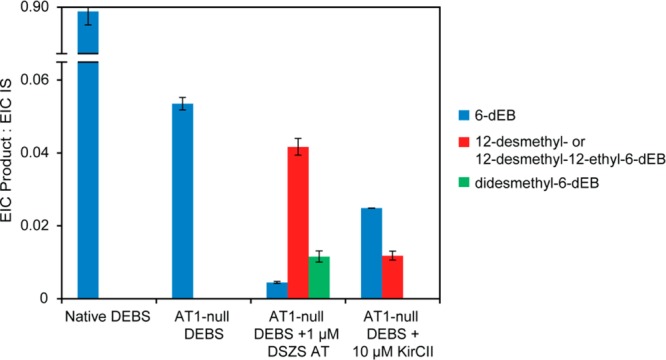
LC-MS product profiles for the native DEBS assembly line and the AT1-null DEBS assembly line with and without trans-AT. Methylmalonate was the only extender unit present in the native DEBS and AT1-null DEBS (without trans-AT) controls. Equimolar concentrations of methylmalonate and malonate (DSZS AT) or methylmalonate and ethylmalonate (KirCII) were present in the complementation reactions. All reactions were allowed to proceed for 30 min. For details regarding the identification of products and EIC quantification, see Experimental Procedures. IS, internal standard, N-Boc-glutamic acid benzyl ester (11.6 μM).
Acknowledgments
The authors thank Dr. Fong Tian Wong for plasmids pFW3, pFW139, and pFW140 and for assistance in ordering codon-optimized constructs. We also thank Dr. Elizabeth Sattely for advice and access to LC–MS instrumentation (NIH Grant R00GM089985 and Stanford School of Engineering).
Glossary
Abbreviations
- PKS
polyketide synthase
- AT
acyltransferase
- CoA
coenzyme A
- ACP
acyl carrier protein
- DEBS
6-deoxyerythronolide B synthase
- DSZS
disorazole synthase
- LC-MS
liquid chromatography–mass spectrometry
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- β-NAD+
β-nicotinamide adenine dinucleotide
- NADH
nicotinamide adenine dinucleotide, reduced form
- α-KGDH
α-ketoglutarate dehydrogenase
- TPP
thiamine pyrophosphate
- EDTA
ethylenediaminetetraacetic acid
- TCEP
tris(2-carboxyethyl)phosphine
- BSA
bovine serum albumin
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced form
- ATP
adenosine triphosphate
- Kirr
kirromycin
- X-CoA
carboxyacyl-CoA
- 6-dEB
6-deoxyerythronolide B
- EIC
extracted ion chromatogram
- IS
internal standard
Supporting Information Available
Ethylmalonyl-CoA synthesis protocol, codon-optimized sequences used for expression of kirromycin proteins, and LC-MS data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ B.J.D. and K.R.W. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
K.R.W. is a recipient of an NIH Institutional Research and Academic Career Development Postdoctoral Award (Grant NIH K12-GM088033). This work was supported by grants from the National Institutes of Health (Grant R01 GM087934 to C.K. and Grant R01 GM022172 to D.E.C.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Walsh C. T. (2008) The chemical versatility of natural-product assembly lines. Acc. Chem. Res. 41, 4–10. [DOI] [PubMed] [Google Scholar]
- Dunn B. J.; Khosla C. (2013) Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J. R. Soc. Interface 10, 20130297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel J. (2010) Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 27, 996–1047. [DOI] [PubMed] [Google Scholar]
- Carvalho R.; Reid R.; Viswanathan N.; Gramajo H.; Julien B. (2005) The biosynthetic genes for disorazoles, potent cytotoxic compounds that disrupt microtubule formation. Gene 359, 91–98. [DOI] [PubMed] [Google Scholar]
- Cheng Y. Q.; Tang G. L.; Shen B. (2003) Type I polyketide synthase requiring a discrete acyltransferase for polyketide biosynthesis. Proc. Natl. Acad. Sci. U S A 100, 3149–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopanik N. B.; Shields J. A.; Buchholz T. J.; Rath C. M.; Hothersall J.; Haygood M. G.; Hakansson K.; Thomas C. M.; Sherman D. H. (2008) In vivo and in vitro trans-acylation by BryP, the putative bryostatin pathway acyltransferase derived from an uncultured marine symbiont. Chem. Biol. 15, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiol E. M.; Hartner T.; Kulik A.; Moldenhauer J.; Piel J.; Wohlleben W.; Weber T. (2011) Supramolecular templating in kirromycin biosynthesis: the acyltransferase KirCII loads ethylmalonyl-CoA extender onto a specific ACP of the trans-AT PKS. Chem. Biol. 18, 438–444. [DOI] [PubMed] [Google Scholar]
- Koryakina I.; McArthur J.; Randall S.; Draelos M. M.; Musiol E. M.; Muddiman D. C.; Weber T.; Williams G. J. (2013) Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem. Biol. 8, 200–208. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Koppisch A. T.; Cane D. E.; Khosla C. (2003) Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J. Am. Chem. Soc. 125, 14307–14312. [DOI] [PubMed] [Google Scholar]
- Wong F. T.; Chen A. Y.; Cane D. E.; Khosla C. (2010) Protein–protein recognition between acyltransferases and acyl carrier proteins in multimodular polyketide synthases. Biochemistry 49, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn B. J.; Cane D. E.; Khosla C. (2013) Mechanism and specificity of an acyltransferase domain from a modular polyketide synthase. Biochemistry 52, 1839–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry B.; Robbins T.; Weng C. H.; O’Brien R. V.; Cane D. E.; Khosla C. (2013) In vitro reconstitution and analysis of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 135, 16809–16812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. Y.; Cane D. E.; Khosla C. (2007) Structure-based dissociation of a type I polyketide synthase module. Chem. Biol. 14, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. Y.; Alekseyev V. Y.; Chen A. Y.; Tang Y.; Cane D. E.; Khosla C. (2004) Reconstituting modular activity from separated domains of 6-deoxyerythronolide B synthase. Biochemistry 43, 13892–13898. [DOI] [PubMed] [Google Scholar]
- Wong F. T.; Jin X.; Mathews I. I.; Cane D. E.; Khosla C. (2011) Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry 50, 6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B. A.; Admiraal S. J.; Gramajo H.; Cane D. E.; Khosla C. (2001) Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291, 1790–1792. [DOI] [PubMed] [Google Scholar]
- Taoka S.; Padmakumar R.; Lai M. T.; Liu H. W.; Banerjee R. (1994) Inhibition of the human methylmalonyl-CoA mutase by various CoA-esters. J. Biol. Chem. 269, 31630–31634. [PubMed] [Google Scholar]
- Weber T.; Laiple K. J.; Pross E. K.; Textor A.; Grond S.; Welzel K.; Pelzer S.; Vente A.; Wohlleben W. (2008) Molecular analysis of the kirromycin biosynthetic gene cluster revealed beta-alanine as precursor of the pyridone moiety. Chem. Biol. 15, 175–188. [DOI] [PubMed] [Google Scholar]
- Kopp M.; Irschik H.; Pradella S.; Muller R. (2005) Production of the tubulin destabilizer disorazol in Sorangium cellulosum: Biosynthetic machinery and regulatory genes. ChemBioChem 6, 1277–1286. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Chen A. Y.; Kim C. Y.; Cane D. E.; Khosla C. (2007) Structural and mechanistic analysis of protein interactions in module 3 of the 6-deoxyerythronolide B synthase. Chem. Biol. 14, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z.; Musiol E. M.; Weber T.; Williams G. J. (2014) Reprogramming Acyl Carrier Protein Interactions of an Acyl-CoA Promiscuous trans-Acyltransferase. Chem. Biol. 21, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiol E. M.; Greule A.; Hartner T.; Kulik A.; Wohlleben W.; Weber T. (2013) The AT(2) domain of KirCI loads malonyl extender units to the ACPs of the kirromycin PKS. ChemBioChem 14, 1343–1352. [DOI] [PubMed] [Google Scholar]
- Alekseyev V. Y.; Liu C. W.; Cane D. E.; Puglisi J. D.; Khosla C. (2007) Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 16, 2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnett S. A.; Rath C. M.; Shareef A. R.; Joels J. R.; Chemler J. A.; Hakansson K.; Reynolds K.; Sherman D. H. (2011) Acyl-CoA subunit selectivity in the pikromycin polyketide synthase PikAIV: Steady-state kinetics and active-site occupancy analysis by FTICR-MS. Chem. Biol. 18, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.; Niederkruger H.; Zimmermann K.; Vagstad A. L.; Moldenhauer J.; Brendel N.; Frank S.; Poplau P.; Kohlhaas C.; Townsend C. A.; Oldiges M.; Hertweck C.; Piel J. (2012) Polyketide proofreading by an acyltransferase-like enzyme. Chem. Biol. 19, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla C.; Tang Y.; Chen A. Y.; Schnarr N. A.; Cane D. E. (2007) Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 76, 195–221. [DOI] [PubMed] [Google Scholar]
- Gokhale R. S.; Lau J.; Cane D. E.; Khosla C. (1998) Functional orientation of the acyltransferase domain in a module of the erythromycin polyketide synthase. Biochemistry 37, 2524–2528. [DOI] [PubMed] [Google Scholar]
- Jencks W. P. (1980) The utilization of binding energy in coupled vectorial processes. Adv. Enzymol. Relat. Areas Mol. Biol. 51, 75–106. [DOI] [PubMed] [Google Scholar]
- Park J. W.; Jung W. S.; Park S. R.; Park B. C.; Yoon Y. J. (2007) Analysis of intracellular short organic acid-coenzyme A esters from actinomycetes using liquid chromatography-electrospray ionization-mass spectrometry. J. Mass Spectrom. 42, 1136–1147. [DOI] [PubMed] [Google Scholar]
- Jung W. S.; Kim E.; Yoo Y. J.; Ban Y. H.; Kim E. J.; Yoon Y. J. (2014) Characterization and engineering of the ethylmalonyl-CoA pathway towards the improved heterologous production of polyketides in Streptomyces venezuelae. Appl. Microbiol. Biotechnol. 98, 3701–3713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.