

Abstract
Developing surface coatings for NaLnF4 nanoparticles (NPs) that provide long-term stability in solutions containing competitive ions such as phosphate remains challenging. An amine-functional polyamidoamine tetraphosphonate (NH2-PAMAM-4P) as a multidentate ligand for these NPs has been synthesized and characterized as a ligand for the surface of NaGdF4 and NaTbF4 nanoparticles. A two-step ligand exchange protocol was developed for introduction of the NH2-PAMAM-4P ligand on oleate-capped NaLnF4 NPs. The NPs were first treated with methoxy-poly(ethylene glycol)-monophosphoric acid (Mn = 750) in tetrahydrofuran. The mPEG750-OPO3-capped NPs were stable colloidal solutions in water, where they could be ligand-exchanged with NH2-PAMAM-4P. The surface amine groups on the NPs were available for derivatization to attach methoxy-PEG (Mn = 2000) and biotin-terminated PEG (Mn = 2000) chains. The surface coverage of ligands on the NPs was examined by thermal gravimetric analysis, and by a HABA analysis for biotin-containing NPs. Colloidal stability of the NPs was examined by dynamic light scattering. NaGdF4 and NaTbF4 NPs capped with mPEG2000–PAMAM-4P showed colloidal stability in DI water and in phosphate buffer (10 mM, pH 7.4). A direct comparison with NaTbF4 NPs capped with a mPEG2000-lysine-based tetradentate ligand that we reported previously (Langmuir 2012, 28, 12861−12870) showed that both ligands provided long-term stability in phosphate buffer, but that the lysine-based ligand provided better stability in phosphate-buffered saline.
Introduction
Lanthanide containing nanoparticles (Ln NPs), e.g., Ln2O3, LnF3, NaLnF4, are employed as imaging agents in biological systems due to their unique luminescent and magnetic properties as well as their low cytotoxicities.1−3 The highest-quality Ln NPs are synthesized at high temperature (between 250 and 330 °C) in high-boiling-point nonpolar solvents such as oleic acid (OA), oleylamine (OM), and 1-octadecene (ODE).4−8 In this way, NPs of small sizes, narrow size distributions and high crystallinities can be prepared. These NPs are coated with hydrophobic ligands OA or OM. For biolabeling applications, the surfaces of these Ln NPs must be modified to render the NPs hydrophilic while maintaining the colloidal stability and physiochemical properties of the NPs.
Various strategies are available to make Ln NPs compatible with (i.e., colloidally stable in) an aqueous environment.9 In ligand substitution approaches, the OA or OM ligands on the as-synthesized NP surface are replaced by more hydrophilic ligands such as citrate, ethylenediamine tetra(methylene phosphonic acid) (EDTMP), poly(acrylic acid) (PAA), poly(N-vinylpyrrolidone) (PVP) and methoxy-poly(ethylene glycol)-phosphoric acid (mPEG-OPO3H2).10−14 The driving force for ligand replacement can be mass action, tighter binding to the surface by the functional group(s) on the new ligand, or cooperative interactions for multidentate ligands. Other strategies include rendering the NP hydrophilic by encapsulation of the original ligands on the NP surface with amphiphilic molecules or polymers such as PEG-phospholipids, poly(maleic anydride-alt-1-octadecene)-co-PEG (PMAO–PEG) and octylamine-grafted PAA.15−17 Alternatively, one can use an inverse microemulsion method to coat the Ln NPs with a thin silica shell.18
For labeling experiments that target specific biomarkers, the NP surface requires additional functional groups for the attachment of bioaffinity agents. This chemistry is well developed for gold NPs and quantum dots, but less developed for Ln NPs. There are examples in which surface functional groups such as amines and maleimides have been incorporated into the surface of Ln NPs and used for biological applications.19,20 Examples include particles used for bioimaging studies in vitro(21) or in vivo,22 for targeted theranostics,23 and for multiplexed bioassays.24
While these studies often demonstrate that their particles can be dispersed in water, the particles were employed for in vitro or in vivo imaging studies without explicitly testing (or reporting) the colloidal stability in buffer-containing media.22,25,26 As we will show later in this contribution, this issue is very important. Most biologically relevant buffers contain phosphate, and phosphate is a strongly competitive ion for the surface of Ln NPs.
For other types of NPs, it is known that particle aggregation can dramatically affect their biological activities. For example, Au NPs capped by zwitterionic ligands can to some extent maintain their colloidal stability in aqueous buffered media but undergo aggregation in a physiological environment with relatively high salt and protein concentrations; this aggregation leads to a drastic increase in their tumor uptake.27 In contrast transferrin-stabilized Au NP aggregates had significantly lowered uptake in HeLa and A549 cells.28 Moreover, recent studies based on Au, Ag, or SiO2 NPs revealed that the density of capping ligands and the position of functional groups on the NP surface also greatly influence their cytotoxicities, in vivo circulation time and targeting ability.29,30 We are unaware of any analogous in-depth studies of Ln NPs.
To improve the performance and reproducibility of Ln NPs as biolabeling agents better surface coating strategies are needed. The goal is to develop surface modification methodologies for Ln NPs that provide functional groups not only for bioaffinity reagent conjugation, but also for robust colloidal stability in a wide range of aqueous buffer media, while also minimizing nonspecific binding of NPs. To reach this goal, one needs more quantitative characterization of the surface coating and more detailed information about the colloidal stability of the coated particles in the presence of competitive buffers and high salt concentrations.
We are interested in Ln NPs as metal-containing reagents for mass cytometry,31 where these issues are particularly important. In this technique, monoclonal antibodies (mAbs) or other bioaffinity agents are labeled with metal isotopes and used for biomarker detection on cells. Signal detection is by inductively coupled plasma mass spectrometry, and sensitivity increases linearly with the number of metal atoms per mAb. In current technology, mAbs are labeled with metal-chelating polymers so that each mAb carries between 100 and 250 metal atoms.32,33 A NaLnF4 NP with a diameter of 10 nm can carry ∼8000 metal atoms, which can in principle increase the sensitivity of the technique by 2 orders of magnitude. Ln NP reagents for mass cytometry not only must show good colloidal stability in physiological buffers, they also must have low nonspecific adsorption to avoid background signal. Up to now, achieving these properties has not been possible.
We recently reported the synthesis, based upon a lysine framework, of an mPEG-diphosphonate bidentate ligand and an mPEG-tetraphosphonate tetradentate ligand, both bearing MPEG = 2000 (PEG2000). These species were used to replace oleate groups on the surface of four different types of NaLnF4 NPs with diameters ranging from 9 to 16 nm.34 The ability of the surface ligands to provide colloidal stability in aqueous media was compared to mPEG2000-phosphate, a monodentate ligand. While mPEG2000-phosphate-stabilized NPs were colloidally stable in water, they rapidly aggregated and precipitated in phosphate-buffered saline (PBS). By eye, the mPEG2000-diphosphonate-coated NPs showed greater colloidal stability in PBS buffer, but aggregation was detected by dynamic light scattering (DLS). Only the mPEG2000-tetraphosphonate-coated NPs showed long-term colloidal stability in PBS buffer, demonstrating the advantage of multidentate ligands.
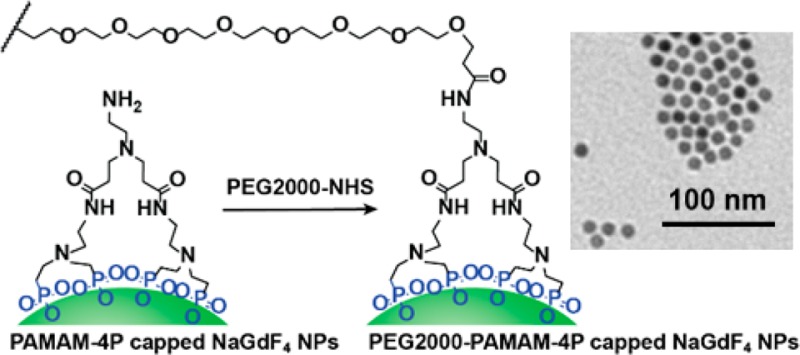
One of the problems with these ligands is that the synthesis was tedious, and modifying the ligand required restarting the synthesis at an early stage. Here we report a new synthesis of a tetraphosphonate ligand, based on a polyamidoamine (PAMAM) dendrimer. Starting with an amine-protected generation-0.5 PAMAM dendrimer, we introduced four phosphonate groups. Then a two-stage exchange protocol was developed to replace oleates on the surface of the as-synthesized NPs to transfer them into water. The NPs had primary amine groups on the surface available for further modification improving the versatility of the synthesis by allowing PEGs of different lengths to be attached to the NPs via the surface -NH2 groups. As we will see below, the footprint of the tetraphosphonate ligands on the MP surface may also be different, due to greater spacing between the phosphonates on the dendrimer. These differences, as well as the difference in the ligand exchange protocol necessary to introduce the tetraphosphonate ligand means that one has to examine carefully the behavior of the PEG-coated NaLnF4 NPs, comparing the behavior of the two different types of surface coatings.
Experimental Section
Instrumentation
Transmission electron microscope (TEM) images were obtained with a Hitachi H-7000 TEM at an accelerating voltage of 100 kV. OA capped NPs dispersed in cyclohexane were directly drop-cast onto hydrophobic Formvar-carbon-coated copper TEM grids. Hydrophilic NPs dispersed in DI-water were drop-cast onto hydrophilic TEM grids pretreated with Triton X-305. 1H-, 13C-, and 31P NMR spectra were recorded on a Bruker Avance III 400 MHz NMR spectrometer at T = 297 K. The accurate masses of all organic compounds were measured on an Agilent 6538 Q-TOF system. DLS measurements were performed on a Malvern Zetasizer Nano ZS instrument at a backscattering angle of 173°. All the NP solutions were passed through a 0.2 μm cellulose syringe filter before DLS measurements. For FT-IR and TGA measurements, the NPs were dried from solution and lyophilized for at least 24 h. FT-IR spectra were recorded on a PerkinElmer Spectrum 1000 infrared spectrometer using the KBr pellet method. TGA measurements were performed on a TA SDT Q600 thermogravimetric analyzer. All of the TGA measurements on NPs were performed in an air atmosphere. The NP samples were heated from room temperature to 100 °C at a ramping rate of 10 °C/min, equilibrated at 100 °C for 30 min to remove any absorbed water, then heated to 800 °C at the same rate of 10 °C/min. UV/vis absorption measurements for the HABA assay employed a PerkinElmer Lambda 35 UV/vis spectrometer. Inductively coupled plasma mass spectrometry (ICP-MS) measurements were performed on a PerkinElmer ELAN 9000 ICP-MS spectrometer. NaLnF4 NPs for ICP-MS measurements were digested in concentrated HNO3 (ICP grade, Seastar Chemical Inc.) by treating an aliquot (0.5 mL) of NP dispersion in water with concentrated HNO3 (4.5 mL) in a capped glass vial and stirring at room temperature overnight.
Materials
PEG derivatives were purchased from JenKem Technology. The Pierce Biotin Quantitation Kit and Slide-A-Lyzer G2 Dialysis Cassettes were purchased from Thermo Scientific. All dialyses employed (10k MWCO, 3 mL capacity) cassettes The mPEG-monophosphates mPEG750-OPO3H2 and mPEG2000-OPO3H2 were synthesized according to a literature procedure.14 All the other chemicals were purchased from Sigma-Aldrich and used without further purification. NaGdF4 and NaTbF4 NPs were synthesized following the method of Li et al.35 with details provided in Supporting Information (SI). The synthesis of the PAMAM dendrimer with an N-Boc-ethylenediamine core (2 in Scheme 1), followed the strategy described by Cao et al. with details provided in SI.36
Scheme 1. Synthesis of the PAMAM-4P Ligand 4 As Its Octasodium Salt.

Synthesis of Amino-PAMAM-Tetraphosphonate, Sodium Salt (PAMAM-4P) (4)
Diethyl vinylphosphonate (3.60 mL, 23.8 mmol) was added to a solution of 2 (1.99 g, 5.1 mmol) in DI-water (10 mL) and stirred at room temperature for 5 days. Then water and unreacted diethyl vinylphosphonate were removed on a rotary evaporator. The crude product was purified by silica column chromatography (eluent: 40:3 CHCl3–CH3OH) to afford compound 3 as a light-yellow liquid. Yield: 1.59 g (30%). 1H NMR (400 MHz, CDCl3): δ 7.89 (2H, br, −CONHCH2−), 5.62 (1H, br, BocNHCH2−), 4.09 (16H, m, −OCHH2CH3), 3.30 (4H, m, −CONHCH2CH2−), 3.17 (2H, d, BocNHCH2−), 2.77–2.82 (12H, m, −CONHCH2CH2– and −CH2CH2PO(OCH2CH3)2−), 2.56 (6H, m, BocNHCH2CH2– and BocNHCH2CH2N(CH2)2−), 2.35 (4H, t, −CH2CH2CONH−), 1.92 (8H, m, −CH2PO(OCH2CH3)2), 1.43 (9H, s, −C(CH3)3), 1.33 (24H, t, −OCHH2CH3). 13C NMR (100 MHz, CDCl3): δ 172.6, 156.1, 78.9, 61.7, 61.6, 52.5, 51.4, 49.9, 46.8, 46.2, 38.4, 36.9, 33.6, 28.5, 23.6, 22.2, 16.5, 16.4. 31P NMR (160 MHz, CDCl3): δ 30.6 (s, −PO(OCH2CH3)2). HRMS m/z for C41H89N6O16P4 ([M + H]+) calculated 1045.5280, found 1045.5270.
Bromotrimethylsilane (TMSBr) (2.0 mL, 14.5 mmol) was added dropwise to a solution of 3 (0.77 g, 0.73 mmol) in anhydrous CH2Cl2 (10 mL). The solution was sealed and stirred at room temperature for 24 h, and then CH2Cl2 and excess TMSBr were removed on a rotary evaporator. The residue was then dissolved in water (5 mL) and stirred for 10 min to dialyze the TMS esters. To the resulting turbid solution, was added 1 M NaOH aq solution dropwise until pH ≈ 10, during which the solution turned clear. Water was removed by vacuum evaporation, and the residue was dissolved in CH3OH (5 mL). The CH3OH solution was filtered to remove any insoluble inorganic salt; then the filtrate was dried again, and the residue was lyophilized overnight to remove any water residue and generate the product 4 as a light-yellow powder. Yield: 0.52 g (79%). 1H NMR (400 MHz, 0.1 M NaOD in D2O): δ 3.24 (4H, t, −CONHCH2CH2−), 2.71 (4H, t, NH2CH2CH2N(CH2)2−), 2.58–2.62 (12H, m, NH2CH2CH2– and −CH2CH2PO(OCH2CH3)2−), 2.51 (4H, t, −CONHCH2CH2−), 2.46 (2H, t, NH2CH2CH2−), 2.33 (4H, t, −CH2CH2CONH−), 1.49 (8H, m, −CH2PO3Na2). 13C NMR (100 MHz, 0.1 M NaOD in D2O): δ 174.9, 54.8, 50.6, 49.2, 48.7, 37.8, 36.7, 32.7, 26.2, 24.9. 31P NMR (160 MHz, 0.1 M NaOD in D2O): δ 19.7 (s, −PO3Na2). HRMS m/z for C20H47N6O14P4 ([M – H]−) calculated 719.2106, found 719.2109.
Two-Step Ligand Exchange
For ligand exchange experiments, freshly purified NaLnF4 NPs (see SI for details) were precipitated with ethanol. The ethanol was decanted to obtain a moist precipitate. A still-moist sample of NaLnF4 NPs (∼30 mg) was redispersed in THF (3 mL) with sonication. A THF solution (1.5 mL) of mPEG750-OPO3H2 (75 mg) was added. The solution was stirred at room temperature for 24 h, then the solution was mixed with 3 mL of DI water. The mixed solution was extracted with hexanes (1 mL) to remove displaced OA, and the aqueous phase was gently warmed on a water bath in vacuo to evaporate THF. The resulting aqueous solution was dialyzed against 3 L of DI-water for 24 h to remove the excess mPEG750-OPO3H2.
To the dialyzed aqueous solution containing mPEG750-OPO3-capped NaLnF4 NPs, an aqueous solution (1 mL) of PAMAM-4P (25 mg) was added. The solution was stirred at room temperature for 24 h, then dialyzed against 3 L of DI-water for 24 h to obtain PAMAM-4P-capped NaLnF4 NPs without excess free ligands. The mPEG2000-OPO3-capped NaGdF4 NPs were prepared in the same way using mPEG2000-OPO3H2 in the ligand exchange reaction.
PEGylation of PAMAM-4P-Capped NaLnF4 NPs
To an aqueous solution (3 mL) containing PAMAM-4P-capped NaLnF4 NPs (∼15 mg) was added 1 mL of 0.5 M NaHCO3 buffer (pH 8.5), followed by addition of an aqueous solution (1 mL) of mPEG2000-NHS (20 mg). The solution was stirred at room temperature for 4 h and then dialyzed against 3 L of DI-water for 48 h to obtain mPEG2000-PAMAM-4P-capped NaLnF4 NPs. To prepare functional biotin–PEG2000–PAMAM-4P capped NaLnF4 NPs, a mixture of mPEG2000-NHS (18 mg) and biotin–PEG2000-NHS (2 mg) was used instead of pure mPEG2000-NHS. The biotin content of these NPs was determined by a HABA colorimetric assay using a Pierce Biotin Quantitation Kit as described in SI.
Comparing Colloidal Stability of NaTbF4 NPs Capped with Two Different mPEG2000-Tetraphosphonate Ligands
Solutions of mPEG2000–PAMAM-4P-capped- and mPEG2000-lysine-4P-capped NaTbF4 NPs were prepared in water, and the particle concentrations were determined by ICP-MS. These samples were transferred to various aqueous media, at a particle concentration of 1.0 mg NaTbF4 NPs/mL. The aqueous media included water, 10 mM sodium phosphate buffer (PB), and 1× phosphate buffered saline (PBS containing 10 mM Na2HPO4, 2.0 mM KH2PO4, 137 mM NaCl, 2.0 mM KCl), and 150 mM PB. All the buffer solutions were adjusted to pH 7.4.
Results and Discussion
NaGdF4 and NaTbF4 NPs were synthesized at 300 °C using the protocol developed by Zhang and co-workers.35,37 The as-prepared NPs could be stored in their original reaction media of OA and ODE for more than a year while maintaining their initial size and morphology. For surface modification experiments, the NPs were first purified to remove excess free OA by precipitation with ethanol, three cycles of washing with a 1:5 THF-EtOH mixture, and redispersion in cyclohexane. Details are presented in SI. The freshly purified NPs were readily dispersible in various organic solvents including cyclohexane, toluene, THF and chloroform to form clear colloidal solutions. TEM images of these NPs are shown in Figure S1 in SI. Both NPs are nearly spherical in shape. Statistical analysis of the TEM images gave average diameters of 11.3 ± 0.6 nm for NaGdF4 and 11.1 ± 0.6 nm for NaTbF4 NPs. DLS CONTIN plots of the NaLnF4 NP dispersed in cyclohexane are presented in Figure S2 in SI. The distributions are symmetric and narrow. By DLS, we obtained mean hydrodynamic radii of 7.4 nm (PDI = 0.03) for NaGdF4 and 7.3 nm (PDI = 0.02) for NaTbF4 NPs in cyclohexane.
Synthesis and Characterization of the PAMAM-4P Surface Ligand
The dendritic ligand PAMAM-4P (4) was synthesized via a four-step strategy in which all the reactions were performed at room temperature (Scheme 1). The first two steps followed the general synthetic method for PAMAM dendrimers.36 The dendrimer 2 obtained in this way was reacted with four equiv of diethyl vinylphosphonate to give the phosphonate-ethyl-ester terminated PAMAM dendrimer 3, whose 1H, 13C, and 31P NMR spectra are presented in Figures S3 and S4 in SI. In the last step of the synthesis, both the Boc group and the eight ethyl groups were cleaved by treatment with TMSBr. Then the residue was hydrolyzed with water to yield free phosphonic acid and amine groups. A stoichiometric amount of NaOH was added to convert the PAMAM-4P to its octasodium salt. After removal of water and lyophilization, the PAMAM-4P-Na8 salt was obtained as a pale-yellow solid. The 1D and 2D NMR spectra of the PAMAM-4P-Na8 salt 4 are shown in Figure S5 and S6 in SI.
Two-Step Ligand Exchange and PEGylation
We prepared PEG-PAMAM-4P-capped NaLnF4 NPs using a two-step ligand exchange and PEGylation strategy, which is described in Scheme 2. The as-prepared NaLnF4 NPs capped with OA were dispersible only in nonpolar organic solvents and semipolar solvents like tetrahydrofuran (THF). In contrast, the PAMAM-4P ligand was soluble only in water, methanol and ethanol. We were unable to find a solvent that is able to disperse the NPs and also dissolve the PAMAM-4P ligand. All attempts to perform ligand exchange experiments in mixed solvents such as THF–CH3OH or CHCl3–CH3OH gave aggregated particles that would not disperse in water. To overcome this problem, we developed a two-step ligand exchange strategy to prepare PAMAM-4P-capped NaLnF4 NPs. In the first step, we treated a dispersion of the NaLnF4 NPs in THF with a solution in THF of mPEG-monophosphoric acid (mPEG750-OPO3H2, MPEG = 750 Da). After stirring at room temperature overnight, the THF solution was diluted with water to give a 1:1 THF–H2O mixture. The solution remained transparent, suggesting that the NaLnF4 NPs were colloidally stable in this solvent mixture. The free OA molecules were removed by a single extraction with hexane. The bottom aqueous phase was separated and warmed gently on a rotary evaporator to evaporate the THF. Then the remaining transparent aqueous solution was dialyzed against DI water to remove excess free mPEG750-OPO3H2. In this way, the OA groups at the NP surface were replaced by mPEG750 chains.
Scheme 2. Surface Modification Strategy for Preparing PEG–PAMAM-4P-Capped NaLnF4 NPs.

The second step involved ligand exchange of mPEG750-OPO3-capped NaLnF4 NPs with PAMAM-4P. Our experimental design relied on the assumption that that the tetradentate PAMAM-4P should bind more tightly to the NP surface than the monodentate mPEG-OPO3. This step was carried out in water at room temperature in the presence of excess PAMAM-4P sodium salt. After 20 h, we assumed that the ligand exchange reaction was complete. The displaced mPEG750-OPO3 ligands and free PAMAM-4P as well as excess sodium ions were removed by dialysis. Taking NaGdF4 NPs as an example, TEM images of both mPEG750-OPO3- and PAMAM-4P-capped NPs (Figure 1A and B) show that the NPs maintained their initial size and morphology, and remained well separated, after each step of the two-step ligand exchange experiment.
Figure 1.

Left column: TEM images of NaGdF4 NPs capped with (A) mPEG750-OPO3 (diameter 11.3 ± 0.5 nm) (B) PAMAM-4P (diameter 10.9 ± 0.5 nm) (C) mPEG2000–PAMAM-4P (diameter 10.8 ± 0.5 nm). Right column: TEM images of mPEG2000-OPO3-capped NaGdF4 NPs (D) immediately after ligand exchange and transfer to water; (E) after 24 h of dialysis against water (F) after 24 h of dialysis against 2.5 mM mPEG2000-OPO3H2 solution.
The product after each ligand exchange reaction was examined by FT-IR spectroscopy (Figure 2). For both mPEG750-OPO3- and PAMAM-4P-capped NaLnF4 NPs, powder samples were prepared by lyophilizing solutions purified by dialysis. In the IR spectra of OA-capped NaGdF4 NPs, the absorption bands at 2920 and 2850 cm–1 (attributed to the stretching vibration of C=C–H bonds) as well as 1560 and 1460 cm–1 (attributed to the stretching vibration of carboxylate C=O bonds) indicated the presence of OA on the NP surface. After the first ligand exchange step, in the IR spectra of mPEG750-OPO3-capped NaGdF4 NPs, all the above bands belonging to OA disappeared, accompanied by the appearance of bands of a C–H stretching vibration at 2880 cm–1 and a C–O stretching vibration at 1110 cm–1, associated with the backbone of the PEG chains. After the second ligand exchange step (PAMAM-4P-capped NaGdF4 NPs), neither band associated with the PEG backbone bond appeared in the IR spectra. The presence of PAMAM on the NP surface was detected by the characteristic amide I and II vibrational bands at 1650 and 1540 cm–1.
Figure 2.

FT-IR spectra of NaGdF4 NPs capped with different surface ligands (a) OA (b) mPEG750-OPO3 (c) PAMAM-4P (d) mPEG2000–PAMAM-4P.
NPs are commonly PEGylated to introduce protein repellency for biological applications.30 Here, we attach PEG molecules onto the NP surface by reacting the −NH2 group of the PAMAM-4P dendrimer ligand with methoxy-poly(ethylene glycol) succinimidyl carboxymethyl ester (mPEG2000-NHS). This step was performed in a pH 8.5 sodium bicarbonate buffer solution, an optimal pH for reactions between amines and NHS esters.38 After the reaction, we removed unreacted free PEG molecules and all excess salts by dialysis against water for 48 h in a 10k MWCO cassette. When the PAMAM-4P-capped NaGdF4 NPs were conjugated with mPEG2000-NHS, the PEG backbone vibrational bands at 2880 and 1110 cm–1 appeared again in the IR spectra of the product, mPEG2000–PAMAM-4P-capped NaGdF4 NPs (Figure 2, trace d).
Complications in Ligand Exchange: Competitive Surface Etching
While the ligand exchange process described above seems simple and straightforward, there are many subtle features of the exchange process that we do not understand. Our first indication of complications in the exchange process was the observation that prolonged exposure of the NPs in water to excess ligand led to particle etching. Figure 1D above shows a TEM image of a sample of NaGdF4 NPs freshly exchanged with excess mPEG2000-OPO3H2. These NPs are characterized by a diameter of 11.1 ± 0.7 nm. After 24 h dialysis against DI water (Figure 1E), the particle size seen by TEM was unchanged (11.3 ± 0.5 nm). After 24 h of dialysis against excess mPEG2000-OPO3H2 ligand (2.5 mM, Figure 1F), the particle size was smaller and more polydisperse (d = 10.1 ± 1.4 nm). Many of the NPs in Figure 1F appear to be more angular and less spherical compared to the NPs in Figure 1D and E. This kind of morphological change suggests that the NaGdF4 NPs underwent surface etching in the presence of excess mPEG2000-OPO3H2 ligand.
In order to examine this etching process more carefully, we carried out additional dialysis experiments against excess ligand, in which we monitored the solution outside the dialysis cartridge using the Arsenazo dye assay39 to quantify the concentration of Gd3+ released into the solution. For details, see SI. Since the dialysis membrane prevents passage of the NPs into the external solution, only free Gd3+ or Gd3+ ions complexed to excess ligand can pass through the membrane and be detected.
In Figure 3A we examine NaGdF4 NPs that were ligand exchanged in THF with mPEG2000-OPO3H2 and then subjected to dialysis. As shown in the lower-most trace, no detectable Gd3+ was found outside the dialysis bag. When these NPs were dialyzed against excess ligand (mPEG2000-OPO3H2), increasing amounts of Gd3+ were detected over a period of 150+ hours, and the rate of metal ion loss from the particles increased with increasing PEG-phosphate in the solution. After 1 week (∼170 h), the concentration of Gd3+ appeared to be leveling off at ∼40 μM in the presence of 1.0 mM mPEG2000-OPO3H2 and at 60 μM when the concentration of PEG-phosphate was 2.5 mM. In Figure 3B, we show a corresponding set of experiments for NaGdF4 NPs that were ligand exchanged in THF with mPEG750-OPO3H2 and then subjected to dialysis. Again there was no detectable loss of metal ions in the presence of DI water, and growing loss of metal ions in the presence of excess mPEG750-OPO3H2. Surprising to us was the finding that the amount of Gd3+ ions lost to the aqueous medium over 1 week in the presence of excess PEG-phosphate was less for mPEG750-OPO3-capped NPs exposed to mPEG750-OPO3H2, than for mPEG2000-OPO3-capped NPs exposed to mPEG2000-OPO3H2.
Figure 3.

Kinetics of Gd3+ release as NaGdF4 NPs were dialyzed against DI water and excess mPEG-phosphate. (A) Particles capped with mPEG2000-OPO3H2 and then dialyzed against DI water or mPEG2000-OPO3H2. (B) Particles capped with mPEG750-OPO3H2 and then dialyzed against DI water or mPEG750-OPO3H2. In each part: dialysis against DI water (bottom trace), against 1.0 mM mPEG-phosphate (middle trace), 2.5 mM mPEG-phosphate (upper trace).
These experiments are important from several points of view. First, they establish that the NPs are stable to metal leaching over a period of days if stored in DI water, but subject to etching if stored in the presence of excess buffer. In separate experiments monitored by TEM, we found that NaGdF4 NPs and NaTbF4 NPs, capped with either mPEG2000-OPO3H2 or mPEG750-OPO3H2, were stable over a period of weeks when stored in DI water. It is known that NaLnF4 NPs capped with PEG-phosphate are colloidally unstable in the presence of phosphate buffer, but it has been reported colloidal stability can be preserved in the presence of excess ligand.14 Our experiments suggest that particle etching can occur under these conditions. Beyond that, the differences seen in the susceptibility to etching in the presence of the two PEG-phosphates that differ only in the length of the PEG chains indicate that there are other factors involved in the ligand exchange process that remain poorly understood.
Characterization of Surface Ligand Density by TGA
Now that we have established conditions for ligand exchange where we are confident that no particle etching has taken place, we turn our attention to characterizing the ligand corona on the NPs. We begin with thermogravimetric analysis (TGA) to characterize the density of organic ligands on the surface of the NPs. Analysis of the data from these measurements presumes that centrifugation followed by redispersion removes excess ligand not chemically bound to the particle surface, that the NaLnF4 NPs maintain their chemical stability as the samples are heated in air, and that mass loss can be attributed to the oxidative degradation of the organic ligands. Under these conditions, the surface ligand density (in molecules/nm2) can be calculated from the mass loss using equation S1 given in the SI. The TGA trace for OA-NaGdF4 NPs (Figure S8, trace A in SI) showed a mass loss of 15.0%. Assuming spherical NPs (r = 5.6 nm) and bulk density (ρ = 5.647 g/cm3) for the inorganic core, the ligand density was calculated to be 4.4 molecules/nm2. This value corresponds to 0.23 nm2/OA, consistent with a literature value for OA-NaYF4:Yb,Er NPs, which is on the order of 0.25 nm2/OA.40
For mPEG750-OPO3-capped NaGdF4 NPs, the mass loss (39.0%, Figure S8, trace B in SI) corresponds to 5.5 molecules/nm2 (i.e., an average area of 0.18 nm2 per molecule). The mass loss for mPEG2000-OPO3-capped NaGdF4 NPs (66.7%, Figure S8, trace C in SI) implied an even higher ligand density (6.4 molecules/nm2), corresponding to 0.16 nm2 per PEG-phosphate ligand. When the capping ligand was mPEG2000–PAMAM-4P, the mass loss was 42.5% (Figure S8, trace D in SI). Since the molar mass of PAMAM-4P (excluding the sodium ions) is 712.46, we used a molecular weight 2710 for mPEG2000–PAMAM-4P to convert the mass loss into surface density. In this way, we obtained a value of 1.7 molecules/nm2. If we assume that the four phosphonates of the mPEG2000–PAMAM-4P ligand all bind to the NP surface, the average area occupied by each phosphonate group would be 0.15 nm2. These average parking area values for the phosphate and phosphonate anchoring groups determined for different PEG-based ligands are similar to one another. This result implies that the average area occupied by the ligand on the NaLnF4 NP surface is determined primarily by the interaction of phosphate and phosphonate groups with Ln ions in the particle surface, rather than by the dimensions of the PEG chains.
For mPEG2000–PAMAM-4P, the value of 1.7 molecules/nm2 corresponds to an average PEG-to-PEG distance d of 0.8 nm. The Flory radius RF of a PEG2000 random coil in a good solvent is estimated to be about 3.4 nm.41,42 Thus, for mPEG2000–PAMAM-4P capped NaGdF4 NPs, the distance between anchoring points of the PEG chains on the particle surface is significantly smaller than 2RF. Under these conditions, the PEG chains on the NP surface become extended and are in the so-called “brush” regime.43 For the PEG monophosphates, the mean distance between PEG chains is even smaller, and the brush should be more extended. Previous studies on PEGylated Au NPs indicate that this high PEG surface density is effective at resisting protein absorption.44 The above results are summarized in Table 1.
Table 1. TGA Percentage Mass Loss, Average Surface Ligand Density and Ligand Parking Area of NaGdF4 NPs Capped with OA, mPEG750-OPO3, mPEG2000-OPO3, or mPEG2000–PAMAM-4P.
| NaGdF4 NPs capped with | OA | mPEG750-OPO3 | mPEG2000-OPO3 | mPEG2000-PAMAM-4P |
|---|---|---|---|---|
| TGA mass loss (%) | 15.0 | 39.0 | 66.7 | 42.5 |
| ligand molecules/nm2 | 4.4 | 5.5 | 6.4 | 1.7 |
| mean area per ligand (nm2) | 0.23 | 0.18 | 0.16 | 0.59 |
Biotin Functionalization and Quantification
Biological assays with NP reagents require surface functionality on the NP surface to attach bioaffinity agents. To introduce surface functionality we used a mixture of mPEG2000-NHS and biotin–PEG2000-NHS with a molar ratio of 9:1 to PEGylate the surface of PAMAM-4P-capped NaTbF4 NPs (Bi-PEG-NPs). We employed a commercial HABA/Avidin colorimetric assay to quantify the average number of biotin groups in the PEG corona, of biotins per NP. As a control we also carried out the HABA/Avidin assay on a sample of mPEG2000–PAMAM-4P-capped NaTbF4 NPs (mPEG-NP) with no biotin functionality. Details are provided in SI, and the results are summarized in Tables S2 and S3. For the Bi-PEG-NP sample, we determined a biotin concentration corresponding to 72 ± 4 biotins/NP. The mPEG-NP control sample gave a signal corresponding to 4 biotins/NP. We take the difference, 68 ± 4, as the mean number of biotins per nanoparticle. For spherical NaTbF4 NPs with d = 11.1 nm, this number corresponds (see SI) to 1.8 ligand/nm2. Since Gd and Tb are adjacent elements in the periodic table, we can assume similar chemistries and compare the ligand density determined by the HABA assay (1.8 ligand/nm2) with that for mPEG2000–PAMAM-4P-capped NaGdF4 NPs determined by TGA (1.7 ligand/nm2). These values are in excellent agreement.
Colloidal Size and Stability of NaGdF4 NPs
PEG and PAMAM-4P Surface Ligands
Solutions of NaGdF4 NPs were examined by DLS to measure their hydrodynamic radii and to assess their colloidal stability. The original OA-capped NaGdF4 NPs in cyclohexane showed a z-average hydrodynamic radius (Rz) of 7.4 nm (Figure S1). After the first ligand exchange with mPEG750-OPO3H2, the NPs can be dispersed in water as a clear colloidal solution. The CONTIN plot for this sample in water showed a monomodal size distribution without any sign of aggregation (Figure 4A). The magnitude of Rz of this sample in water increased to 12 nm. When transferred to 10 mM pH 7.4 sodium phosphate buffer (PB), the sample aggregated almost immediately (Figure 4A) as noted by the shift in the still-monomodal CONTIN plot to a Rz of 54 nm. Within 30 min, the NPs precipitated.
Figure 4.

CONTIN plots of the hydrodynamic radius distribution of 1.0 mg NP/mL solutions of NaGdF4 NPs capped with (A) mPEG750-OPO3; (B) PAMAM-4P in water and in 10 mM phosphate buffer (PB, pH 7.4); (C) mPEG2000–PAMAM-4P in different aqueous media, all adjusted to pH 7.4. The mPEG750-OPO3-capped NPs in PB aggregated immediately to give the plot shown, but then precipitated over the subsequent 30 min. For the mPEG2000–PAMAM-4P-capped NPs in (C), all of the measurements were performed immediately after mixing the NP stock solution in DI water with the concentrated buffer solution to obtain the final buffer concentrations shown in the Figure. During the measurement, the sample in 150 mM PB buffer became slightly turbid, and this turbidity increased upon standing afterward.
After the surface ligands of the mPEG750-OPO3-capped NaGdF4 NPs were exchanged to PAMAM-4P, the solution in water remained clear, and the particles remained colloidally stable. The DLS CONTIN plot was bimodal, with a main peak at 16 nm, accompanied by a weaker peak at 90 nm (Figure 4B). In the presence of PB buffer, the aggregate peak increased in intensity in the CONTIN plot, with a shift in magnitude to lower hydrodynamic radius (∼60 nm). After storage in 10 mM PB for about 1 week, the PAMAM-4P-capped NaGdF4 NPs precipitated.
Corresponding CONTIN plots for mPEG2000–PAMAM-4P capped NaGdF4 NPs are presented in Figure 4C. Their hydrodynamic size distribution in water was monomodal. When these NPs were dispersed in either 10 mM PB or PBS at pH 7.4, they retained their monomodal size distribution. The peak values shifted to larger size, accompanied by a small increase in polydispersity. The Rz values increased from 12.0 nm (in water) to 20.6 nm (in 10 mM PB) and to 21.2 nm (in PBS). These results demonstrate that the PAMAM-4P ligand, whether PEGylated or not, can provide colloidal stability to NaGdF4 NPs in phosphate buffer under conditions where the PEG-monophosphate-capped NPs precipitate. In a more strongly competitive buffer, 150 mM PB, mPEG2000–PAMAM-4P was no longer able to stabilize the NPs, and the DLS CONTIN plot showed severe particle aggregation (Figure 4C).
For studies of particle aggregation, the most meaningful data to examine are intensity distributions. We remind the reader that these data require an ill-posed inverse Laplace transform of the autocorrelation function, and the long-time tail in the autocorrelation decay can appear as a peak at large Rh in the CONTIN plot. It is sometimes tempting to transform such data into volume or number distributions. The technical notes from Malvern45 and other user guides to DLS measurements46 point out that these conversions of DLS data invoke Mie theory and involve some serious assumptions, namely that all particles are spherical, that all particles are homogeneous, that the optical properties of the particles are known (i.e., the real and imaginary components of the refractive index), and that there is no error in the intensity distributions. For aggregate formation, these assumptions are unlikely to be satisfied. In Figure S10 we compare the intensity distribution plot of the mPEG2000-PAMAM-4P capped NaGdF4 NP sample dispersed in 150 mM PB as shown in Figure 4C with the calculated number distribution plot from the same set of DLS data. In the number distribution, the peak of the aggregates disappeared.
Comparing mPEG-lysine-4P and mPEG–PAMAM-4P Surface Ligands
In the Introduction section of this paper, we pointed out that in the mPEG-lysine-tetraphosphonate ligand34 (PEG-lysine-4P) the pairs of phosphonate groups are spaced differently (1 methylene) from the mPEG–PAMAM-4P ligand (2 methylenes). The chemical structures of these two tetraphosphonate ligands are presented in Figure 5A. It is important to test whether the differences in structure affect the ability of the ligands to promote and maintain colloidal stability in phosphate-buffered media. To address this issue, we carried out parallel kinetic studies by DLS on a sample of NaTbF4 NPs capped with each ligand.
Figure 5.

(A) Structure of mPEG2000–PAMAM-4P and mPEG2000-lysine-4P ligands. The phosphonates on the dendrimer are shown as ionized as a reminder that ligand exchange was carried out in water with the octasodium salt of the ligand. The phosphonates on the lysine-based ligand are shown as protonated as a reminder that ligand exchange was carried out in THF solution with the fully protonated form of the ligand. (B and C): Comparison of the temporal evolution of z-average hydrodynamic radii of 1 mg NP/mL solutions of NaTbF4 NPs capped with the two different PEG-tetraphosphonate ligands in either (B) 10 mM PB or (C) PBS, both pH 7.4. The corresponding CONTIN plots are shown in Figures S11 and S12 in SI. The z-average hydrodynamic radii and PDI values are summarized in Table S4 in SI.
One set of experiments was carried out in 10 mM phosphate buffer, pH 7.4 (Figure 5B). In the initial solution (t = 0) both samples showed monomodal CONTIN plots with Rz = 19.3 nm for the mPEG2000-lysine-4P-capped NPs and 18.6 nm for the mPEG2000–PAMAM-4P-capped NPs. For the both samples, there was essentially no change in size over 20 h and no formation of aggregates detected by DLS (Figure S11 in SI). The lysine-4P sample showed similar behavior in PBS buffer (Figure 5C), which contains 12 mM phosphate, 2.7 mM KCl and 137 mM NaCl, whereas striking differences in behavior were found for the mPEG2000–PAMAM-4P-capped NPs. Here a shift in the peak maximum could be seen after only 2 h, with substantial peak broadening in the CONTIN plot after 4 h (Figure S12 in SI). After 20 h, there was a well-defined aggregate peak in the CONTIN plot centered at ∼300 nm, along with a residual peak of the size of the initial sample. The Malvern Zetasizer software calculated an Rz value of 100 nm, and this is the value shown for this sample at 20 h in Figure 5C. This point should be taken as a sign of colloidal instability.
The difference in colloidal stability for the NPs capped with the two different tetradentate ligands was unexpected. This result raises questions that go beyond the small differences in the chemical structure of the ligands. For example, there are differences in the ligand exchange protocols. Ligand exchange in THF involves the fully protonated phosphate (for mPEG-monophosphate) or tetraphosphonate (for mPEG-lysine-4P). We imagine that each phosphate or phosphonate group may replace one oleate, which carries away one proton to form oleic acid. Thus, each phosphate/phosphonate group carries (on average) one negative charge, with which to form a P–O–Ln bond. In contrast, when mPEG-monophosphate is displaced from the NPs with the dendrimer tetraphosphonate PAMAM-4P, this ligand exchange involves the fully deprotonated dendrimer as its octasodium salt. These differences may affect how the phosphonate groups of the two different tetradentate ligands are bound to the particle surface.47 Whatever differences there are in how the two tetradentate ligands bind to the NP surface, they have a prominent effect on the colloidal stability of the NPs in PBS buffer.
Summary and Conclusion
We synthesized an amine-functional PAMAM dendrimer tetraphosphonate and developed a two-stage ligand exchange protocol to attach it to the surface of NaGdF4 and NaTbF4 NPs both with mean diameters of ∼11 nm. Ligand exchange with mPEG750-OPO3H2 in THF allowed the NPs to be dispersed in water, where they were treated with an excess of the dendrimer tetraphosphonate (8 Na+ salt). The amino groups on the surface of the particles dispersed in water are available for reaction with various reagents such as PEG-NHS esters. Thus, the dendrimer has great potential as a multidentate ligand for modifying the surface of NaLnF4 NPs in a way that further functionalization is possible with amine-reactive moieties.
While NaGdF4 and NaTbF4 NPs in water, stabilized by PEG-monophosphate or by the dendrimer ligand PAMAM-4P, appear to be chemically as well as colloidally stable for days or weeks, the PEG-phosphate-stabilized nanoparticles degrade when stored in the presence of excess ligand. For NaGdF4 NPs, we observed surface etching by TEM and Gd3+ ion loss to the aqueous medium.
Thermogravimetric analysis of one set of samples (NaGdF4 NPs capped with oleate, mPEG750-phosphate, and mPEG2000-phosphate) indicated that the area occupied per phosphate (0.16–0.18 nm2/phosphate) was somewhat smaller than the area occupied per oleate (0.23 nm2/oleate). This result is surprising because we expected that repulsion between the PEG corona chains would play a role in determining the number of ligands per nanoparticle. We have no definitive explanation for this result. One could speculate that there were PEG-phosphate chains associated with the NPs but not attached to the surface via their phosphate groups, and that dialysis was not effective at removing them from the sample.
The main motivation for synthesizing the dendrimer tetraphosphonate PAMAM-4P was as a ligand to provide colloidal stability for lanthanide nanoparticles in phosphate-containing media. We are concerned about factors that can affect the short and long-term stability of lanthanide nanoparticles in phosphate-buffered media. The most surprising result in this study was the finding of differences in colloidal stability of NaTbF4 NPs with mPEG2000–PAMAM tetraphosphonate groups at the surface compared to the same NPs with the mPEG2000-lysine tetraphosphonate groups. While both ligands provided long-term colloidal stability in 10 mM phosphate buffer at pH 7.4, the presence of salt in PBS buffer had a deleterious effect on the colloidal stability of the NPs with the PEG–PAMAM-4P. In contrast, the NPs with the PEG-lysine-tetraphosphonate maintained its colloidal stability in PBS buffer.
The origin of this difference may be related to structural differences that affect placement of the phosphonate groups on the NP surface, or it may be related to differences in the ligand exchange protocol. The synthetic versatility of the dendrimer ligand is an important advantage. Studies to understand the sensitivity of NPs stabilized with the PEG dendrimer are ongoing in our laboratories.
Acknowledgments
The authors thank NSERC Canada, DVS Sciences, and the Office of Aids Research at the National Institutes of Health for their financial support.
Supporting Information Available
Additional experimental details, additional results and discussion, 1H, 13C and 31P NMR spectra, DLS CONTIN plots of OA-capped NaLnF4 NPs in cyclohexane, DLS autocorrelation curves of NaGdF4 NPs capped with mPEG750-OPO3, PAMAM-4P or mPEG2000-PAMAM-4P and CONTIN plots of NaTbF4 NPs capped with mPEG2000-PAMAM-4P or mPEG2000-lysine-4P in PBS or 10 mM PB. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lin M.; Zhao Y.; Wang S.; Liu M.; Duan Z.; Chen Y.; Li F.; Xu F.; Lu T. Recent Advances in Synthesis and Surface Modification of Lanthanide-Doped Upconversion Nanoparticles for Biomedical Applications. Biotechnol. Adv. 2012, 30, 1551–1561. [DOI] [PubMed] [Google Scholar]
- Na H. B.; Song I. C.; Hyeon T. Inorganic Nanoparticles for MRI Contrast Agents. Small 2009, 21, 2133–2148. [Google Scholar]
- Wang F.; Banerjee D.; Liu Y.; Chen X.; Liu X. Upconversion Nanoparticles in Biological Labeling, Imaging, and Therapy. Analyst 2010, 135, 1839–1854. [DOI] [PubMed] [Google Scholar]
- Mai H.-X.; Zhang Y.-W.; Si R.; Yan Z.-G.; Sun L.; You L.-P.; Yan C.-H. High-Quality Sodium Rare-Earth Fluoride Nanocrystals: Controlled Synthesis and Optical Properties. J. Am. Chem. Soc. 2006, 128, 6426–6436. [DOI] [PubMed] [Google Scholar]
- Boyer J.-C.; Vetrone F.; Cuccia L. A.; Capobianco J. A. Synthesis of Colloidal Upconverting NaYF4 Nanocrystals Doped with Er3+, Yb3+ and Tm3+, Yb3+ via Thermal Decomposition of Lanthanide Trifluoroacetate Precursors. J. Am. Chem. Soc. 2006, 128, 7444–7445. [DOI] [PubMed] [Google Scholar]
- Wang F.; Han Y.; Lim C. S.; Lu Y.; Wang J.; Xu J.; Chen H.; Zhang C.; Hong M.; Liu X. Simultaneous Phase and Size Control of Upconversion Nanocrystals through Lanthanide Doping. Nature 2010, 463, 1061–1065. [DOI] [PubMed] [Google Scholar]
- Ye X.; Collins J. E.; Kang Y.; Chen J.; Chen D. T. N.; Yodh A. G.; Murray C. B. Morphologically Controlled Synthesis of Colloidal Upconversion Nanophosphors and Their Shape-Directed Self-Assembly. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22430–22435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski A. D.; Chan E. M.; Gargas D. J.; Katz E. M.; Han G.; Schuck P. J.; Milliron D. J.; Cohen B. E. Controlled Synthesis and Single-Particle Imaging of Bright, Sub-10 Nm Lanthanide-Doped Upconverting Nanocrystals. ACS Nano 2012, 6, 2686–2692. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Liu Z.; Li F. Upconversion Nanophosphors for Small-Animal Imaging. Chem. Soc. Rev. 2012, 41, 1323–1349. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Zhu X.; Peng J.; Li F. Core–Shell Lanthanide Upconversion Nanophosphors as Four-Modal Probes for Tumor Angiogenesis Imaging. ACS Nano 2013, 7, 11290–11300. [DOI] [PubMed] [Google Scholar]
- Peng J.; Sun Y.; Zhao L.; Wu Y.; Feng W.; Gao Y.; Li F. Polyphosphoric Acid Capping Radioactive/upconverting NaLuF4:Yb,Tm,153Sm Nanoparticles for Blood Pool Imaging in Vivo. Biomaterials 2013, 34, 9535–9544. [DOI] [PubMed] [Google Scholar]
- Liu C.; Wang H.; Li X.; Chen D. Monodisperse, Size-Tunable and Highly Efficient β-NaYF4:Yb,Er(Tm) Up-Conversion Luminescent Nanospheres: Controllable Synthesis and Their Surface Modifications. J. Mater. Chem. 2009, 19, 3546–3553. [Google Scholar]
- Johnson N. J. J.; Sangeetha N. M.; Boyer J.-C.; Veggel F. C. J. M. van. Facile Ligand-Exchange with Polyvinylpyrrolidone and Subsequent Silica Coating of Hydrophobic Upconverting B-NaYF4: Yb3+/Er3+ Nanoparticles. Nanoscale 2010, 2, 771–777. [DOI] [PubMed] [Google Scholar]
- Boyer J.-C.; Manseau M.-P.; Murray J. I.; van Veggel F. C. J. M. Surface Modification of Upconverting NaYF4 Nanoparticles with PEG-Phosphate Ligands for NIR (800 Nm) Biolabeling within the Biological Window. Langmuir 2009, 26, 1157–1164. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zhao Z.; Jiang M.; Que D.; Shi S.; Zheng N. Preparation and Photodynamic Therapy Application of NaYF4:Yb, Tm-NaYF4:Yb, Er Multifunctional Upconverting Nanoparticles. New J. Chem. 2013, 37, 1782–1788. [Google Scholar]
- Jiang G.; Pichaandi J.; Johnson N. J. J.; Burke R. D.; van Veggel F. C. J. M. An Effective Polymer Cross-Linking Strategy To Obtain Stable Dispersions of Upconverting NaYF4 Nanoparticles in Buffers and Biological Growth Media for Biolabeling Applications. Langmuir 2012, 28, 3239–3247. [DOI] [PubMed] [Google Scholar]
- Yi G.-S.; Chow G.-M. Water-Soluble NaYF4: Yb,Er(Tm)/NaYF4/Polymer Core/Shell/Shell Nanoparticles with Significant Enhancement of Upconversion Fluorescence. Chem. Mater. 2007, 19, 341–343. [Google Scholar]
- Abdul Jalil R.; Zhang Y. Biocompatibility of Silica Coated NaYF4 Upconversion Fluorescent Nanocrystals. Biomaterials 2008, 29, 4122–4128. [DOI] [PubMed] [Google Scholar]
- Wang M.; Hou W.; Mi C.-C.; Wang W.-X.; Xu Z.-R.; Teng H.-H.; Mao C.-B.; Xu S.-K. Immunoassay of Goat Antihuman Immunoglobulin G Antibody Based on Luminescence Resonance Energy Transfer between Near-Infrared Responsive NaYF4: Yb, Er Upconversion Fluorescent Nanoparticles and Gold Nanoparticles. Anal. Chem. 2009, 81, 8783–8789. [DOI] [PubMed] [Google Scholar]
- Zako T.; Nagata H.; Terada N.; Utsumi A.; Sakono M.; Yohda M.; Ueda H.; Soga K.; Maeda M. Cyclic RGD Peptide-Labeled Upconversion Nanophosphors for Tumor Cell-Targeted Imaging. Biochem. Biophys. Res. Commun. 2009, 381, 54–58. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Wang X.; Chen F.; Zhang Q.; Dong B.; Yang H.; Liu G.; Zhu Y. Functionalization of Upconverted Luminescent NaYF4:Yb/Er Nanocrystals by Folic Acid–Chitosan Conjugates for Targeted Lung Cancer Cell Imaging. J. Mater. Chem. 2011, 21, 7661–7667. [Google Scholar]
- Wang Y.-F.; Liu G.-Y.; Sun L.-D.; Xiao J.-W.; Zhou J.-C.; Yan C.-H. Nd3+-Sensitized Upconversion Nanophosphors: Efficient In Vivo Bioimaging Probes with Minimized Heating Effect. ACS Nano 2013, 7, 7200–7206. [DOI] [PubMed] [Google Scholar]
- Yang D.; Kang X.; Ma P.; Dai Y.; Hou Z.; Cheng Z.; Li C.; Lin J. Hollow Structured Upconversion Luminescent NaYF4:Yb3+,Er3+ Nanospheres for Cell Imaging and Targeted Anti-Cancer Drug Delivery. Biomaterials 2013, 34, 1601–1612. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Haushalter R. C.; Haushalter R. W.; Shi Y.; Zhang Y.; Ding K.; Zhao D.; Stucky G. D. Rare-Earth Upconverting Nanobarcodes for Multiplexed Biological Detection. Small 2011, 7, 1972–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; Gu Y.-J.; Man C. W.-Y.; Cheng J.; Xu Z.; Zhang Y.; Wang H.; Lee V. H.-Y.; Cheng S. H.; Wong W.-T. Polymer-Coated NaYF4:Yb3+, Er3+ Upconversion Nanoparticles for Charge-Dependent Cellular Imaging. ACS Nano 2011, 5, 7838–7847. [DOI] [PubMed] [Google Scholar]
- Shen J.-W.; Yang C.-X.; Dong L.-X.; Sun H.-R.; Gao K.; Yan X.-P. Incorporation of Computed Tomography and Magnetic Resonance Imaging Function into NaYF4:Yb/Tm Upconversion Nanoparticles for in Vivo Trimodal Bioimaging. Anal. Chem. 2013, 85, 12166–12172. [DOI] [PubMed] [Google Scholar]
- Liu X.; Chen Y.; Li H.; Huang N.; Jin Q.; Ren K.; Ji J. Enhanced Retention and Cellular Uptake of Nanoparticles in Tumors by Controlling Their Aggregation Behavior. ACS Nano 2013, 7, 6244–6257. [DOI] [PubMed] [Google Scholar]
- Albanese A.; Chan W. C. W. Effect of Gold Nanoparticle Aggregation on Cell Uptake and Toxicity. ACS Nano 2011, 5, 5478–5489. [DOI] [PubMed] [Google Scholar]
- Kim S. T.; Saha K.; Kim C.; Rotello V. M. The Role of Surface Functionality in Determining Nanoparticle Cytotoxicity. Acc. Chem. Res. 2013, 46, 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokerst J. V.; Lobovkina T.; Zare R. N.; Gambhir S. S. Nanoparticle PEGylation for Imaging and Therapy. Nanomedicine 2011, 6, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall S. C.; Nolan G. P.; Roederer M.; Chattopadhyay P. K. A Deep Profiler’s Guide to Cytometry. Trends Immunol. 2012, 33, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majonis D.; Herrera I.; Ornatsky O.; Schulze M.; Lou X.; Soleimani M.; Nitz M.; Winnik M. A. Synthesis of a Functional Metal-Chelating Polymer and Steps toward Quantitative Mass Cytometry Bioassays. Anal. Chem. 2010, 82, 8961–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illy N.; Majonis D.; Herrera I.; Ornatsky O.; Winnik M. A. Metal-Chelating Polymers by Anionic Ring-Opening Polymerization and Their Use in Quantitative Mass Cytometry. Biomacromolecules 2012, 13, 2359–2369. [DOI] [PubMed] [Google Scholar]
- Cao P.; Tong L.; Hou Y.; Zhao G.; Guerin G.; Winnik M. A.; Nitz M. Improving Lanthanide Nanocrystal Colloidal Stability in Competitive Aqueous Buffer Solutions Using Multivalent PEG-Phosphonate Ligands. Langmuir 2012, 28, 12861–12870. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang Y. An Efficient and User-Friendly Method for the Synthesis of Hexagonal-Phase NaYF4:Yb, Er/Tm Nanocrystals with Controllable Shape and Upconversion Fluorescence. Nanotechnology 2008, 19, 345606. [DOI] [PubMed] [Google Scholar]
- Cao W.; Zhu L. Synthesis and Unimolecular Micelles of Amphiphilic Dendrimer-like Star Polymer with Various Functional Surface Groups. Macromolecules 2011, 44, 1500–1512. [Google Scholar]
- Li Z.; Zhang Y.; Jiang S. Multicolor Core/Shell-Structured Upconversion Fluorescent Nanoparticles. Adv. Mater. 2008, 20, 4765–4769. [Google Scholar]
- Hirsch L. R.; Halas N. J.; West J. L.. Whole-Blood Immunoassay Facilitated by Gold Nanoshell-Conjugate Antibodies. In NanoBiotechnology Protocols; Rosenthal S. J., Wright D. W., Eds.; Methods in Molecular Biology, Vol. 303; Humana Press: Totowa, NJ, 2005; pp 101–111. [DOI] [PubMed] [Google Scholar]
- Fritz J. S.; Oliver R. T.; Pietrzyk D. J. Chelometric Titrations Using Azoarsonic Acid Indicator. Anal. Chem. 1958, 30, 1111–1114. [Google Scholar]
- Liu Y.; Ai K.; Lu L. Designing Lanthanide-Doped Nanocrystals with Both Up- and Down-Conversion Luminescence for Anti-Counterfeiting. Nanoscale 2011, 3, 4804–4810. [DOI] [PubMed] [Google Scholar]
- Jeppesen C.; Wong J. Y.; Kuhl T. L.; Israelachvili J. N.; Mullah N.; Zalipsky S.; Marques C. M. Impact of Polymer Tether Length on Multiple Ligand-Receptor Bond Formation. Science 2001, 293, 465–468. [DOI] [PubMed] [Google Scholar]
- Hansen P. L.; Cohen J. A.; Podgornik R.; Parsegian V. A. Osmotic Properties of Poly(Ethylene Glycols): Quantitative Features of Brush and Bulk Scaling Laws. Biophys. J. 2003, 84, 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuberger M.; Drobek T.; Spencer N. D. Interaction Forces and Morphology of a Protein-Resistant Poly(ethylene glycol) Layer. Biophys. J. 2005, 88, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundqvist M.; Stigler J.; Elia G.; Lynch I.; Cedervall T.; Dawson K. A. Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynamic Light Scattering - Common Terms Defined. http://www.malvern.com/en/support/resource-center/Whitepapers/WP111214DLSTermsDefined.aspx (accessed May 21, 2014).
- Shaw R. Dynamic Light Scattering Training: Achieving Reliable Nano Particle Sizing. http://149.171.168.221/partcat/wp-content/uploads/Malvern-Zetasizer-LS.pdf (accessed May 22, 2014).
- In measurements carried out on a different sample of NaGdF4 NPs, the area per molecule for mPEG2000-lysine-4P was determined to be 1.5/nm2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.