

Background: Cell surface levels of glucose transporter Glut4 are tightly controlled in adipocytes.
Results: The effects of insulin and differentiation on the trafficking kinetics of Glut4, the transferrin receptor, and LRP1 were measured to identify regulatory steps.
Conclusion: Six independent steps determine cell surface Glut4; insulin stimulates three of these.
Significance: These results provide a framework for functionally mapping treatments/proteins that affect Glut4 translocation.
Keywords: Adipocyte, Endocytosis, Exocytosis, Fibroblast, Glucose, Glucose Transport, Glucose Transporter Type 4 (Glut4), Insulin, Transferrin Receptor
Abstract
The trafficking kinetics of Glut4, the transferrin (Tf) receptor, and LRP1 were quantified in adipocytes and undifferentiated fibroblasts. Six steps were identified that determine steady state cell surface Glut4: (i) endocytosis, (ii) degradation, (iii) sorting, (iv) sequestration, (v) release, and (vi) tethering/docking/fusion. Endocytosis of Glut4 is 3 times slower than the Tf receptor in fibroblasts (ken = 0.2 min−1 versus 0.6 min−1). Differentiation decreases Glut4 ken 40% (ken = 0.12 min−1). Differentiation also decreases Glut4 degradation, increasing total and cell surface Glut4 3-fold. In fibroblasts, Glut4 is recycled from endosomes through a slow constitutive pathway (kex = 0.025–0.038 min−1), not through the fast Tf receptor pathway (kex = 0.2 min−1). The kex measured in adipocytes after insulin stimulation is similar (kex = 0.027 min−1). Differentiation decreases the rate constant for sorting into the Glut4 recycling pathway (ksort) 3-fold. In adipocytes, Glut4 is also sorted from endosomes into a second exocytic pathway through Glut4 storage vesicles (GSVs). Surprisingly, transfer from endosomes into GSVs is highly regulated; insulin increases the rate constant for sequestration (kseq) 8-fold. Release from sequestration in GSVs is rate-limiting for Glut4 exocytosis in basal adipocytes. AS160 regulates this step. Tethering/docking/fusion of GSVs to the plasma membrane is regulated through an AS160-independent process. Insulin increases the rate of release and fusion of GSVs (kfuseG) 40-fold. LRP1 cycles with the Tf receptor and Glut4 in fibroblasts but predominantly with Glut4 after differentiation. Surprisingly, AS160 knockdown accelerated LRP1 exocytosis in basal and insulin-stimulated adipocytes. These data indicate that AS160 may regulate trafficking into as well as release from GSVs.
Introduction
Insulin stimulation of glucose uptake in muscle and adipose tissue is crucial for the regulation of blood glucose homeostasis. In adipocytes, very little glucose is taken up into the cells under basal/fasting conditions. Insulin stimulates glucose transport 10–40-fold in these cells. Glucose uptake is rate-limited by the number of facilitative glucose transporters present in plasma membranes. Insulin stimulates glucose uptake by altering the subcellular distribution of glucose transporter 4 (Glut4) from intracellular stores to the plasma membrane, a process known as Glut4 translocation (1, 2). Defects in this process are observed in insulin resistance, type II diabetes mellitus, and metabolic syndrome.
Glut4 traffics through the general endocytic recycling pathway as well as through specialized compartments unique to its function (3–6). Under basal conditions, less than 5% of Glut4 is found in the plasma membrane of primary adipocytes (7). Internal Glut4 is found predominantly in small tubulo-vesicular elements and vesicles that lack endosomal markers. However, 12% of the Glut4 is co-localized with endocytosed albumin in “early” and “late” endosomes under these conditions. Consistent with this steady state subcellular distribution, Glut4 is found in two kinetically distinct pools in basal 3T3-L1 adipocytes: 70–90% of the Glut4 is in a sequestered non-cycling/very slowly cycling pool known as Glut4 storage vesicles (GSVs3; also referred to as insulin-responsive vesicles), whereas 10–30% remains in an actively cycling pool. Insulin increases the amount of Glut4 in the plasma membrane by as much as 40-fold through two effects on Glut4 trafficking: 1) insulin decreases the proportion of Glut4 sequestered in the GSVs, and 2) insulin increases the overall rate constant of exocytosis (kex) of the cycling Glut4 (8–15). Insulin has little or no effect on Glut4 endocytosis (16, 17).
In adipocytes, intracellular Glut4 partially co-localizes with the transferrin (Tf) receptor, a marker for the early endosomes. However, under basal conditions, the intracellular Tf receptor traffics 5–15 times more rapidly than Glut4 (16). The rate constant of endocytosis of the Tf receptor is also 5 times faster than Glut4. These data indicate that although Glut4 and the Tf receptor show some overlap, they are largely trafficked via different pathways/mechanisms in adipocytes. Intracellular Glut4 also co-localizes with LRP1 (low density lipoprotein receptor-related protein 1), the receptor for α2-macroglobulin (α2-M) expressed in adipocytes. Like Glut4, there are two kinetically distinct pools of LRP1 in adipocytes, a rapidly cycling pool, and a slowly cycling/non-cycling pool (16). Immunoprecipitation studies showed that LRP1 is enriched with Glut4 and insulin-regulated aminopeptidase in the insulin-responsive GSVs (18). However, LRP1 is less efficiently packaged into the sequestered GSV pool than Glut4 (40% LRP1 versus 70–90% Glut4) (16). In contrast, only a small percentage of the Tf receptor is detected in these compartments (4).
Glut4 is not expressed in 3T3-L1 fibroblast cells prior to induction of adipocyte differentiation (12). When Glut4 is exogenously expressed in these cells, it cycles constitutively and is not directed into highly regulated GSV compartments. There is only a modest 1.5–3-fold increase of exogenously expressed Glut4 at the plasma membrane in 3T3-L1 fibroblast cells (12). In contrast, insulin increases cell surface Glut4 10–20-fold in differentiated 3T3-L1 adipocytes. Although other cargo proteins found in the insulin-responsive GSVs, such as insulin-regulated aminopeptidase and LRP1, are normally expressed in fibroblasts, these proteins are also not trafficked to highly insulin-responsive compartments until differentiation induces expression of adipocyte-specific factors (12, 19). Thus, comparisons of trafficking and localization of proteins in 3T3-L1 cells before and after differentiation can yield important insights into the proteins and pathways that contribute to the specialized, highly regulated trafficking of Glut4 observed in adipocytes. For example, two proteins that play important roles in Glut4 trafficking, sortilin (20) and AS160 (21), are expressed at high levels only in adipocytes, not in fibroblasts. Co-expression of sortilin with exogenous Glut4 in fibroblasts is sufficient to reconstitute the effects of this protein on Glut4 stability and sorting that were observed in adipocytes (20) (data not shown). Expression of AS160 with sortilin in fibroblasts further alters Glut4 trafficking (21). However, trafficking of Glut4 to highly insulin-responsive GSV compartments has not yet been fully reconstituted in fibroblast cells.
Many proteins have been identified that affect the plasma membrane levels of Glut4 in adipocytes (5, 6). For some proteins, their function in Glut4 trafficking remains unclear. We have developed high throughput quantitative flow cytometric assays to measure the trafficking kinetics of Glut4, the Tf receptor, and LRP1 (15, 16). We are using these assays to functionally map proteins to specific steps in Glut4 trafficking. For example, these assays were used to examine the specific effects of knockdown of AS160 (a negative regulator of Glut4 translocation) on Glut4 trafficking in adipocytes (22). Careful analysis of the effect of AS160 knockdown on Glut4 trafficking kinetics revealed that release of Glut4 from sequestration in GSVs involves two sequential insulin-regulated rate-limiting steps. AS160 regulates the first of these; in the AS160 knockdown cells, Glut4 accumulated at a second insulin- and Akt-regulated rate-limiting step that lies downstream of AS160 but upstream of fusion with the plasma membrane. Thus, our kinetics assays allowed for the identification of a novel insulin-regulated step in the pathway.
The purpose of this study was to identify additional novel sites of regulation of Glut4 trafficking. We have identified six steps that contribute to the unique trafficking of Glut4 relative to the constitutively recycling Tf receptor. Mathematical modeling and simulations were used to estimate the rate constants for flux through these steps. Interestingly, these models were also able to accurately simulate the trafficking kinetics and relative subcellular distribution of Glut4 previously measured in primary adipocytes (7, 9, 23, 24). These analyses revealed that three steps in the Glut4 trafficking itinerary are highly regulated by insulin. They also underscore the importance of non-insulin-regulated steps, such as endocytosis and degradation, in controlling the total amount of Glut4 at the cells surface and hence in regulation of overall glucose homeostasis.
EXPERIMENTAL PROCEDURES
Tissue Culture
3T3-L1 cells were obtained from ATCC and passaged as fibroblasts in 10% calf serum in DMEM complete medium (high glucose DMEM supplemented with 2 mm l-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin). Fibroblasts were plated at just subconfluence (they reached confluence with 24 h). They were then refed in complete medium with 10% calf serum and used for experiments 1–2 days postconfluence. Cells were differentiated into adipocytes for use in experiments as described previously (15).
Viral Infections
The lentiviral HA-Glut4/GFP reporter protein was prepared and transduced into fibroblasts as described previously (15). Cells were infected at a viral titer that resulted in ∼50% of the cells expressing the construct. At this titer, the majority of infected cells contained only one virion, and no cytopathic effects were detectable (15, 22). The uninfected cells serve as internal controls to correct for cellular autofluorescence and nonspecific antibody binding/uptake. This reporter has been carefully characterized; when expressed at the low levels observed in our infected cells, the HA-Glut4/GFP reporter traffics with endogenous Glut4 (14, 15, 25, 26).
Antibodies and Reagents
HA.11 monoclonal antibody (α-HA; Covance) was purchased as ascites and purified using a 1-ml rProteinA-FF column (GE Healthcare) as described previously (15). Purified antibody was labeled with an AlexaFluor647 (AF647) protein labeling kit according to the manufacturer's instructions (Invitrogen), and free dye was removed by desalting twice into PBS using 10-ml Zeba spin columns (7000 molecular weight cut-off, ThermoScientific) (16). Human α2-M was purchased from AssayPro and labeled using an AF647 monoclonal antibody labeling kit according the manufacturer's instructions (Invitrogen) (16). α2-M was activated using methylamine as described previously (27). Free dye and excess methylamine were removed from labeled and activated α2-M using a 5-ml HiTrap desalting column (GE Healthcare). Protein concentrations and labeling efficiency were determined by absorption spectroscopy (15).
Glut4 Translocation Assay/Surface Glut4 Labeling
Cells expressing HA-Glut4/GFP were incubated for 2 h in low serum medium (LSM; DMEM complete medium and 0.5% fetal bovine serum). Cells were then incubated with or without 100 nm insulin for 45 min. To label surface Glut4, cells were rapidly chilled on an ice-water slurry and labeled with LSM containing 50 μg/ml AF647-α-HA for 1 h on ice. Antibody was removed, and cells were washed, collagenase-digested, and analyzed by flow cytometry as described below (16, 22).
Steady-state Anti-HA Antibody (α-HA) Uptake Assay
Cells were serum-starved in LSM as described above. For insulin-stimulated uptake, some cells were incubated with 100 nm insulin for the last 45 min of starvation. At various times, the incubation medium was replaced with 30 μl of warm LSM containing 50 μg/ml AF647-α-HA with or without 100 nm insulin, and incubation continued. At the end of the time course, cells were chilled, washed, collagenase-digested, and analyzed by flow cytometry (16, 22). The data show an increase in AF647-α-HA labeling from an initial level to a maximum plateau. Under the conditions of this assay, cell surface HA-Glut4 is nearly instantaneously labeled, and the antibody remains bound throughout the assay. Thus, at t = 0 (extrapolated), the labeling is equal to the steady state level of Glut4 at the cell surface, and the increase in labeling with time measures the exocytosis of intracellular Glut4 at a rate corresponding to the overall exocytosis due to all exocytic processes (13). The plateau level measures the size of the total pool of Glut4 that is cycling between the cell surface and the intracellular pools. The size of this pool is dependent on both the total amount of HA-Glut4/GFP expressed in the cell and the proportion of the total that is in actively cycling pools versus non-cycling pools. To correct for differences in the level of expression of the reporter construct in different cell types, mean fluorescence ratio (MFR) values were calculated for each sample (mean fluorescence of AF647/mean fluorescence of GFP). A difference in the maximum value of MFR (MFRmax) between cell types or experimental conditions indicates that there are differences in the proportion of the total cellular Glut4 that is cycling. The non-cycling pools include both Glut4 that can be mobilized (i.e. in the sequestered GSV pool) and Glut4 that cannot be mobilized with insulin (in the biosynthetic or degradative compartments). The α-HA data are expressed as the proportion of the total Glut4 that can be labeled in insulin-stimulated cells that is labeled at each time point (MFR/MFRmax insulin). The exocytosis rate constants, kex, were determined from fits of mathematical models to these data.
Glut4 Transition Experiments
Cells were serum-starved for 2 h as described above. For the basal to insulin transition, 100 nm insulin was added to cells for increasing amounts of time. For the transition from the insulin-stimulated to the phosphatidylinositol 3-kinase (PI3K)-inhibited state, insulin was added for the final 45 min of preincubation in LSM, and then the PI3K inhibitor LY294002 (LYi, 50 μm; EMD Biochemicals) was added for increasing amounts of time. Cells were placed on ice; surface Glut4 was labeled as described above; and then cells were washed, collagenase-digested, and analyzed by flow cytometry. In this protocol, the data indicate the level of Glut4 present in the plasma membrane at each time point and measure the rate of transition from one steady state to a new steady state. The kinetics of these transitions are determined by a combination of both exocytotic and endocytotic processes acting in the cells (e.g. kobs = kex + ken, for a two-rate/two-compartment model of the process). However, PI3K inhibitors inhibit Glut4 exocytosis ∼90% with no effect on endocytosis (16). Thus, in the insulin + LYi transition experiments, the rate of transition observed is largely determined by endocytosis alone, and fits of single exponential decay functions to this data yield estimates of the endocytic rate constant, ken. This method for measuring ken has been validated using two independent measures (16). Mathematical models were fit to the data as described below.
Transferrin Efflux
Uninfected cells were labeled with 5 μg/ml AF647-holo-Tf for 30 min at 37 °C. Cells were quickly washed and then incubated at 37 °C in LSM with 500 μg/ml unlabeled holo-Tf for increasing amounts of time. Samples were then rapidly chilled in an ice-water slurry, collagenase-treated at 4 °C, and analyzed. Incubation of cells in excess unlabeled holo-Tf was sufficient to remove all surface label. In these experiments, iron-loaded Tf (holo-Tf) binds with high affinity to its receptor at the cell surface. Once it is internalized, the Tf loses its bound iron in the low pH of the endosomes, but the iron-depleted Tf (apo-Tf) remains bound to its receptor as it is recycled from the endosomes back to the plasma membrane. Once it reaches the cell surface, the apo-Tf dissociates from its receptor and is displaced by unlabeled holo-Tf. Thus, the loss of AF647 labeling measures the rate of exocytosis of a bolus of labeled Tf. Data were fitted using a single exponential decay function, the rate of which was the exocytosis rate constant, kex (16). The endocytic rate constant, ken, was calculated from the measured kex and the partition coefficient, P (P = kex/ken = PM/(1 − PM)) as described previously (10, 16).
α2-M Uptake and Surface Labeling
Cells were incubated in serum-free DMEM (DMEM complete medium and 0.5% BSA) for 2 h and then incubated with or without 100 nm insulin for 30 min. Cells were then incubated in 4 μg/ml AF647-α2-M for increasing amounts of time. Cells were placed on ice, and additional wells on the same plate were incubated with 4 μg/ml AF647-α2-M for 90 min to label surface LRP1. Cells were gently washed with 200 μl of cold PBS and collagenase-digested for 10 min at 37 °C, allowing surface-bound AF647-α2-M to be internalized to prevent loss during analysis. Cells were analyzed by flow cytometry as described below. In these experiments, α2-M binds with high affinity to its receptor (LRP1) at the cell surface but dissociates from its receptor when it reaches the low pH of the endosomes. LRP1 recycles back to the plasma membrane, where it binds additional ligand and is reinternalized. The labeled α2-M is retained within the cell and delivered to the lysosomes for degradation. Thus, the linear increase of AF647 with time measures the rate of endocytosis of LRP1. The rate constant of LRP1 endocytosis (ken) was determined according to the method of Wiley and Cunningham (28). The internal to surface (IN/SUR) ratio for each time point was determined; the slope of the IN/SUR versus time plot is equal to ken. Data were fit using linear equations. The exocytic rate constant, kex, was calculated from ken and the partition coefficient, P, as described previously (16). Relative surface levels of α2-M binding/LRP1 receptor were verified in independent samples by labeling with biotin-conjugated anti-LRP1 antibody (clone 8G1, Fitzgerald Industries International), followed by AF647 streptavidin (Invitrogen).
Flow Cytometry
Flow cytometry, gating, and analysis were performed essentially as described (15, 16, 22). Labeled cells in 96-well plates on ice were washed three times with 200 μl of ice-cold PBS. Adipocytes were incubated with 20 μl of collagenase (Type III; 1 mg/ml in PBS with 2% BSA; Worthington) at 37 or 4 °C for 10 min and then resuspended in 180 μl of PBS. For experiments with fibroblasts, cells were resuspended in 180 μl of PBS, 0.5 mm EDTA (after incubation for an additional 10 min at 37 °C). Resuspended cells were gently filtered through a 100-μm cell strainer to remove clumps of cells and analyzed on an Accuri C6 cytometer. Detection thresholds for adipocytes were set at 1,000,000 for forward scatter height (FSC-H) and 500,000 for side scatter height (SSC-H). For fibroblast analysis, the sole threshold was 750,000 for FSC-H. Log intensities of scattered light (forward and side scatter) and fluorescence (FL1, 488 nm excitation/533 nm emission; FL2, 488 nm excitation/585 nm emission; FL3, 488 nm excitation/>670 nm emission; FL4, 640 nm excitation/675 nm emission) were collected for each cell. For analysis of adipocytes, selective gating using CFlow Plus software (Accuri) was used to analyze only the adipocytes in the sample, excluding any residual fibroblasts and cellular debris as described previously (15). For analysis of fibroblasts, cells were gated in the two-dimensional histogram of FSC-H versus SSC-H. For either cell type, cells infected with HA-Glut4/GFP could be distinguished from uninfected cells in a two-dimensional histogram of FL1-H (GFP) versus FL3-H (autofluorescence). The geometric mean fluorescence of gated populations was determined using FCS Express (De Novo Software). The mean fluorescence values of uninfected cells are a measure of cellular autofluorescence (FL1) and nonspecific antibody labeling (FL4) and were subtracted from the mean values of FL1 and FL4 measured for the infected cells in the same sample, before the MFR values were calculated (22). For analysis of α2-M or Tf uptake or binding, adipocytes or fibroblasts were gated as described above, and fluorescence levels were quantified as the geometric mean of FL4 (AF647); the mean autofluorescence measured in unlabeled cells was subtracted from these values.
Data Analysis
Fits of models to the data were performed using GraphPad Prism version 5.0 for Windows/Mac (GraphPad Software, San Diego, CA) as well as custom code implemented in Matlab (MATLAB and Statistics Toolbox Release 2013a, The MathWorks, Inc., Natick, MA). All fits were done on combined data sets, including the data points from each of n = 5–19 individual experiments. The rate constants obtained from the fits of the combined data were compared with the average of the rate constants obtained from fits of each independent experiment and were always in good agreement. The statistical significance of observed differences between steady state distributions under different experimental conditions was assessed by either t test or two-way analysis of variance (Figs. 1, 5A, and 6A), whereas differences between experimental conditions for the kinetics data were assessed by comparing fits of the data sets using either single exponents (Figs. 2 and 3) or lines (Figs. 5 (C and D) and 6 (C and D)) and calculating the p value for the null hypothesis that both sets of data were best fit by the same function (***, p ≤ 0.0001; **, p ≤ 0.001; *, p ≤ 0.01).
FIGURE 1.
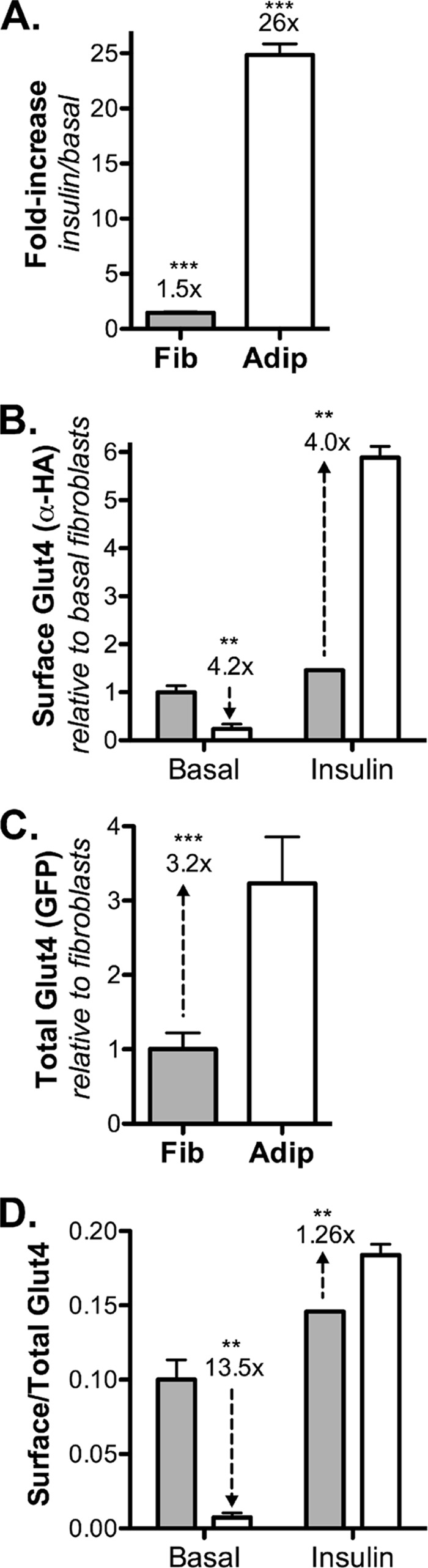
Differentiation increases Glut4 stability, promotes basal intracellular retention, and enhances Glut4 translocation after insulin stimulation. 3T3-L1 fibroblasts (Fib; gray) or adipocytes (Adip, white) expressing HA-Glut4/GFP were incubated with or without 100 nm insulin and placed on ice, surface Glut4 was labeled with AlexaFluor647-conjugated α-HA antibody (AF647-α-HA), and cells were analyzed by flow cytometry. Data are the means ± S.D. (error bars) of n = 4–10 independent experiments. A, -fold increase of surface Glut4 (insulin/basal for each cell type). B, surface Glut4. Data are mean AF647-α-HA, standardized to fibroblast basal values. Fibroblasts: basal, 1 ± 0.13; insulin, 1.5 ± 0.002. Adipocytes: basal, 0.24 ± 0.1; insulin, 5.9 ± 0.23. C, total HA-Glut4/GFP. Data are mean GFP, standardized to the average fibroblast value. Fibroblasts, 1.0 ± 0.22; adipocytes, 3.2 ± 0.6. D, surface/total Glut4; surface AF647-α-HA/total maximum AF647-α-HA uptake after insulin stimulation (Fig. 3). Fibroblasts: basal, 0.1 ± 0.01; insulin, 0.146 ± 0.002. Adipocytes: basal, 0.0074 ± 0.003; insulin, 0.184 ± 0.007. A, ***, p ≤ 0.0001, basal versus insulin. B–D, **, p < 0.001; ***, p ≤ 0.0001, fibroblasts versus adipocytes.
FIGURE 5.

Differentiation decreases cell surface LRP1/α2-M receptor and α2-M uptake by decreasing kex in both basal and insulin-stimulated cells. 3T3-L1 fibroblasts (gray) or adipocytes (white) were incubated with or without 100 nm insulin for 30 min (circles, basal; squares, +insulin). AF647-α2-M was added (with or without insulin) for the times indicated, and then cells were placed on ice and analyzed by flow cytometry. To label surface LRP1, additional wells were incubated with AF647-α2-M for 90 min at 4 °C. Data are the average of means ± S.D. (error bars) (A and C) or S.E. (error bars) (B and D) of n = 4 (fibroblasts) or n = 5 (adipocytes) independent experiments. A, surface/total LRP1, estimated by comparing the LRP1 surface binding to total labeling after chloroquine treatment (16). Fibroblasts: basal, 0.22 ± 0.02; insulin, 0.24 ± 0.02. Adipocytes: basal, 0.069 ± 0.006; insulin, 0.11 ± 0.01. Relative surface levels of LRP1 were verified by anti-LRP1 binding (data not shown). B, AF647-α2-M uptake. Lines, linear fits of the data. C and D, endocytic rate constants (ken) were calculated from the slope of the IN/SUR versus time plots (28). Fibroblasts: basal, ken = 0.41 ± 0.05 min−1; insulin, ken = 0.41 ± 0.03 min−1. Adipocytes: basal, ken = 0.28 ± 0.02 min−1; insulin, ken = 0.34 ± 0.05 min−1. E, exocytic rate constants (kex) were estimated using the partition coefficient (16); kex = (PMLRP1 × ken)/(1 − PMLRP1). Fibroblasts: basal, kex = 0.10 min−1; insulin, kex = 0.11 min−1. Adipocytes: basal, kex = 0.021 min−1; insulin, kex = 0.043 min−1. F, AF647-α2-M uptake. Lines, simulations using the model, dynamic retention with Tf receptor recycling (Table 3 and supplemental Fig. 1F). *, p ≤ 0.01; **, p < 0.001 fibroblasts versus adipocytes.
FIGURE 6.

AS160 knockdown in adipocytes increases surface LRP1/α2-M receptor and α2-M uptake by increasing kex in both basal and insulin-stimulated cells. Control adipocytes expressing a nonspecific shRNA (white) or AS160 knockdown adipocytes (black) were treated and analyzed as described in Fig. 5. Data are the average means ± S.D. (error bars) (A and C) or S.E. (error bars) (B) of n = 7 independent experiments. A, surface/total LRP1. Control: basal, 0.06 ± 0.005; insulin, 0.13 ± 0.01. AS160 KD: basal, 0.10 ± 0.009; insulin, 0.15 ± 0.02. B, AF647-α2-M uptake. Lines, linear fits of the data. C and D, endocytic rate constants (ken) were calculated from the slope of the IN/SUR versus time plots (28). Control: basal, ken = 0.27 ± 0.01 min−1; insulin, ken = 0.31 ± 0.02 min−1. AS160 KD: basal, ken = 0.39 ± 0.03 min−1; insulin, ken = 0.41 ± 0.03 min−1. E, estimated exocytic rate constants (kex). Control: basal, kex = 0.017 min−1; insulin, kex = 0.046 min−1. AS160 KD: basal, kex = 0.043 min−1; insulin, kex = 0.072 min−1. F, AF647-α2-M uptake. Lines, simulations (Table 3 and supplemental Fig. 1F). *, p ≤ 0.01; **, p < 0.001, control versus AS160 KD.
FIGURE 2.
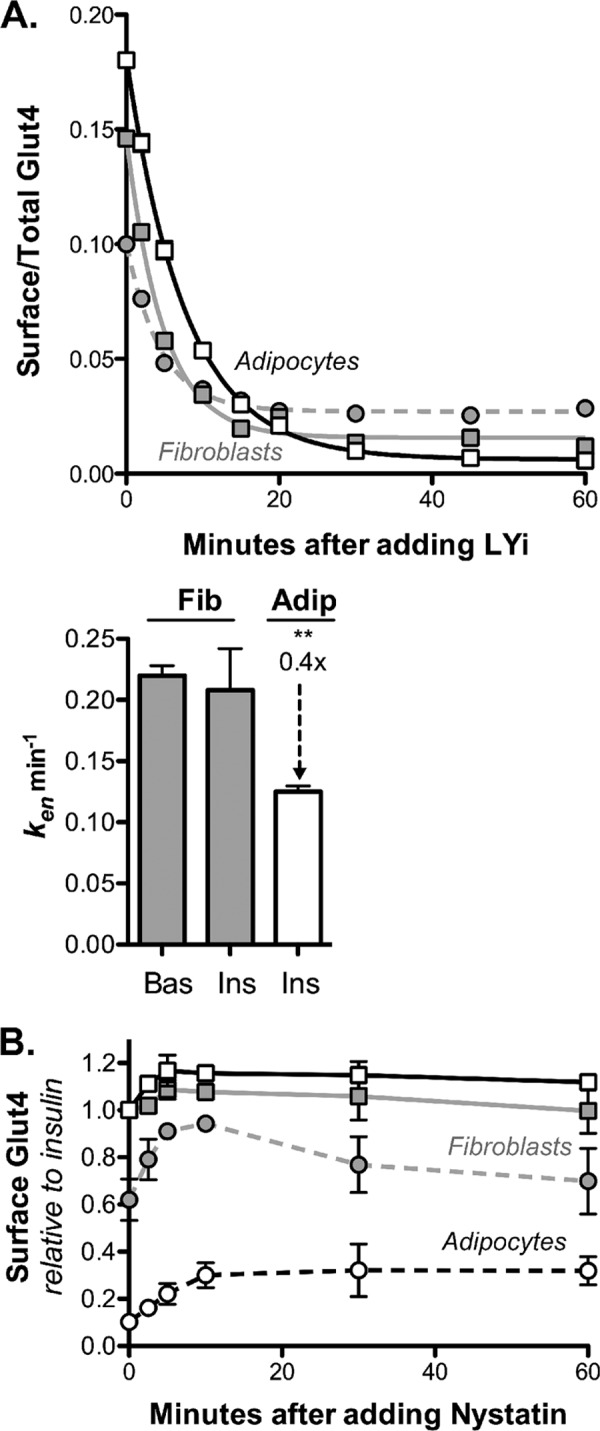
Differentiation decreases the rate constant of Glut4 endocytosis (ken) by 40%. A, 3T3-L1 fibroblasts (gray) or adipocytes (white) expressing HA-Glut4/GFP were incubated with (Ins; squares) or without (Bas; circles) 100 nm insulin for 45 min, and then either the PI3K inhibitor LYi (50 μm) (A) or nystatin (50 μg/ml) (B) was added for the indicated times (LYi inhibits exocytosis, and nystatin inhibits endocytosis of Glut4). Cells were placed on ice, labeled with AF647-α-HA, and analyzed by flow cytometry. A, data points are the average surface/total Glut4 ± S.E. (error bars) for n = 3–6 (fibroblast) or 16 (adipocyte) independent experiments. Lines, single exponential fits (kobs ≈ ken) (16) Fibroblasts: basal, ken = 0.22 ± 0.01 min−1; insulin, ken = 0.21 ± 0.03 min−1; adipocytes: insulin, ken = 0.12 ± 0.005 min−1). **, p < 0.001, fibroblasts versus adipocytes. B, data are the average of means ± S.E. of surface Glut4, standardized to control insulin, from two independent experiments. p < 0.001 control versus +nystatin in both basal fibroblasts and adipocytes at the maximal effect of nystatin treatment (5–10 min fibroblasts, 20–60 min adipocytes; n = 4–12 samples).
FIGURE 3.
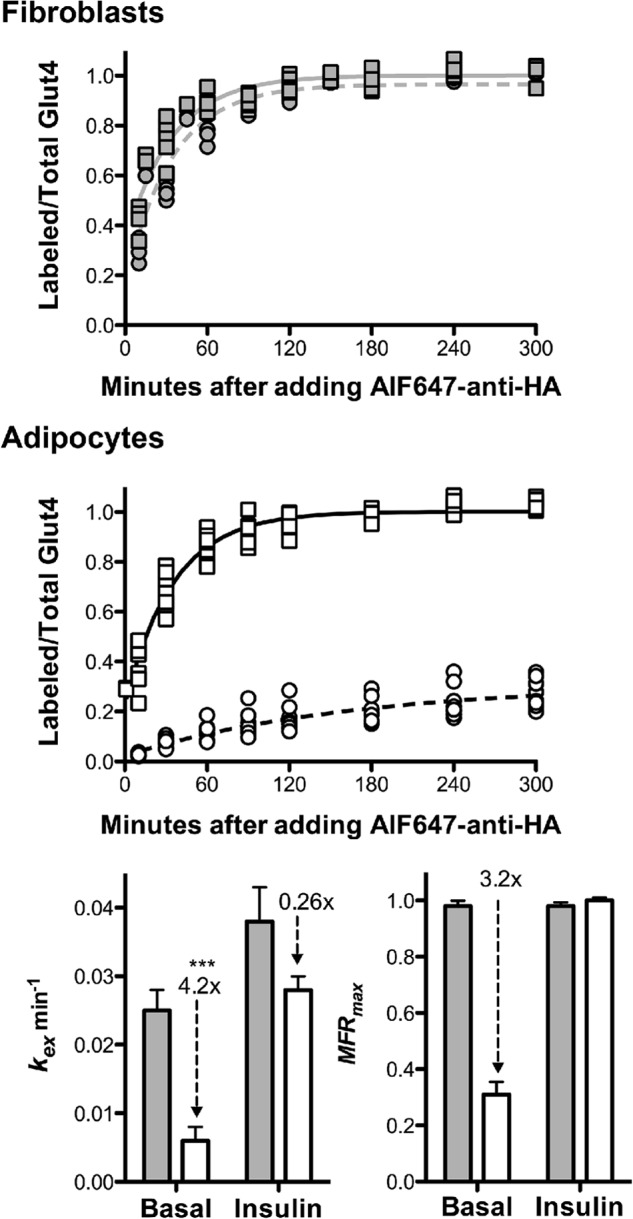
Differentiation decreases the rate constant of Glut4 exocytosis (kex) and sequesters Glut4 in basal adipocytes. 3T3-L1 fibroblasts (gray) or adipocytes (white) expressing HA-Glut4/GFP were incubated with (squares) or without (circles) insulin (100 nm) for 45 min and then incubated with AF647-α-HA (with or without insulin) at 37 °C for the times indicated. Individual data points from n = 6 (fibroblasts) or n = 9 (adipocytes) independent experiments are shown. Data points are the average MFR (AF647/GFP) of n = 2–3 samples, ∼1000 cells/sample, standardized to MFRmax insulin for each independent experiment (Labeled/Total Glut4). Lines, single exponent to MFRmax fits of the combined data sets (kobs = kex; MFRmax = cycling pool). Fibroblasts: basal, kex = 0.025 ± 0.003 min−1, MFRmax = 0.98 ± 0.02; insulin, kex = 0.038 ± 0.005 min−1, MFRmax = 0.98 ± .014. Adipocytes: basal, kex = 0.006 ± 0.002 min−1, MFRmax = 0.31 ± 0.05; insulin, kex = 0.027 ± 0.002 min−1, MFRmax = 1.0 ± 0.02. ***, p < 0.0001 basal adipocytes versus insulin-stimulated adipocytes or fibroblasts. Error bars, S.D.
Three simple mathematical models of Glut4 exocytosis were compared for each cell type (fibroblasts versus adipocytes) and experimental condition (basal versus insulin-stimulated) (supplemental Fig. 1): 1) dynamic equilibrium, single exponent to 1 (a single exocytic pathway with a single intracellular pool of Glut4) (11, 14); 2) static retention, single exponent to MFRmax (a single exocytic pathway with two pools of Glut4, an actively cycling pool and a sequestered (non-cycling) pool) (12, 13); and 3) two exocytic pathways, two exponents to 1 (two intracellular pools of Glut4 with independent exocytic pathways). In dynamic equilibrium, the total amount of Glut4 that is cycling is insulin-independent, whereas in static retention, the total amount of Glut4 that is cycling is insulin-dependent. In the two-exocytic pathway model, the distribution of Glut4 between the two intracellular pools is insulin-dependent, but the total is insulin-independent. To compare the models, the α-HA uptake data in Fig. 3 were fit in PRISM using the following mathematical functions: dynamic equilibrium, a single exponent constrained to a reach a plateau of 1 (but not constrained to start at 0); static retention, a single exponent with a variable plateau (not constrained to start at 0); and two exocytic pathways, two exponents constrained to reach a combined plateau of 1 (not constrained to start at 0). For these fits, the Akaike information criterion method was used to select the model most likely to have generated the data.
To compare the ability of more complicated models to simulate the behavior of Glut4, simultaneous fits of four data sets were done. The data sets included two α-HA uptake experiments (basal and insulin; Fig. 3) and two transition experiments (basal to insulin and insulin + LYi; Figs. 2 and 4) for each cell type. Systems of coupled ordinary differential equations were identified for each model explicitly representing the output of each type of experiment (see supplemental material and supplemental Tables 1 and 2). These were then fitted using a combination of numerical integration and nonlinear least squares regression to optimize the parameter values simultaneously over the entire range of experiments and experimental data.
FIGURE 4.
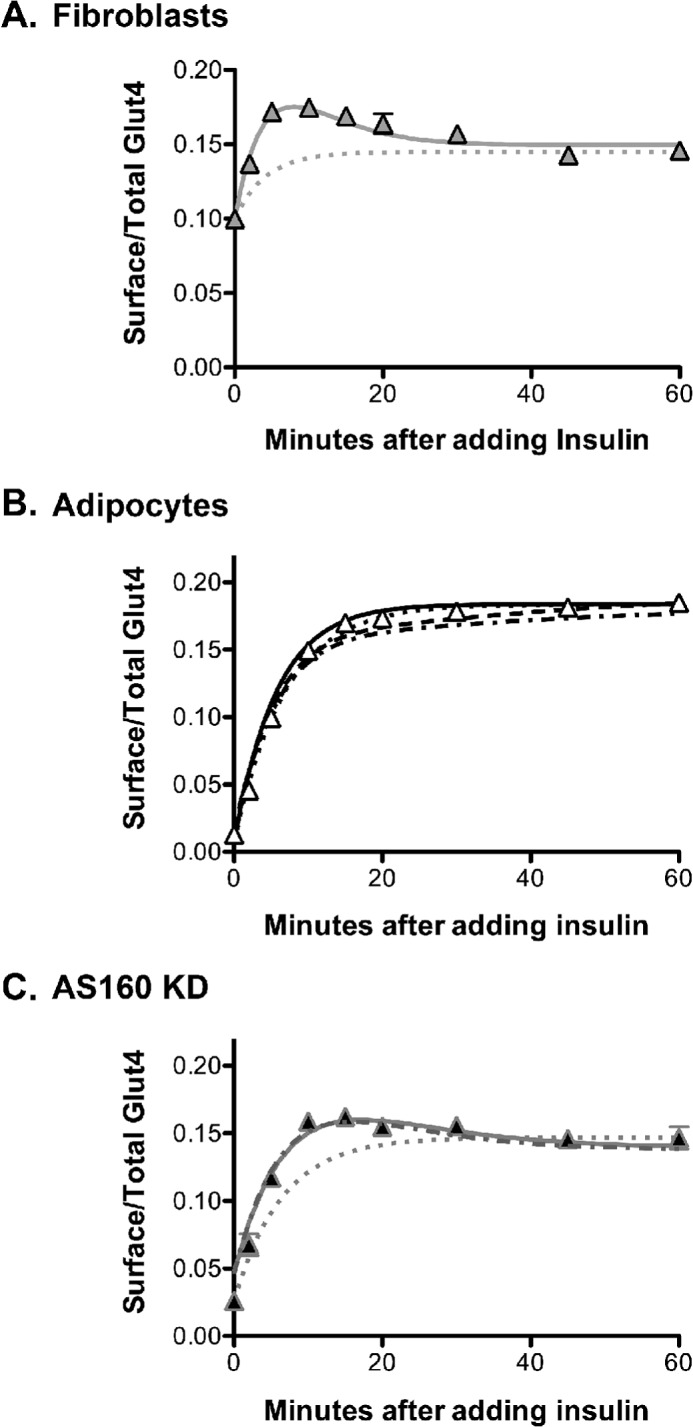
The kinetics of the transition from basal to insulin-stimulated levels of surface Glut4 are different in adipocytes and fibroblasts. 3T3-L1 fibroblasts (A), control adipocytes (B), or AS160 KD adipocytes expressing HA-Glut4/GFP (C) were stimulated with insulin for the indicated times and then placed on ice and labeled with AF647-α-HA. Data are the average mean ± S.E. (error bars) of surface/total Glut4 from n = 10 (fibroblasts), n = 19 (adipocytes), or n = 5 (AS160 KD) independent experiments. Lines, simulations using different models. Dotted lines, static retention; solid lines, three-step model; dashed lines, dynamic retention, constrained (fibroblast) fits; dotted/dashed lines, dynamic retention, alternate (primary adipocyte) fits (Table 2 and supplemental material).
The coupled ordinary differential equation systems were solved using a one-step explicit Dormand-Prince (Runge-Kutta) method (29), and the nonlinear regression fitting these to the data employed a reflective trust region least-squares algorithm (30–34) implemented in Matlab. From this, both estimates of the parameter values and the 95% confidence intervals could be determined. In performing a nonlinear regression to data, the width of the confidence interval in S.E. values is related to the percentage of observations falling within the confidence interval. To estimate the S.E., Student's t inverse cumulative distribution function was used to calculate the number of S.E. values spanned by the confidence interval (35). Finally, the confidence interval was divided by the number of S.E. values spanned by the confidence interval to find the S.E. for the parameter.
RESULTS
Differentiation Increases Glut4 Protein Stability and Alters Its Subcellular Distribution
Glut4 is not expressed in undifferentiated 3T3-L1 fibroblasts. Differentiation induces the expression of not only Glut4 but also the proteins required for its specialized insulin-dependent trafficking (12, 20, 21). This is clear when an exogenously expressed HA-Glut4/GFP reporter construct is expressed in both cell types (Fig. 1). In fibroblasts, insulin has only a modest effect on the distribution of HA-Glut4/GFP (a transient 1.8-fold increase, which decreases to a stable 1.5-fold increase; Figs. 1A and 4). In contrast, insulin increases cell surface HA-Glut4/GFP 26-fold in adipocytes.
Whereas -fold stimulation shows insulin responsiveness, the total amount of Glut4 at the plasma membrane controls the rate of glucose transport in adipocytes and muscle. As expected, differentiation decreased total Glut4 at the plasma membrane 4.2-fold in basal adipocytes compared with fibroblasts (Fig. 1B). Unexpectedly, differentiation increased Glut4 at the plasma membrane 4-fold after insulin stimulation.
The increase in cell surface Glut4 in insulin-stimulated adipocytes relative to fibroblasts is largely due to an increase in Glut4 expression. Differentiation increased the total amount of the HA-Glut4/GFP reporter in cells 3.2-fold (Fig. 1C). This is due to an increase in the stability of the Glut4 protein and not due to transcriptional regulation. The phosphoglycerate kinase 1 promoter used to express the exogenous reporter does not show differentiation dependence when used to express GFP alone (data not shown). In contrast, the widely used CMV promoter is highly suppressed in adipocytes relative to the fibroblasts when used for the stable expression of proteins in the lentiviral vector (data not shown). An increase in Glut4 protein will lead to a proportional increase in cell surface Glut4, independent of effects on trafficking.
In 3T3-L1 fibroblasts, 10% of the total Glut4 is at the plasma membrane in basal cells, and 14.6% is at the plasma membrane after insulin stimulation (Fig. 1D and Table 1). After adipocyte differentiation, 0.7% of the total is at the plasma membrane in basal cells, whereas 18.4% is at the plasma membrane after insulin stimulation. Thus, differentiation caused a 13.5-fold decrease in the percentage of total Glut4 in the plasma membrane in basal adipocytes relative to fibroblasts and a 26% increase in the percentage of total Glut4 in the plasma membrane after insulin stimulation (Fig. 1D). A change in the relative distribution of Glut4 between the cell surface and intracellular pools requires effects on Glut4 trafficking. Therefore, differentiation affects Glut4 trafficking under both basal and insulin-stimulated conditions.
TABLE 1.
Summary of rate constants (k) and distribution of proteins (fraction of total) in the PM and sequestered/noncycling GSVs
Rate constants and distributions were measured or calculated (calc) from the following equations as described (Figs. 1–5): GSV = 1 − MFRmax; P = kex/ken; PMcalc = MFRmax × (P/1 + P); ken(calc) = kex × ((1 − PM)/PM); kex(calc) = ken × (PM/(1 − PM)).
| GLUT4 |
Tf receptor |
α2-MR (LRP1) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PM | ken | kex | GSV | PMcalc | PM | ken(calc) | kex | PM | ken | kex(calc) | |
| min−1 | min−1 | min−1 | min−1 | min−1 | min−1 | ||||||
| Fibroblasts | |||||||||||
| Basal | 0.10 | 0.2 | 0.025 | 0.11 | 0.25 | 0.6 | 0.2 | 0.22 | 0.41 | 0.10 | |
| Insulin | 0.146 | 0.2 | 0.038 | 0.16 | 0.25 | 0.6 | 0.2 | 0.24 | 0.41 | 0.11 | |
| Adipocytes | |||||||||||
| Basal | 0.007 | 0.12 | 0.006 | 0.69 | 0.015 | 0.17 | 0.6 | 0.12 | 0.069 | 0.28 | 0.021 |
| Insulin | 0.184 | 0.12 | 0.027 | 0.18 | 0.25 | 0.6 | 0.2 | 0.112 | 0.34 | 0.043 | |
Differentiation Slows the Rate of Glut4 Endocytosis in both Basal and Insulin-stimulated Cells
Glut4 is actively cycling between the plasma membrane and intracellular pools under both basal and insulin-stimulated conditions. The percentage of Glut4 at the plasma membrane is dependent on the rate of insertion into the plasma membrane (exocytosis) and rate of retrieval from the plasma membrane (endocytosis) as well as the size of the actively cycling pool.4 Therefore, if the proportion of the total Glut4 at the plasma membrane changes after differentiation, then differentiation must be affecting one of these elements. The rates of endocytosis and exocytosis are dependent on the intrinsic rate constants of endocytosis (ken) and exocytosis (kex) as well as the amount of Glut4 in the plasma membrane and intracellular cycling pool. The distribution of Glut4 in the plasma membrane versus the intracellular cycling pool as well as the overall rate constants of endocytosis and exocytosis can be estimated via two independent assays (16).
To measure Glut4 endocytosis, cells were treated with the PI3K inhibitor LYi to inhibit exocytosis, and the loss of cell surface Glut4 with time was measured (Fig. 2A). In this assay, the rate constant for the relaxation to the new steady state distribution of surface Glut4, kobs, is approximately equal to the endocytosis rate constant, ken. Differentiation caused a 40% decrease in ken, from 0.2 min−1 in fibroblasts to 0.12 min−1 in adipocytes (Table 1). Insulin had no effect on this rate constant in either cell type (Fig. 2A) (16). Thus, Glut4 is internalized significantly more slowly in adipocytes than in fibroblasts. A decrease in ken, with no effect on kex, will increase the amount of Glut4 on the cell surface. Interestingly, using similar assays, the endocytic rate constant measured in primary adipocytes was very slow, ∼0.053 min−1 (0.047–0.059 min−1) (9, 23, 24).
In adipocytes and muscle, Glut4 is internalized via two different routes: one that is nystatin-sensitive and one that is nystatin-insensitive and AP2- and clathrin-dependent (5, 17, 36, 37). We therefore examined whether Glut4 is internalized via a nystatin-sensitive endocytic pathway in fibroblasts as well as in adipocytes. Nystatin transiently increased cell surface Glut4 1.5-fold in basal fibroblasts and 3-fold in basal adipocytes (Fig. 2B). This increase is due to a transient 40% inhibition of ken in basal fibroblasts and a more sustained 70% inhibition in basal adipocytes. Thus, Glut4 is internalized via a nystatin-dependent pathway in fibroblasts as well as in adipocytes. Therefore, the difference in endocytosis between fibroblasts and adipocytes is not due to differential expression of the nystatin-dependent pathway. In addition, the nystatin-sensitive pathway is the major pathway in basal cells, whereas the nystatin-insensitive pathway is the predominant pathway after insulin stimulation in both cell types. Despite this shift in mechanism of internalization, however, insulin has little to no effect on the ken for Glut4 in either cell type, indicating that the two pathways have similar rate constants (16, 17).
Differentiation Slows the Rate of Glut4 Exocytosis, but Only under Basal Conditions
To measure Glut4 exocytosis, basal or insulin-stimulated cells were incubated at 37 °C with AF647-labeled α-HA, and the accumulation of labeled antibody was measured with time (Fig. 3) (16, 22). Within the time resolution in these experiments, Glut4 is instantly labeled. Because Glut4 remains labeled as it cycles through the cell, the rate constant for antibody uptake, kobs, is the overall exocytosis rate constant, kex, due to all processes bringing Glut4 to the plasma membrane.
In fibroblasts, α-HA uptake rapidly approached a plateau value, with a t½ ≈ 30 min in basal cells and t½ ≈ 20 min after insulin stimulation. The proportion of the total that is labeled did not change after insulin stimulation in these cells (the MFRmax did not change with insulin stimulation in fibroblasts; MFR = AF647-α-HA/GFP). In insulin-stimulated adipocytes, α-HA uptake also rapidly approached a plateau value, with a t½ ≈ 25 min. Thus, differentiation had little effect on exocytosis of Glut4 after insulin stimulation. Differentiation also had little effect on the proportion of the total cellular Glut4 that is labeled after insulin stimulation (MFRmax was not statistically different in fibroblasts and insulin-stimulated adipocytes; data not shown). Therefore, there is not a significant difference in the two cell types in the proportion of Glut4 accumulated in non-cycling pools (i.e. in the biosynthetic or degradative pathways). The major effect of differentiation on Glut4 trafficking was a large decrease in the rate of exocytosis of Glut4 in basal adipocytes. Less than 25% of the total HA-Glut4/GFP in the cell that can be labeled after insulin stimulation was labeled in basal adipocytes after a 5-h incubation. α-HA uptake had not reached a plateau value by the end of the experiment in basal cells.
Two Kinetically Distinct Pathways Are Required to Accurately Simulate the Basal Trafficking of Glut4 after Differentiation
The α-HA uptake data for fibroblasts and for adipocytes after insulin stimulation is well fitted using a single exponent (dynamic equilibrium, single exponent to 1; Supplemental Fig. 1A). Using single exponential fits, the estimated values for kex for Glut4 were 0.025 min−1 in basal fibroblasts, 0.038 min−1 in insulin-stimulated fibroblasts, and 0.027 min−1 in insulin-stimulated adipocytes (Table 1). These values are similar to the estimated value for kex for Glut4 in primary adipocytes after insulin stimulation (0.032 min−1), when measured at steady state under conditions similar to our assays (cells maintained at 37 °C in the continuous presence of insulin) (9).
In contrast, the α-HA uptake data in basal 3T3-L1 adipocytes can only be well fit by a single exponent if the model includes a non-cycling (static) sequestered pool (static retention, single exponent to MFRmax; supplemental Figs. 1B and 2B) (12, 13, 15, 16). In this model, the total amount of Glut4 that is cycling is insulin-dependent. Fits of the basal data with this model yield kex = 0.006 min−1 and MFRmax = 0.31 (∼70% of the Glut4 is sequestered in non-cycling GSVs; Table 1). The basal adipocyte uptake data are not well fit if the model is forced to reach a plateau of 1 (supplemental Fig. 2A) (12, 13, 15, 16). Comparing the two models using the Akaike information criterion, dynamic equilibrium was rejected in favor of static retention (86.5% probability that static retention is the model that generated the observed data, with a ratio of probabilities = 6.4). The two models yield nearly identical fits of the fibroblast data and the adipocyte data after insulin stimulation.
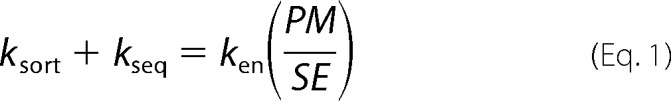
The data in Figs. 1–3 were analyzed using the static retention model to describe the trafficking of the actively cycling Glut4. Mathematically, this is a two-step/two-compartment model with two rate constants: ken (plasma membrane to endosomes) and kex (endosomes to plasma membrane), with the total amount of Glut4 dependent on insulin. The ratio of Glut4 in the plasma membrane (PM) versus sorting endosomes (SE) is the partition coefficient, P, which is equal to the ratio of the rate constants for exocytosis and endocytosis (P = kex/ken = PM/SE) (10, 22). The amount of Glut4 at the plasma membrane is also dependent on the size of the cycling pool, MFRmax. Thus, PMcalc = MFRmax × (P/(1 + P)).
As previously observed (16, 22), there is good agreement between the measured values and the calculated expected values of Glut4 in the plasma membrane in 3T3-L1 fibroblasts and adipocytes after insulin stimulation (Table 1). Application of this same analysis to the data from primary adipocytes after insulin stimulation also shows excellent agreement between calculated and measured values (P = 0.032/0.053; PMcalc = 38%, PMmeasured = 39–42%) (7, 9, 10, 22). In contrast, the calculated value for surface Glut4 in basal adipocytes is twice as high as the observed value (PMcalc = 0.015; PMmeasured = 0.007; Table 1). Therefore, the static retention model is insufficient to fully account for the basal data.
Alternatively, the basal α-HA uptake data can be fit using two exponents. In this model, the intracellular Glut4 is present in two separate intracellular pools with different exocytic rate constants. Two-exponent fits of the basal data yield a small pool that is exocytosed rapidly (Total1 = 0.09, kex1 = 0.032 min−1) and a large pool that is exocytosed very slowly (Total2 = 0.91, kex2 = 0.0007 min−1). This model is the preferred model relative to static retention for the basal uptake data (80% probability that observed data were generated by a process with two pools with different exocytic rate constants rather than a cycling pool and a non-cycling static pool; ratio of probabilities = 4). In contrast, the two-exponent model is rejected for the uptake data from the fibroblasts and from adipocytes after insulin stimulation (>99.99% probability that the one-exponent model is correct). Thus, in basal adipocytes, the data can be best fit by a model with two pathways for exocytosis: a constitutive pathway with kinetics similar to the pathway observed in fibroblasts (the endosomal recycling pathway through endosomal recycling intermediate compartments (ERC) (kfuseE = 0.032 min−1, total in ERC = 9%) and a specialized pathway through GSVs that is very highly regulated by insulin (kfuseG = 0.0007 min−1, total in GSVs = 91%; supplemental Figs. 1C and 2C). In contrast, in fibroblasts and in adipocytes after insulin stimulation, Glut4 is either predominantly exocytosed via a single pathway or through multiple pathways with similar rate constants.
Although the exocytic rate constants and relative amounts of Glut4 in each pathway can be determined from two-exponential fits of the basal α-HA uptake data, the expected level of Glut4 at the plasma membrane cannot be calculated from these fits. A more complicated mathematical model that includes pathways for delivery of Glut4 into the two pools from the plasma membrane was needed to simulate the Glut4 trafficking data. In order to help build this model, the kinetics of trafficking of Glut4 in fibroblasts and adipocytes was examined in more detail.
Three Sequential Steps Are Required to Accurately Simulate the Trafficking of Glut4 in Fibroblasts and AS160 Knockdown (KD) Adipocytes
The cell surface, α-HA uptake, and insulin + LYi transition data yield information about the rates of insertion and retrieval of Glut4 from the plasma membrane. However, these assays cannot resolve individual steps in the intracellular trafficking pathways; the measured rate constants, ken and kex, are the net result of all steps in multiple trafficking pathways. In contrast, measuring the kinetics of the transition from the basal to insulin-stimulated state can resolve intermediate steps in the intracellular pathways (10, 22). For example, previous work using cells in which AS160 was knocked down showed that there are at least two sequential insulin-regulated steps in the Glut4 exocytic pathway in adipocytes (22). There is an overshoot in the basal to insulin transition experiment in adipocytes after AS160 knockdown that cannot be simulated using a model with a single rate-limiting step in the exocytic pathway.
There is also an overshoot observed in fibroblasts in the basal to insulin transition experiment (Fig. 4). Glut4 transiently increases at the plasma membrane 1.8-fold within 5 min after insulin stimulation. This increase then gradually decays to only 1.5-fold by 30–40 min. A mathematical model with a single rate-limiting step in exocytosis is insufficient to account for this overshoot (dotted line). Thus, the basal to insulin transition data show that Glut4 traffics through multiple kinetically distinct sequential steps in fibroblasts.
To determine the minimal model that accurately describes the observed fibroblast data, simultaneous fits to a three-step model (supplemental Fig. 1D) were done for the fibroblast data in Figs. 2–4 (four different experiments: two α-HA uptake experiments, basal and insulin, and two transition experiments, insulin + LYi and basal to insulin). The three-step model includes three sequential rate constants: ken (PM to SE), ksort (SE to ERC), and kfuseE (ERC to PM). The overall exocytic rate constant, kex, is a function of both ksort and kfuseE. Three hypotheses were utilized to model the effect of insulin on the system: ksort regulated by insulin (Δksort); kfuseE regulated by insulin (ΔkfuseE); and both ksort and kfuseE regulated by insulin (Δksort and ΔkfuseE), with the other rates held constant (insulin-independent; Table 2 and supplemental Table 3, a and b). To model the transitions, the system is assumed to instantly change to the new rate constants upon application of insulin or LYi. The total recycling pool was constrained to be insulin-independent.
TABLE 2.
Rate constants used for simulations
Rate constants were generated for models from simultaneous fits of basal and insulin anti-HA uptake, + LYi transition, and basal to insulin transition data for 3T3-L1 fibroblasts and adipocytes (Figs. 2–4) or from steady state subcellular distribution data in primary adipocytes (7). See supplemental Table 3 for 95% confidence bounds, S.E. values, and data on the goodness of fit.
| ken | ksort |
kfuseE |
kseq |
kfuseG |
||||
|---|---|---|---|---|---|---|---|---|
| Basal | Insulin | Basal | Insulin | Basal | Insulin | |||
| Three-step: Δkfuse | ||||||||
| 3T3-L1 fibroblasts, free fita | 0.18 | 0.053 | 0.033 | 0.080 | ||||
| 3T3-L1 adipocytes, free fita | 0.11 | 0.350 | 0.001 | 0.027 | ||||
| AS160 KD adipocytes, free fita | 0.12 | 0.055 | 0.006 | 0.032 | ||||
| Dynamic retention: two exocytic pathways, GSV and ERC | ||||||||
| 3T3-L1 adipocytes, constrained fita | 0.12 | 0.042 | 0.025 | 0.088 | 0.021 | 0.059 | 0.0007 | 0.025 |
| Primary adipocytes, calculatedb | 0.05 | 0.013 | 0.018 | 0.078 | 0.006 | 0.143 | 0.0005 | 0.05 |
| 3T3-L1 adipocytes, alternate fitc | 0.11 | 0.013 | 0.018 | 0.078 | 0.006 | 0.143 | 0.0006 | 0.028 |
| AS160 KD adipocytes, alternate fitc | 0.12 | 0.013 | 0.018 | 0.078 | 0.018 | 0.030 | 0.005 | 0.028 |
a Determined from free fits or constrained fits (ksort and kfuseE from fibroblasts) of the kinetics data.
b Calculated from the steady state distributions using Equations 1–3.
c Determined from constrained fits using ksort, kfuseE, and kseq from primary adipocytes instead of fibroblasts. For AS160 KD, kseq was estimated from LRP1 surface data (Table 3) as described.
In order to simulate the observed overshoot in fibroblasts, the third rate constant, kfuseE, must be rate-limiting in basal cells, whereas the second rate constant, ksort, must be rate-limiting after insulin stimulation. The fibroblast data can be well simulated using a three-step model with only kfuseE regulated by insulin (Fig. 4A, solid line). This fit yielded well defined rate constants (with S.E. values <10%) for all three steps and an excellent goodness of fit (R2 = 0.95; supplemental Table 3a). An unconstrained free fit of the data, allowing both kfuseE and ksort to change with insulin, gave nearly the same fit as allowing only kfuseE to change, except that both basal and insulin ksort were poorly defined (they had very large S.E. values; see supplemental Table 3b). This analysis indicates that there are two rate-limiting steps in Glut4 trafficking through the constitutive pathway in fibroblasts and that insulin increases the rate of transit through the second step, with little or no effect on the first.
In contrast to fibroblasts, there was no overshoot observed in the basal to insulin transitions in adipocytes. In an unconstrained simultaneous fit of the adipocyte data, the three-step model is essentially reduced to a two-step model, with ksort ≫ kfuse (Table 2). In contrast to the fits from fibroblast data, ksort remained poorly defined (supplemental Table 3c). Although this model yields a good fit of the transition data (Fig. 4B, solid line), it is not a good fit of the basal α-HA uptake data in adipocytes (supplemental Fig. 2D). This model requires an additional non-cycling (static) pool to accurately simulate the kinetics data in adipocytes.
As observed in fibroblasts, the three-step model yields an excellent fit of the transition experiment in AS160 KD adipocytes (Fig. 4C, solid line). The fusion step is rate-limiting in basal AS160 KD cells, whereas the trafficking step is rate-limiting after insulin stimulation (Table 2). However, the fusion step is much slower in the basal AS160 KD adipocytes than in fibroblasts, and it is more highly regulated by insulin. In these fits, the three rate constants were well defined, and there was an excellent goodness of fit (R2 = 0.96; see supplemental Table 3d). However, although the Three-Step model can also simulate basal α-HA uptake in AS160 KD cells (data not shown), a model with two exocytic pathways is preferred relative to the single exponent, three-step model (80% probability that the observed basal uptake data were generated by a process with two separate pools with different exocytic rate constants rather than a single cycling pool; ratio of probabilities = 4). Two exponent fits of the basal AS160 KD α-HA uptake data indicate that AS160 knockdown has two effects; it accelerates the rate of cycling of the GSV pool and also expands the size of the “constitutive” cycling pool of Glut4 (kfuseG = 0.004 min−1, total in GSV = 65%; kfuseE = 0.032 min−1, total in ERC = 35%). However, the majority of the Glut4 continues to cycle through the specialized GSV pathway in basal AS160 KD cells. Thus, the transition data indicate that the GSV pathway also has multiple rate-limiting steps (22).
Dynamic Retention, a Mathematical Model with Two Exocytic Pathways, Accurately Simulates the Trafficking of Glut4 in Adipocytes
Our mathematical analysis and simulations suggest that Glut4 traffics through two kinetically distinct cycling pools in basal adipocytes: a constitutive (fibroblast-like) endosomal pathway and a very slowly cycling specialized GSV pool. Furthermore, they suggest that both of these pathways have at least two sequential rate-limiting steps. Based on this, we created a mathematical model that combines the best fits from the fibroblast data while incorporating a second exocytic route via the GSVs (dynamic retention; supplemental Fig. 1E). In this model, Glut4 is internalized via a single pathway with a single rate constant, ken, and is delivered to an SE. This rate constant is insulin-independent. Sorting endosomes are defined as the compartments where Glut4 is either sorted into constitutive ERC with a single rate constant (ksort) or sequestered into GSVs through an independent process with a second rate constant (kseq). Glut4 in the ERC is delivered to the PM with a single rate constant, kfuseE, that is insulin-dependent. The release of GSVs from sequestration and fusion to the plasma membrane is also controlled by a single rate constant, kfuseG, that is regulated by insulin. Although similar models have been proposed in previous studies (4–6, 11, 26), neither mathematical simulations nor fits of kinetics data using this model have been reported previously.
To determine if dynamic retention can accurately describe the observed kinetics data, simultaneous fits to this five-step/four-compartment model were done for the adipocyte data in Figs. 2–4. Data from four different experiments were analyzed: two α-HA uptake experiments (basal and insulin), and two transition experiments (the insulin + LYi transition and the basal to insulin transition), utilizing various hypotheses for the effects of insulin on the system (supplemental Table 3e) (data not shown). The rate constants determined from the simultaneous fits of the adipocyte data (Table 2, dynamic retention, 3T3-L1 adipocytes, constrained fit) yield an excellent simulation of the basal to insulin transition experiment (Fig. 4B, dashed line) and both the basal and insulin α-HA uptake experiments in control adipocytes (supplemental Fig. 2E, solid lines), with an excellent goodness of fit (R2 = 0.99; supplemental Table 3e). Thus, the kinetics data in adipocytes can be very well represented using a model in which a small amount of the Glut4 continues to traffic through its default (fibroblast) pathway in adipocytes, whereas the majority of the Glut4 is redirected into a highly regulated specialized pathway after differentiation. In this model, differentiation has no effect on the constitutive pathway (Table 2). The major effect of differentiation is the formation of the highly regulated GSV pathway. The GSV pathway is rate-limited by the fusion of GSVs, kfuseG, which increases 38-fold with insulin. The two pathways diverge in sorting endosomes and converge at the plasma membrane.
One of the main goals of this study was to develop a model that would allow us to test hypotheses about where proteins/treatments affect Glut4 trafficking. We have a good idea of the overall structure of the trafficking pathways from our assay conditions and data fitting to different mathematical models. However, in fits of the data with the dynamic retention model, the rate constants for trafficking from the sorting endosomes into the two exocytic pathways, ksort and kseq, remain poorly defined, as evidenced by the large S.E. values (supplemental Table 3e). These steps are not rate-limiting under the conditions of our experiments, so they can vary widely in the simulations with little effect on the overall rate constants of exocytosis. However, the ratio of these two rate constants is important; kseq must be slow relative to ksort under basal conditions to avoid overaccumulation of Glut4 in GSVs and fast after insulin to maintain flux through the GSV pathway.
We independently verified our estimates for the rate constants of endocytosis, ken, and overall exocytosis, kex, by comparing the measured values for the relative amounts of Glut4 in the plasma membrane with the expected values calculated from these rate constants based on the partition coefficients (Table 1). Likewise, if the relative distribution of Glut4 between the PM, SE, ERC, and GSVs were known, it would be possible to independently verify the estimates for ksort, kseq, kfuseE, and kfuseG that were derived from the fits using the measured value for ken. The inferred steady state values of Glut4 in each compartment can be calculated as a function of the rate constants (supplemental Table 1). Rearranging these equations, we find that although the relative amounts of Glut4 in each compartment do not give definitive values for each of the rate constants, they do place constraints on what these rate constants can be.
 |
 |
 |
The relative distribution of Glut4 in these functionally defined compartments in 3T3-L1 adipocytes is unknown. However, the steady state subcellular distribution of Glut4 has been carefully examined and quantified in primary adipocytes using electron microscopy (7). Based on the electron microscopy data and the known functions of these morphological compartments from many studies, the steady state data in primary adipocytes were used to estimate the amounts of Glut4 in our kinetically defined compartments: PM, 2.3% basal, 42.1% insulin; SE, 6.5% basal, 14.3% insulin; GSV, 86% basal, 41% insulin; and ERC, 5.2% basal, 2.4% insulin.5 Based on this distribution, we obtained estimated values for ksort, kseq, kfuseE, and kfuseG in primary adipocytes (Table 2). The inferred values for kfuseE and kfuseG from the steady state distributions in primary adipocytes were similar to those obtained in the optimized fits of the 3T3-L1 adipocyte data. The values for ksort and kseq were quite different, however. The primary adipocyte data show that the overall rate constant for exit of Glut4 from early/sorting endosomes must be slower in basal adipocytes (ksort + kseq = 0.019 min−1; Equation 1) than in fibroblasts (ksort = 0.053 min−1). This suggests that differentiation inhibits transport of Glut4 out of endosomes.
Strikingly, the distribution of Glut4 in primary adipocytes shows that insulin must increase the overall rate constant for exit of Glut4 from “early endosomes” 8-fold in the primary adipocytes (ksort + kseq = 0.156 min−1 after insulin stimulation; Equation 1).6 The 20-fold increase in Glut4 at the plasma membrane after insulin stimulation is expected to cause a 20-fold increase in the amount of Glut4 in the next downstream compartment (early/sorting endosomes) unless there is also a change in the rate constant for exit from the compartment. The amount of Glut4 in the early/sorting endosomes increases only 2.2-fold, however. Thus, the exit of Glut4 from early/sorting endosomes is a newly identified insulin-regulated step in Glut4 trafficking in adipocytes. Using the calculated values for ksort, kseq, and kfuseE from primary adipocytes in simulations of trafficking in 3T3-L1 adipocytes (Table 2, dynamic retention, 3T3-L1 adipocytes, alternate fit) yields excellent fits of the basal to insulin transition data and the α-HA uptake data in both basal and insulin-stimulated cells (Fig. 4B and supplemental Fig. 2E, dotted /dashed lines). In fact, these rate constants gave the same goodness of fit (R2 = 0.99) as the fits where kseq was unconstrained (supplemental Table 3, e and f).
Glut4 Cycles through a Different Pathway than the Transferrin Receptor in Fibroblasts
What is the nature of the constitutive pathway that Glut4 cycles through in fibroblasts? Previous work had shown that in adipocytes, Glut4 predominantly trafficked through compartments that were kinetically distinct from the rapid recycling pathway followed by the Tf receptor (16). However, based on co-localization studies, it has been suggested that Glut4 might be following the same pathways as the Tf receptor in fibroblasts (38). If this were true, it is expected that the rate constants of endocytosis and exocytosis would be similar for these two proteins in fibroblasts but would differ after differentiation. Inconsistent with this hypothesis, however, the rates of both endocytosis and exocytosis of Glut4 are significantly slower than the Tf receptor in fibroblasts as well as in adipocytes (Table 1). The rate constant of endocytosis of the Tf receptor was 3-fold higher than Glut4 in fibroblasts (ken = 0.6 min−1 versus 0.2 min−1). The rate constant of exocytosis was 5–8-fold higher for the Tf receptor than for Glut4 in fibroblasts (kex = 0.2 min−1 versus 0.025–0.04 min−1). Differentiation had very little effect on the rate of cycling of the Tf receptor. Therefore, even in fibroblast cells, a cell type that normally does not express Glut4, Glut4 exhibits specialized trafficking relative to constitutive endosomal proteins. Thus, the constitutive cycling pathway for Glut4 in both fibroblasts and adipocytes is different than the fast Tf cycling pathway, despite the overlap in distribution of these proteins. These data show that fibroblasts have both a slow and a fast recycling pathway and that Glut4 has intrinsic sorting sequences that target it to the slow pathway in fibroblasts. This is consistent with microscopy studies showing that although these two proteins overlap, they are largely exocytosed via different vesicles (26, 39, 40).
It is interesting that there is a 5-fold difference in the rate constants of endocytosis of the Tf receptor and Glut4, even under conditions where both proteins are internalized predominantly via clathrin-coated pits (i.e. in adipocytes after insulin stimulation). This difference indicates that there are differences in the mechanisms or possibly in the efficiency of packaging of these two proteins into clathrin-dependent pathways at the cell surface. The differences in the mechanisms of internalization between Glut4 and the Tf receptor (nystatin-sensitive versus nystatin-insensitive, slow clathrin-dependent versus fast clathrin-dependent) are observed in cells that normally do not express Glut4. Therefore, both the adipocytes and the non-differentiated cells have multiple pathways for endocytosis, and Glut4 has intrinsic sorting sequences that target it to the slow internalization pathways.
Differentiation Slows Both Endocytosis and Exocytosis of LRP1
LRP1, an α2-M receptor expressed in adipocytes, is highly co-localized with Glut4 and is sequestered with Glut4 in GSVs (18). Consistent with this, we have previously reported that both Glut4 and LRP1 cycle significantly more slowly than the Tf receptor in adipocytes (16). LRP1 has a trafficking itinerary different from that of the Tf receptor. Unlike the Tf receptor, which releases its cargo (iron) in mildly acidic early endosomes, LRP1 carries its cargo into the more acidic later endosomal compartments for delivery to lysosomes and is recycled from a later compartment (41). Therefore, we hypothesized that the default pathway for Glut4 exocytosis in fibroblasts might be through this alternative recycling pathway.
To determine the rate constants for the internalization and exocytosis of LRP1, the kinetics of the uptake of its ligand, α2-M, were measured (Fig. 5). In fibroblasts, LRP1 was cycling at a rate that was intermediate between the values for Glut4 and the Tf receptor (ken = 0.41 min−1, kex = 0.125 min−1; Table 1). These results suggest that in fibroblasts, LRP1 could be distributed in two cycles: the fast (Tf receptor) and slow (Glut4) endocytic and recycling pathways. The overall observed endocytic or exocytic rate constants, kobs LRP1, would be a weighted average of the individual rate constants for the two different pathways (kobs = XkTf + (1 − X)kGlut4, where X is the proportion of LRP1 internalized and recycled via the fast Tf receptor pathways). If this model is correct, then 60–65% of the LRP1 is internalized and recycled with the Tf receptor in fibroblasts, whereas 35–40% is internalized and recycled through the slower Glut4 pathway in these cells (Table 3).
TABLE 3.
Modeling LRP1 trafficking through fast (Tf receptor) and slow (Glut4) cycling pathways
| ken(obs) | Endocytic pathwaya |
Basal/Insulin |
||||
|---|---|---|---|---|---|---|
| Fast (Tf receptor) | Slow (Glut4) | PMobs | PMsim | krec | ||
| % | % | |||||
| Fibroblasts | ||||||
| LRP1 | 0.4 | 65 | 35 | 0.22/0.24 | 0.22/0.24 | 0.24/0.15 |
| Glut4 | 0.2 | 30 | 70 | 0.10/0.15 | 0.10/0.16 | |
| Adipocytes | ||||||
| LRP1 | 0.3 | 40 | 60 | 0.06/0.13 | 0.06/0.13 | 0.20/0.12 |
| Glut4 | 0.12 | 10 | 90 | 0.007/0.18 | 0.01/0.18 | |
| AS160 KD adipocytes | ||||||
| LRP1 | 0.4 | 65 | 35 | 0.10/0.15 | 0.10/0.15 | 0.20/0.12 |
| Glut4 | 0.12 | 10 | 90 | 0.03/0.15 | 0.046/0.14 | |
a Percentage of the total internalized through each pathway. Xfast = (kobs − kslow)/kfast − kslow), Xslow = 1 − Xfast. ken(fast) = 0.6 min−1, ken(slow) = 0.053 min−1. ken(obs), PMobs, measured values from Figs. 1, 2, and 5 (fibroblasts) and Fig. 6 (adipocytes and AS160 KD adipocytes). PMsim, simulations of the three-step model: Δkfuse (fibroblasts) or dynamic retention, alternate fit (control and AS160 KD adipocytes) models (Table 2) with an additional fast direct recycling step from sorting endosomes to the PM with a single rate constant, krec, added to the models (supplemental Fig. 1F). krec(TfR) = 0.12 min−1 basal, 0.2 min−1 insulin; ksort(TfR) and kseq(TfR) = 0.
Differentiation decreased cell surface LRP1 in both basal and insulin-stimulated cells 2–3-fold (Fig. 5A). Differentiation also led to a 3–5-fold decrease in the rate of uptake of labeled α2-M in adipocytes versus fibroblasts (Fig. 5B). As with Glut4, there was a small (30%) decrease in the rate constant of endocytosis of LRP1 with differentiation (Fig. 5, C and D). However, this small decrease in ken is insufficient to account for the 3–5-fold decrease in α2-M uptake that is observed between fibroblasts and adipocytes. The decrease is due primarily to the decrease in the amount of LRP1 at the cell surface. A decrease in ken would increase cell surface LRP1, not decrease it. Therefore, differentiation must also be decreasing the overall rate constant of LRP1 exocytosis 3–5-fold, decreasing the number of cell surface receptors (Fig. 5E).
LRP1 is highly co-localized with Glut4 in adipocytes, including in the specialized GSVs (18). However, it is less efficiently sequestered than Glut4 in these cells. We hypothesized that this is due to the fact that LRP1 can be recycled from the endosomes either through the fast Tf receptor pathway or through the slow, regulated Glut4 pathways. To test this hypothesis, a mathematical model was built with an additional fast recycling pathway from the sorting endosomes to the plasma membrane, with a single rate constant, krec (dynamic retention with Tf receptor recycling; supplemental Fig. 1F). Using our estimates for the other rate constants (Table 2), the values of krec required to generate the observed cell surface levels of LRP1 in each cell type under basal and insulin-stimulated conditions were determined (Table 3). krec ranged between 0.12 and 0.24 min−1 in both adipocytes and fibroblasts, very close to the exocytic rate constants measured for the Tf receptor in these cells (0.12–0.2 min−1). The model was then used to simulate α2-M uptake, yielding an excellent fit of the data (Fig. 5F). Thus, the difference in trafficking between LRP1 and Glut4 can be very well described by a model with LRP1, but not Glut4, recycled to the cell surface with the Tf receptor from sorting endosomes. In this model, the sorting endosome is the compartment where three exocytic pathways diverge.
AS160 Knockdown in Adipocytes Accelerated Exocytosis of LRP1 under Both Basal and Insulin-stimulated Conditions
To verify that LRP1 traffics with Glut4 through both the constitutive and the highly regulated GSV pathways, the effects of AS160 knockdown on α2-M uptake and LRP1 subcellular distribution were determined (Fig. 6). AS160 knockdown increases cell surface Glut4 in basal adipocytes 3–5-fold (Table 3) (22, 42–44). This increase is due to an increase in the exocytosis of GSVs, with little effect on Glut4 endocytosis (supplemental Fig. 2F) (22, 43, 44). Therefore, release of LRP1 from the GSVs with Glut4 was expected to increase cell surface LRP1 in basal AS160 KD cells, through an increase in LRP1 exocytosis. This was what was observed. AS160 KD increased cell surface LRP1 1.7-fold in basal adipocytes (Fig. 6A). As expected, there was an increase in α2-M uptake in basal AS160 knockdown cells (Fig. 6B). This was due to the increase in cell surface LRP1, a small increase (30–40%) in the rate constant of endocytosis of LRP1 (Fig. 6, C and D), and a 2.5-fold increase in LRP1 kex (Fig. 6E).
Unexpectedly, however, there was also a significant increase in α2-M uptake after insulin stimulation in AS160 knockdown cells relative to control cells, with a small (15%) increase in cell surface Glut4 (Fig. 6, A and B). In contrast, AS160 knockdown inhibits Glut4 translocation 17–20% in insulin-stimulated cells (Table 3) (22, 43, 44). The increase in α2-M uptake was due in part to the small increase in ken, but there was also a 1.6-fold increase in kex for LRP1 in AS160 knockdown cells relative to control cells after insulin stimulation (Fig. 6E). Thus, AS160 knockdown accelerated the overall rate constant of exocytosis of LRP1 in insulin-stimulated cells. In contrast, AS160 knockdown inhibited Glut4 kex in insulin-stimulated adipocytes, with no effect on Glut4 endocytosis (22, 43, 44).
In previous studies, the inhibition of maximal translocation of Glut4 observed in AS160 KD cells after insulin stimulation was assumed to be due to effects of AS160 knockdown on the maximal rate of fusion of GSVs to the plasma membrane. However, this would decrease, not increase, LRP1 exocytosis. In our mathematical model, any treatments that increase the amount of Glut4 and LRP1 in the sorting endosomes would be expected to increase the rate constant of exocytosis of LRP1, but not Glut4. An increase in the residency time of LRP1 in endosomes would allow for greater efflux of LRP1 through the fast Tf receptor pathway, accounting for the acceleration of LRP1 exocytosis. In contrast, Glut4 cannot exit through the fast Tf receptor pathway and would remain rate limited by the slow Glut4 pathways. Therefore, we hypothesized that AS160 knockdown affected the rate constant of trafficking of Glut4 from sorting endosomes into sequestered GSVs (kseq) as well as the rate constant of fusion of GSVs to the plasma membrane (kfuseG). We tested this hypothesis using mathematical simulations.
The effect of AS160 knockdown in basal adipocytes was modeled as a partial increase in the rate of release and fusion of GSVs, kfuseG (Table 2, dynamic retention, AS160 KD adipocytes, alternative fit), as described previously (22). This was sufficient to account for the increase in basal α-HA uptake observed in these cells (supplemental Fig. 2E) (22). An additional small increase in kseq was required in basal AS160 KD cells to prevent overaccumulation of LRP1 at the cell surface (Table 3). To account for both the decrease in the exocytic rate constant for Glut4 and in the increase for LRP1, the effect of AS160 knockdown in insulin-stimulated cells was modeled as an inhibition of kseq, with all other rate constants held the same as in control adipocytes. Inhibition of sequestration was sufficient to account for both the elevated surface levels of LRP1 and the decreased surface levels of Glut4 observed in the AS160 KD cells after insulin stimulation (Table 3). Simulations of trafficking using these rate constants yielded excellent fits of the α-HA uptake data (supplemental Fig. 2E) and the α2-M uptake data (Fig. 6F) in both basal and insulin-stimulated AS160 KD adipocytes. Furthermore, they yielded an excellent fit of the complex basal to insulin transition observed in AS160 KD cells (Fig. 4C, dotted/dashed line). There was no significant difference in the goodness of fit to the Glut4 data in AS160 KD cells between a model with both kseq and kfuseG affected by knockdown and the previously used model with effects on only kfuseG (supplemental Table 3, g and h; R2 = 0.96). Therefore, the data and simulations suggest not only that AS160 regulates the release of Glut4 and LRP1 from sequestration in GSVs but that it may also regulate the efficient packaging of Glut4 and LRP1 into the GSVs from the endosomes.
DISCUSSION
Differentiation of 3T3-L1 fibroblasts into adipocytes induces the expression of both Glut4 and proteins that regulate the subcellular distribution of Glut4. Differentiation affects surface Glut4 in both basal and insulin-stimulated cells. In basal adipocytes, there is 4.2-fold less total Glut4 at the plasma membrane than in basal fibroblasts, whereas in insulin-stimulated adipocytes, there is 4-fold more Glut4 at the plasma membrane than in insulin-stimulated fibroblasts (Fig. 1B). There are four possible independent ways of affecting the amount of Glut4 at the plasma membrane: 1) change the rate constant of endocytosis; 2) change the overall rate constant of exocytosis; 3) change the total amount of Glut4 expressed in the cells; or 4) change the distribution of Glut4 in actively cycling versus sequestered non-cycling/very slowly cycling compartments. Using kinetics assays that allow each of these to be measured, it was determined that differentiation affects all of these processes. In fact, six steps were identified that account for the distinctive trafficking of Glut4 relative to the constitutive, rapid recycling of the Tf receptor in adipocytes (Fig. 7).
FIGURE 7.
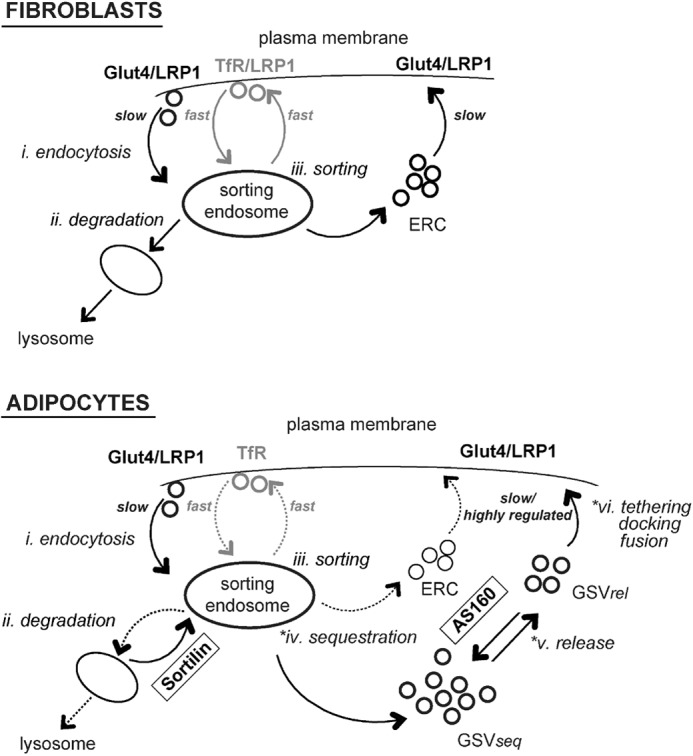
Dynamic retention. Six steps account for the unique trafficking of Glut4 compared with the constitutively cycling Tf receptor. *, three of these steps are highly insulin-regulated in adipocytes, increasing 10–30-fold with insulin. i, endocytosis; there are multiple pathways for endocytosis in both fibroblasts and adipocytes, the fast (Tf receptor) pathway, and a slower (Glut4) pathway. ii, degradation; differentiation induces the expression of proteins, including sortilin, that redirect Glut4 from lysosomal degradation pathways. iii, sorting; there are two pathways for constitutive recycling from sorting endosomes in both in fibroblasts and adipocytes, the fast (Tf receptor) pathway and a slower (Glut4) recycling pathway. *iv, sequestration; differentiation induces a second recycling pathway for Glut4 in basal adipocytes, a highly regulated non-cycling/very slowly cycling pathway through GSVs (GSVseq, sequestered GSVs). Unexpectedly, insulin increases the rate constant for transfer from sorting endosomes into GSVs. *v, release; the GSVs are rapidly mobilized in response to insulin (GSVrel, released GSVs), through a process regulated by AS160. Our data and modeling suggest that AS160 may also be required for formation of GSVs. *vi, tethering/docking/fusion; GSV exocytosis is regulated by a second, AS160-independent, Akt- and PI3K-dependent mechanism.
Endocytosis
In 3T3-L1 adipocytes, the rate constant for endocytosis of Glut4 (ken = 0.12 min−1) is slow relative to the Tf receptor (ken = 0.6 min−1) (9, 23, 24). This rate constant is even slower in primary adipocytes (ken = 0.053 min−1). This 5–11-fold difference in the rate constants of endocytosis is consistent with the difference in the function of these two proteins. Due to this slow endocytic rate constant, Glut4 will tend to remain at the cell surface, which is where it is active, once inserted into the plasma membrane. In contrast, the Tf receptor is rapidly internalized and recycled to deliver its cargo. Interestingly, chronic insulin stimulation of primary adipocytes inhibits Glut4 translocation by increasing ken, with little effect on kex (ken = 0.1 min−1, kex = 0.03 min−1, PM = 23%) (24). Thus, possible effects on ken should be considered for treatments that affect Glut4 translocation, despite the fact that acute insulin stimulation has little effect on this process.
Further supporting an important role for endocytosis in the regulation of glucose homeostasis, differentiation of 3T3-L1 cells causes a 40% decrease in the rate constant of endocytosis of Glut4 (from 0.2 to 0.12 min−1; Fig. 2 and Table 1). We do not know the basis of this decrease in ken. A simple explanation for the observed difference would be that there are two pathways for endocytosis, a fast one (ken(fast) = 0.6 min−1) and a slow one (ken(slow) = 0.053 min−1), and that differentiation shifts the distribution of Glut4 between these two pathways (30% fast in 3T3-L1 fibroblasts, 10% fast in 3T3-L1 adipocytes, 0% fast in primary adipocytes; Table 3). This mechanism can also explain the change in endocytic rate constants measured for LRP1. Perhaps differentiation changes the expression or function of adaptor proteins that target Glut4 and LRP1 into the fast versus slow pathways. Likewise, any treatments (e.g. mutations in Glut4) that change the amount of Glut4 internalized via the fast pathway versus the slow pathway will affect cell surface Glut4 levels, despite the fact that the fast endocytic pathway has little role in normal Glut4 trafficking in adipocytes (45–47).
Retrieval of Glut4 from the Degradative Lysosomal Pathway
The 4-fold increase in total surface Glut4 observed after insulin stimulation in adipocytes compared with fibroblasts can be largely accounted for by an increase in total Glut4 protein. Differentiation increased the total amount of the Glut4 reporter 3.2-fold (Fig. 1C). This increase is due to an increase in protein stability and not a change in protein expression. Therefore, differentiation induces the expression of proteins that protect Glut4 from degradation in lysosomes. The increase in Glut4 stability can be phenocopied in fibroblasts by the expression of sortilin, a protein whose expression increases significantly during differentiation (20). Co-expression of sortilin with a Glut4 reporter protein in fibroblasts increased Glut4 levels 3-fold, whereas shRNA-mediated knockdown of sortilin in adipocytes decreased HA-Glut4/GFP levels 30% (20) (data not shown). Although sortilin expression affected the total amount of Glut4 in the cell and thus the total amount of Glut4 at the cell surface, it had little effect on the kinetics of Glut4 trafficking or the relative distribution of Glut4 between the intracellular compartments and the cell surface (data not shown). Similarly, any treatments that affect Glut4 stability will affect the total amount of Glut4 protein at the cell surface, even if they have no effect on Glut4 trafficking kinetics. Thus, it is important to measure the effects of treatments on both the total amount of Glut4 expressed in cells and the proportion of the total Glut4 that is at the cell surface.
The total amount of surface Glut4 controls the rate of glucose transport into adipocytes and muscle. The amount of Glut4 at the plasma membrane is directly proportional to the total amount of Glut4. Therefore, retrieval of Glut4 from degradation is another trafficking step important in overall glucose homeostasis that is not acutely regulated by insulin.
Sorting into Intermediate Compartments (ERC) for Recycling from Endosomes
In fibroblasts, Glut4 is exocytosed 5–8 times more slowly than the Tf receptor. Therefore, even in non-specialized cells, there are multiple routes for recycling from the endocytic pathway, and there are intrinsic signals in Glut4 that target it to the slow recycling pathway. In our model, we designate this as the ERC, but the exact nature of the kinetic intermediate between the early/sorting endosomes and the plasma membrane that represents the rate-limiting step observed in our kinetics data is unclear. The fact that LRP1 also cycles through this slow pathway indicates that it may be a recycling intermediate on the late endosome/lysosome degradative pathway. This is consistent with electron microscopy data (7, 48). The fact that there are both fast and slow exocytic pathways means that a possible mechanism to increase cell surface Glut4 (or LRP1) would be to redirect Glut4 from the slow constitutive recycling pathway through the ERC into the more rapidly recycling Tf receptor pathway. Therefore, although the fast Tf recycling pathway is not part of normal Glut4 trafficking, any treatments (including mutations in Glut4) that redirect Glut4 from the slow recycling pathway into the fast recycling pathway would increase cell surface Glut4. Thus, this mechanism should be considered when evaluating treatments/mutations that affect “sequestration” of Glut4. Treatments/mutations that increase Glut4 exocytosis through the Tf receptor pathway will affect Glut4 surface levels in both fibroblasts and adipocytes. They are also expected to accelerate Glut4 exocytosis (and increase cell surface Glut4) in both basal and insulin-stimulated cells.
Sequestration of Glut4 into the Specialized, Highly Regulated GSVs
In basal adipocytes, Glut4 cycles through two kinetically distinct pools, the endosomal cycling pool and a highly regulated sequestered pool in GSVs (15, 22). Thus, differentiation induces expression of proteins that redirect Glut4 from its constitutive recycling pathway into the highly regulated GSVs. There are two models that can fit the observed data. The first is that the Glut4 in GSVs is static, not cycling (static retention; supplemental Fig. 1B) (12, 13). In this model, the rate of exocytosis in the constitutive cycling pool is also affected by differentiation. It is inhibited in basal cells (kex = 0.006 min−1, t½ ≈ 2 h) so that it is 4 times slower than the kinetics of exocytosis of the cycling pool in fibroblasts (kex = 0.025 min−1). Insulin stimulates the quantal release of Glut4 from GSVs, and it also increases the rate constant of exocytosis from the constitutive pathway. This is the model used to analyze the α-HA uptake data (Fig. 3 and Table 1) and yields an excellent fit of the kinetics data in both control and AS160 knockdown adipocytes (22).
An alternative model that fits the data equally well is that in basal adipocytes, the GSVs are cycling at a very slow rate (kex ≤ 0.0007, t½ ≥ 16 h), and there is a small pool (∼10%) of Glut4 that is cycling through a compartment with kinetics similar to the fibroblast pathway (dynamic retention; supplemental Fig. 1E). The observed exocytic rate constant measured in the α-HA uptake assay would be a weighted average from these two pathways. This model gives an excellent fit of the basal to insulin transitions and uptake data in both control and AS160 KD adipocytes (Fig. 4, B and C, dotted/dashed lines, and supplemental Fig. 2E). The major difference between these two models is whether differentiation affects the endosomal ERC recycling pathway as well as the GSV pathway (in “static retention,” both must be affected, whereas in dynamic retention, only the GSV pathway is highly regulated).
Interestingly, when either model is used to analyze the steady state distribution of Glut4 in primary adipocytes, it is revealed that the movement of Glut4 into GSVs must be regulated by insulin. It must be slow (or 0) in basal cells, to allow some Glut4 to remain cycling through the endosomal pathway, and it must increase at least 8-fold after insulin stimulation to clear Glut4 from the endosomes and to repopulate the GSVs. Any treatments that inhibit the increase in the rate constant of trafficking of Glut4 from the early/sorting endosomes into the GSVs (kseq) will decrease Glut4 at the cell surface in insulin-stimulated cells but only in adipocytes.
In contrast to Glut4, it is expected that treatments that decrease trafficking of Glut4 and LRP1 into the GSV pathway (increase their concentration in the endosomal cycling pathway) will accelerate the exocytosis rate of LRP1 in insulin-stimulated cells. We have also observed that treatments that inhibit Glut4 sequestration in GSVs (increase its concentration in endosomes) increase the degradation of Glut4 in lysosomes (e.g. AS160 knockdown decreases total Glut4 protein levels by 25%) (22).
Release of the Sequestered GSVs
AS160 is required for sequestration of Glut4; shRNA-mediated knockdown of AS160 in adipocytes causes the redistribution of the Glut4 from GSVs into the constitutive cycling pool (22). However, exogenous expression of AS160 in fibroblasts is insufficient to cause Glut4 sequestration (data not shown). Co-expression of AS160 and sortilin is also not sufficient to induce sequestration (data not shown). As observed for AS160 knockdown, any treatments that affect packaging and retention of Glut4 in GSVs will affect Glut4 levels at the plasma membrane. Consistent with this, knockdown of the AS160 substrates Rab10 and Rab14 affect sequestration of Glut4 and decrease cell surface Glut4 in adipocytes.7 Modeling and simulations of this data indicate that Rab10 is required for release of Glut4 from sequestration in GSVs, whereas Rab14 is required in packaging/sorting of proteins from endosomes into GSVs, consistent with recent microscopy studies (40, 48). Interestingly, although Glut4 traffics through specialized GSVs in muscle cells, kinetics studies show that this compartment is not as tightly regulated as in adipocytes (49, 50). It remains to be determined whether the difference between adipocytes and muscle is due to different pathways for trafficking of Glut4 in the two cell types or to different signal transduction pathways with different stringency of regulation of the trafficking through the same pathways in both cell types.
Tethering/Docking/Fusion
AS160 knockdown releases Glut4 from the GSVs, but there is an additional step that regulates the rate of fusion of the released vesicles (22). The proteins that control this step remain unidentified but are regulated by insulin in part through Akt and phosphatidylinositol 3-phosphates (22). This step affects GSVs downstream from AS160. Therefore, the minimal mathematical model that can account for all of the kinetics data is a six-step, five-pool model, with two insulin-regulated steps in the GSV pathway: the AS160-dependent priming step and the AS160-independent tethering/docking/fusion step (supplemental Fig. 3). This step is implicitly, not explicitly, represented in our four-pool model (supplemental Fig. 1, E and F). Release and fusion affect a single rate constant, kfuseG. In a simultaneous free fit of the adipocyte data, the six-step/five-pool model is reduced to a five-step/four-pool model, with the fusion step much faster than the release step (kfuseG ≫ krel; supplemental Table 3, f and i). Additional data are required to estimate krel and kfuseG. Likewise, it is likely that other steps in the kinetics model are actually multiple independent steps (e.g. “GSVs” are a collection of four different morphological compartments) (7).5 Additional data will be required to resolve these steps.
The rate constant of tethering/docking/fusion of GSVs to the plasma membrane (kfuseG) is very highly regulated in adipocytes, whereas transport to the plasma membrane from the ERC is less highly regulated. However, treatments that inhibit the exocytosis from either pathway will decrease cell surface Glut4. Treatments that affect exocytosis from the ERC will affect trafficking in both fibroblasts and adipocytes, whereas treatments that affect exocytosis of GSVs will only reduce surface Glut4 levels in adipocytes. In addition, inhibition of the tethering/docking/fusion step is expected to slow down the basal to insulin transition (22). Thus, inhibition of the basal to insulin transition in adipocytes, with no effect in fibroblasts, is a defining phenotype for treatments that inhibit the final fusion of GSVs.
In the “dynamic retention” model, differentiation changes the expression or function of proteins that inhibit GSV but not ERC exocytosis. Therefore, it is expected that affecting these regulatory proteins would affect trafficking only in adipocytes, not in fibroblasts. In contrast, the “static retention” model predicts that differentiation must change the expression/function of proteins that regulate exocytosis from the constitutive ERC as well as GSVs. These proteins would effect Glut4 trafficking in both fibroblasts and adipocytes. It remains to be determined which model is correct.
Conclusions
These observations support the dynamic retention model depicted in Fig. 7. Six steps account for the unique trafficking of Glut4 compared with the Tf receptor: endocytosis, degradation, sorting, sequestration, release, and tethering/docking/fusion (numbered i–vi in Fig. 7). Three of these steps are highly regulated by insulin. There are two pathways of endocytosis in both fibroblasts and adipocytes, the fast (Tf receptor) pathway and a much slower (Glut4) pathway. LRP1 is internalized through both pathways in both cell types. Differentiation induces the expression of proteins, including sortilin, that redirect Glut4 from lysosomal degradation, increasing its stability. In fibroblasts, there are two pathways for protein recycling from endosomes to the plasma membrane, a fast pathway utilized by the Tf receptor and a slower pathway utilized by Glut4. LRP1 recycles through both pathways in fibroblasts. Differentiation redistributes LRP1 into the slow Glut4 cycling pathways. Differentiation induces a second kinetically distinct recycling pathway for Glut4 in basal adipocytes, the sequestered non-cycling/very slowly cycling GSV pathway. LRP1 is also sorted into the GSV compartments. Unexpectedly, we found that the rate constant for trafficking into the GSVs in adipocytes is a novel regulatory step in Glut4 traffic; insulin increases kseq >8-fold. The GSVs are rapidly mobilized in response to insulin. AS160 is essential for sequestration of Glut4 in the GSVs; shRNA knockdown of AS160 releases Glut4 from sequestration. Our data and simulations suggest that AS160 inhibits the release of GSVs from sequestration and also regulates GSV formation. Differentiation induces the expression of proteins that regulate vesicle fusion independently of AS160. These proteins are regulated in part through Akt and PI3K.
This model provides a framework for mapping the sites of action of proteins/treatments on Glut4 trafficking. In addition, we have described the phenotypes observed or expected when each of these six steps is perturbed. These phenotypes are being used to identify additional proteins required for each of these steps and to help interpret the effects of treatments/proteins on Glut4 translocation in adipocytes.
Supplementary Material
Research supported by grants from the American Diabetes Association (7-08-RA-100, 1-12-BS-132, and 1-12-BS-16) and from the National Institutes of Health (Grant T32 DK007058 to E. N. H.)

This article contains supplemental Figs. 1–3 and Tables 1–3.
Glut4 that is not cycling (i.e. that is accumulated in biosynthetic or degradative compartments) or that is sequestered in non-cycling GSVs does not traffic to the plasma membrane or contribute to cell surface Glut4.
PM, plasma membrane and clathrin-coated pits/coated vesicles associated with the PM; SE, sorting endosomes, “early endosomes” (vacuolar structures containing the endocytic marker albumin but no cathepsin D) with an associated network of recycling tubules; GSV, the trans-Golgi region (TGR) and three classes of small tubular-vesicular elements not associated with other organelles; ERC, endosomal recycling intermediate compartments, “late endosomes” (multivesicular vacuoles containing albumin and cathepsin D, with an associated network of recycling tubules) and non-coated invaginations of the plasma membrane. In primary adipocytes, insulin increased Glut4 in the plasma membrane, clathrin-coated pits/vesicles, non-coated plasma membrane invaginations, and “early endosomes.” Insulin decreased Glut4 in the “late endosomes,” the TGR, and all three classes of small tubular-vesicular compartments (7).
In a unidirectional cycling membrane system with one compartment feeding into the next one, changes in the Glut4 levels in the compartments are caused by changes in the rate of delivery of Glut4 to the compartment and/or rate of exit from the compartment. The rates of entry into and exit from a compartment are function of both the amount/concentration of Glut4 in each compartment as well as the rate constants for each step.
P. D. Brewer, E. N. Habtemichael, I. Romenskaia, C. C. Mastick, and A. C. F. Coster, submitted for publication.
- GSV
- Glut4 storage vesicle
- α2-M
- α2-macroglobulin
- α-HA
- anti-HA antibody
- AF647
- AlexaFluor 647
- ERC
- endosomal recycling intermediate compartment(s)
- IN/SUR
- internal/surface
- ken
- kex, and kobs, endocytic, exocytic, and observed relaxation rate constant, respectively
- ksort
- kseq, kfuseE, and kfuseG, rate constant for sorting from endosomes into ERC, sorting from endosomes into GSVs (sequestration), and transport to the plasma membrane from either ERC or GSVs (fusion), respectively
- LSM
- low serum medium
- LYi
- PI3K inhibitor LY294002
- MFR
- mean fluorescence ratio (AF647/GFP; labeled Glut4/total cellular Glut4)
- Tf
- transferrin
- KD
- knockdown
- PM
- plasma membrane
- SE
- sorting or “early” endosome(s)
- FSC-H and SSC-H
- forward and side scatter height, respectively.
REFERENCES
- 1. Karnieli E., Zarnowski M. J., Hissin P. J., Simpson I. A., Salans L. B., Cushman S. W. (1981) Insulin-stimulated translocation of glucose transport systems in the isolated rat adipose cell. Time course, reversal, insulin concentration dependency, and relationship to glucose transport activity. J. Biol. Chem. 256, 4772–4777 [PubMed] [Google Scholar]
- 2. Kono T., Suzuki K., Dansey L. E., Robinson F. W., Blevins T. L. (1981) Energy-dependent and protein synthesis-independent recycling of the insulin-sensitive glucose transport mechanism in fat cells. J. Biol. Chem. 256, 6400–6407 [PubMed] [Google Scholar]
- 3. Muretta J. M., Mastick C. C. (2009) How insulin regulates glucose transport in adipocytes. Vitam. Horm. 80, 245–286 [DOI] [PubMed] [Google Scholar]
- 4. Kandror K. V., Pilch P. F. (2011) The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic 12, 665–671 [DOI] [PubMed] [Google Scholar]
- 5. Foley K., Boguslavsky S., Klip A. (2011) Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50, 3048–3061 [DOI] [PubMed] [Google Scholar]
- 6. Stöckli J., Fazakerley D. J., James D. E. (2011) GLUT4 exocytosis. J. Cell Sci. 124, 4147–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slot J. W., Geuze H. J., Gigengack S., Lienhard G. E., James D. E. (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113, 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang J., Holman G. D. (1993) Comparison of GLUT4 and GLUT1 subcellular trafficking in basal and insulin-stimulated 3T3-L1 cells. J. Biol. Chem. 268, 4600–4603 [PubMed] [Google Scholar]
- 9. Satoh S., Nishimura H., Clark A. E., Kozka I. J., Vannucci S. J., Simpson I. A., Quon M. J., Cushman S. W., Holman G. D. (1993) Use of bismannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose cells. Evidence that exocytosis is a critical site of hormone action. J. Biol. Chem. 268, 17820–17829 [PubMed] [Google Scholar]
- 10. Holman G. D., Lo Leggio L., Cushman S. W. (1994) Insulin-stimulated GLUT4 glucose transporter recycling. A problem in membrane protein subcellular trafficking through multiple pools. J. Biol. Chem. 269, 17516–17524 [PubMed] [Google Scholar]
- 11. Karylowski O., Zeigerer A., Cohen A., McGraw T. E. (2004) GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol. Biol. Cell 15, 870–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Govers R., Coster A. C., James D. E. (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell Biol. 24, 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coster A. C., Govers R., James D. E. (2004) Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic 5, 763–771 [DOI] [PubMed] [Google Scholar]
- 14. Martin O. J., Lee A., McGraw T. E. (2006) GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J. Biol. Chem. 281, 484–490 [DOI] [PubMed] [Google Scholar]
- 15. Muretta J. M., Romenskaia I., Mastick C. C. (2008) Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J. Biol. Chem. 283, 311–323 [DOI] [PubMed] [Google Scholar]
- 16. Habtemichael E. N., Brewer P. D., Romenskaia I., Mastick C. C. (2011) Kinetic evidence that Glut4 follows different endocytic pathways than the receptors for transferrin and α2-macroglobulin. J. Biol. Chem. 286, 10115–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Antonescu C. N., Foti M., Sauvonnet N., Klip A. (2009) Ready, set, internalize: mechanisms and regulation of GLUT4 endocytosis. Biosci. Rep. 29, 1–11 [DOI] [PubMed] [Google Scholar]
- 18. Jedrychowski M. P., Gartner C. A., Gygi S. P., Zhou L., Herz J., Kandror K. V., Pilch P. F. (2010) Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J. Biol. Chem. 285, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi J., Huang G., Kandror K. V. (2008) Self-assembly of Glut4 storage vesicles during differentiation of 3T3-L1 adipocytes. J. Biol. Chem. 283, 30311–30321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi J., Kandror K. V. (2005) Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev. Cell 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 21. Hatakeyama H., Kanzaki M. (2011) Molecular basis of insulin-responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic 12, 1805–1820 [DOI] [PubMed] [Google Scholar]
- 22. Brewer P. D., Romenskaia I., Kanow M. A., Mastick C. C. (2011) Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J. Biol. Chem. 286, 26287–26297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clark A. E., Holman G. D., Kozka I. J. (1991) Determination of the rates of appearance and loss of glucose transporters at the cell surface of rat adipose cells. Biochem. J. 278, 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pryor P. R., Liu S. C., Clark A. E., Yang J., Holman G. D., Tosh D. (2000) Chronic insulin effects on insulin signalling and GLUT4 endocytosis are reversed by metformin. Biochem. J. 348, 83–91 [PMC free article] [PubMed] [Google Scholar]
- 25. Dawson K., Aviles-Hernandez A., Cushman S. W., Malide D. (2001) Insulin-regulated trafficking of dual-labeled glucose transporter 4 in primary rat adipose cells. Biochem. Biophys. Res. Commun. 287, 445–454 [DOI] [PubMed] [Google Scholar]
- 26. Lampson M. A., Schmoranzer J., Zeigerer A., Simon S. M., McGraw T. E. (2001) Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol. Biol. Cell 12, 3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonias S. L., Reynolds J. A., Pizzo S. V. (1982) Physical properties of human α2-macroglobulin following reaction with methylamine and trypsin. Biochim. Biophys. Acta 705, 306–314 [DOI] [PubMed] [Google Scholar]
- 28. Wiley H. S., Cunningham D. D. (1982) The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J. Biol. Chem. 257, 4222–4229 [PubMed] [Google Scholar]
- 29. Dormand J. R., Prince P. J. (1980) A family of embedded Runge-Kutta formulae. J. Comp. Appl. Math. 6, 19–26 [Google Scholar]
- 30. Morè J. J., Sorensen D. C. (1983) Computing a trust region step. SIAM J. Sci. Stat. Comput. 3, 553–572 [Google Scholar]
- 31. Branch M. A., Coleman T. F., Li Y. (1999) A subspace, interior, and conjugate gradient method for large-scale bound-constrained minimization problems. SIAM J. Sci. Stat. Comput. 21, 1–23 [Google Scholar]
- 32. Byrd R. H., Schnabel R. B., Shultz G. A. (1988) Approximate solution of the trust region problem by minimization over two-dimensional subspaces. Math. Progam. 40, 247–263 [Google Scholar]
- 33. Coleman T. F., Verma A. (2001) A preconditioned conjugate gradient approach to linear equality constrained minimization. Comput. Optim. Appl. 20, 61–72 [Google Scholar]
- 34. Sorensen D. C. (1997) Minimization of a large-scale quadratic function subject to a spherical constraint. SIAM J. Optim. 7, 141–161 [Google Scholar]
- 35. Student (Gosset, W. S.) (1908) On the probable error of the mean. Biometrika 6, 1–25 [Google Scholar]
- 36. Ros-Baro A., Lopez-Iglesias C., Peiro S., Bellido D., Palacin M., Zorzano A., Camps M. (2001) Lipid rafts are required for GLUT4 internalization in adipose cells. Proc. Natl. Acad. Sci. U.S.A. 98, 12050–12055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blot V., McGraw T. E. (2006) GLUT4 is internalized by a cholesterol-dependent nystatin-sensitive mechanism inhibited by insulin. EMBO J. 25, 5648–5658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeigerer A., Lampson M. A., Karylowski O., Sabatini D. D., Adesnik M., Ren M., McGraw T. E. (2002) GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol. Biol. Cell 13, 2421–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y., Zhang J., Chen Y., Jiang L., Ji W., Xu T. (2009) Characterization of GLUT4-containing vesicles in 3T3-L1 adipocytes by total internal reflection fluorescence microscopy. Sci. China C Life Sci. 52, 665–671 [DOI] [PubMed] [Google Scholar]
- 40. Chen Y., Wang Y., Zhang J., Deng Y., Jiang L., Song E., Wu X. S., Hammer J. A., Xu T., Lippincott-Schwartz J. (2012) Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J. Cell Biol. 198, 545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taguchi T. (2013) Emerging roles of recycling endosomes. J. Biochem. 153, 505–510 [DOI] [PubMed] [Google Scholar]
- 42. Larance M., Ramm G., Stöckli J., van Dam E. M., Winata S., Wasinger V., Simpson F., Graham M., Junutula J. R., Guilhaus M., James D. E. (2005) Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 280, 37803–37813 [DOI] [PubMed] [Google Scholar]
- 43. Eguez L., Lee A., Chavez J. A., Miinea C. P., Kane S., Lienhard G. E., McGraw T. E. (2005) Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2, 263–272 [DOI] [PubMed] [Google Scholar]
- 44. Zeigerer A., McBrayer M. K., McGraw T. E. (2004) Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol. Biol. Cell 15, 4406–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Piper R. C., Tai C., Kulesza P., Pang S., Warnock D., Baenziger J., Slot J. W., Geuze H. J., Puri C., James D. E. (1993) GLUT-4 NH2 terminus contains a phenylalanine-based targeting motif that regulates intracellular sequestration. J. Cell Biol. 121, 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blot V., McGraw T. E. (2008) Molecular mechanisms controlling GLUT4 intracellular retention. Mol. Biol. Cell 19, 3477–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Corvera S., Chawla A., Chakrabarti R., Joly M., Buxton J., Czech M. P. (1994) A double leucine within the GLUT4 glucose transporter COOH-terminal domain functions as an endocytosis signal. J. Cell Biol. 126, 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reed S. E., Hodgson L. R., Song S., May M. T., Kelly E. E., McCaffrey M. W., Mastick C. C., Verkade P., Tavaré J. M. (2013) A role for Rab14 in the endocytic trafficking of GLUT4 in 3T3-L1 adipocytes. J. Cell Sci. 126, 1931–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fazakerley D. J., Holman G. D., Marley A., James D. E., Stöckli J., Coster A. C. (2010) Kinetic evidence for unique regulation of GLUT4 trafficking by insulin and AMP-activated protein kinase activators in L6 myotubes. J. Biol. Chem. 285, 1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foster L. J., Li D., Randhawa V. K., Klip A. (2001) Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J. Biol. Chem. 276, 44212–44221 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.