

Background: Global MGAT2 knock-out mice are protected from obesity.
Results: Intestine-specific MGAT2 knock-out mice showed increased energy expenditure and were protected against excess weight gain and metabolic disorders induced by high fat feeding.
Conclusion: Intestinal triacylglycerol metabolism is crucial in regulating systemic energy balance.
Significance: Intestinal MGAT2 may be a feasible intervention target for diseases associated with excess caloric intake.
Keywords: Diacylglycerol, Diet, Energy Metabolism, Lipid Absorption, Triacylglycerol
Abstract
The absorption of dietary fat involves the re-esterification of digested triacylglycerol in the enterocytes, a process catalyzed by acyl-CoA:monoacylglycerol acyltransferase (MGAT) 2. Mice without a functional gene encoding MGAT2 (Mogat2−/−) are protected from diet-induced obesity. Surprisingly, these mice absorb normal amounts of dietary fat but increase their energy expenditure. MGAT2 is expressed in tissues besides intestine, including adipose tissue in both mice and humans. To test the hypothesis that intestinal MGAT2 regulates systemic energy balance, we generated and characterized mice deficient in MGAT2 specifically in the small intestine (Mogat2IKO). We found that, like Mogat2−/− mice, Mogat2IKO mice also showed a delay in fat absorption, a decrease in food intake, and a propensity to use fatty acids as fuel when first exposed to a high fat diet. Mogat2IKO mice increased energy expenditure although to a lesser degree than Mogat2−/− mice and were protected against diet-induced weight gain and associated comorbidities, including hepatic steatosis, hypercholesterolemia, and glucose intolerance. These findings illustrate that intestinal lipid metabolism plays a crucial role in the regulation of systemic energy balance and may be a feasible intervention target. In addition, they suggest that MGAT activity in extraintestinal tissues may also modulate energy metabolism.
Introduction
Dietary fat is absorbed in the intestine where its digested components trigger hormone release and neural output (1). These intestinal signals can in turn regulate food intake and coordinate metabolism, preparing the rest of the body for efficient use and storage of incoming nutrients (1–3). Acyl-CoA:monoacylglycerol acyltransferase (MGAT)3 in the intestine is thought to be important for the absorption of dietary fat (4, 5). The enzyme is involved in the resynthesis of digested triacylglycerol (TAG), catalyzing the esterification of monoacylglycerol (MAG). Its product, diacylglycerol (DAG), serves as substrate for the final step catalyzed by acyl-CoA:diacylglycerol acyltransferase (DGAT) (6). This resynthesis of TAG is crucial for the assembly and secretion of chylomicrons, which deliver absorbed fat to other tissues (7–9). MGAT2 is one of the three known MGATs that share sequence homology (10–13) and is highly expressed in both mouse and human intestine. In contrast, MGAT1 is not expressed in the intestine, whereas MGAT3 is expressed in human but not mouse intestine.
Consistent with the concept that intestinal fat metabolism regulates energy balance, MGAT2 enhances systemic metabolic efficiency. Mice with a deletion of the gene encoding the enzyme (Mogat2−/−) are protected from obesity and associated metabolic disorders induced by high fat feeding (14). Despite the putative role of MGAT2 in fat absorption, Mogat2−/− mice consume and absorb normal amounts of dietary fat. Interestingly, they exhibit an increase in energy expenditure. This difference is exacerbated with high fat feeding, but the increase in energy expenditure persists even when the mice are fed a fat-free diet (15). Deficiency of MGAT2 protects mice from obesity caused by agouti mutation-induced hyperphagia as well as from excess weight gain induced by a diet rich in refined carbohydrate (15). These findings are consistent with the idea that MGAT2 normally enhances metabolic efficiency and mediates positive energy balance in response to a surplus of dietary calories.
MGAT2 is expressed in tissues besides intestine, including the kidney and adipose tissue in both mice and humans (11, 12, 14). Recently, we found that reintroducing MGAT2 expression exclusively in the small intestine is not sufficient to restore metabolic efficiency in Mogat2−/− mice completely (16), implicating a role of extraintestinal MGAT2 in modulating energy balance. To test the hypothesis that intestinal MGAT2 is necessary for maximizing metabolic efficiency, in this study, we examined whether inactivating MGAT2 exclusively in the small intestine protects mice from excessive accretion of body fat and associated comorbidities induced by high fat feeding.
EXPERIMENTAL PROCEDURES
Mice
To generate mice lacking Mogat2 specifically in the small intestine, embryonic stem cells bearing a targeted Mogat2 allele were obtained from the Knockout Mouse Project (KOMP) repository (EPD0093_1_A12), injected into blastocysts, and implanted into pseudopregnant mice. The offspring (C57BL/6N-Mogat2tm1a(KOMP)Wtsi) carry a trapping cassette “SA-geo-pA” flanked by the flippase (FLP) recombinase target (FRT) sites upstream from exon 2 of the Mogat2 allele (Fig. 1A, Mogat2 Targeted). After crossing with a mouse (B6.Cg-Tg(ACTFLPe)9205Dym/J) expressing the FLP1 recombinase under the control of the human β-actin promoter (The Jackson Laboratory) to excise the trapping cassette, offspring carrying a “floxed” Mogat2 allele with exon 2 flanked by Cre recombinase target sites (loxP) were produced. Mice possessing the floxed Mogat2 allele without the FLP1 gene (Mogat2f/f) in the C57BL/6 genetic background were then produced through intercrossing. Next, Mogat2f/f mice were bred with a mouse expressing the Cre transgene under the control of the mouse villin 1 promoter (B6.SJL-Tg(Vil-cre)997Gum/J; The Jackson Laboratory), which drives expression in all four types of intestinal epithelia beginning by postcoital day 12.5 (17); through intercrossing, mice with an intestine-specific deletion of Mogat2 (Mogat2IKO mice) were produced.
FIGURE 1.
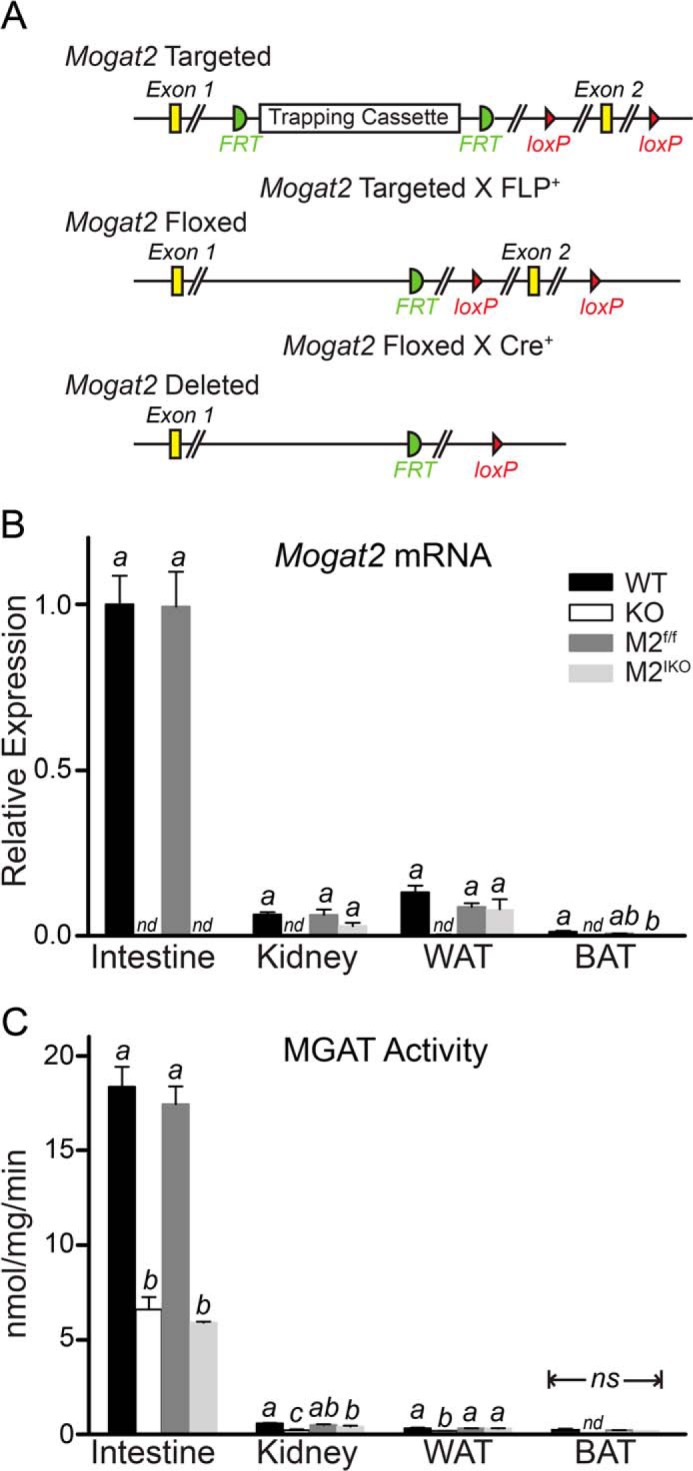
Generation of intestine-specific MGAT2-deficient mice. A, illustration of the Mogat2 targeted allele with a trapping cassette (flanked by the FLP recombinase target site (FRT)) that introduces two Cre recombinase target loxP sites flanking exon 2, the Mogat2 floxed allele after removal of the trapping cassette by FLP recombination, and the Mogat2 deleted allele after removal of exon 2 by Cre recombination. Efficiency and tissue specificity of MGAT2 ablation were confirmed by measuring Mogat2 mRNA expression level (B) and MGAT activity (C) in jejunum (Intestine), kidney, inguinal fat pad (WAT), and interscapular fat pad (BAT) of wild type (WT), Mogat2−/− (KO), Mogat2f/f (M2f/f), and Mogat2IKO (M2IKO) mice. n = 5–11/genotype. Within each tissue, differences between bars without the same letter are statistically significant. Error bars represent S.E. nd, not detected; ns, not statistically significant.
For experiments, Mogat2f/f mice were bred with Mogat2IKO mice to produce Mogat2IKO and Mogat2f/f littermate controls. To generate mice deficient in MGAT2 in all tissues, Mogat2+/− mice were also bred to produce Mogat2−/− and wild type littermate controls as described (14). Differences in metabolic phenotypes were most pronounced in males; therefore, adult male mice were used for experiments. Female mice were used in a few key experiments as indicated to verify whether the effects were sex-dependent. Mice were housed at 22 °C on a 12-h light, 12-h dark cycle. Weighing of mice and changes of diets and cages were performed between 3 and 6 p.m. All animal procedures were approved by the University of Wisconsin-Madison Animal Care and Use Committee and were conducted in conformity with the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Genotyping
Genotypes of mice were determined by PCR. To determine the presence of the Cre recombinase transgene, the following four primers were used: Cre forward, 5′-CCCGGCAAAACAGGTAGTTA-3′; Cre reverse, 5′-TGCCAGGATCAGGGTTAAG-3′; positive control forward, 5′-CCTTTAGCCTGGTCTAGGCAGAG-3′; and positive control reverse, 5′-CAGCAAAGCCCCCTCCTGAATCTCTC-3′. This reaction produces a 194-bp amplicon and a 381-bp amplicon from the transgene and the internal control gene, respectively. To determine the presence of the wild type or floxed Mogat2 allele, the following two primers were used: M2 forward, 5′-GTATGCCACCTGGTGGTAC-3′; and M2 reverse, 5′-GCAGTCCTATACCAGTACAG-3′. This reaction produces a 478-bp amplicon and a 512-bp amplicon from the wild type allele and the allele with the addition of a 34-bp loxP site, respectively.
Diets
Mice were fed a complete, fixed formula chow (8604, Teklad, Madison, WI). A series of semipurified (defined) diets containing 10, 45, or 60% calories from fat (D12450B, D12451, and D12492, Research Diets, New Brunswick, NJ) were used to examine the effect of dietary fat, replacing refined carbohydrate, on food intake and energy expenditure in metabolic chambers and during long term feeding experiments. These defined diets contained 20% calories from protein (casein) and fixed amounts of micronutrients and fiber per calorie, but they varied in metabolizable energy (3.8, 4.7, or 5.2 kcal/g, corresponding to the fat content).
Real Time Quantitative PCR Analysis
To assess the levels of Mogat2 mRNA, tissues from age-matched mice fed a 10 or 60 kcal % fat diet for 12–16 weeks were homogenized, and total RNA was extracted with the AurumTM Total RNA Mini kit (Bio-Rad) and purified by on-column digestion of DNA with DNase I to eliminate residual genomic DNA. Total RNA was used for cDNA synthesis (iScriptTM cDNA synthesis kit, Bio-Rad). Real time quantitative PCR was performed with iTaqTM SYBR Green Supermix (Bio-Rad) and analyzed with the ABI PRISM® 7000 Sequence Detection System (Applied Biosystems, Grand Island, NJ). Relative expression levels were calculated by the comparative CT (cycle of threshold detection) method as outlined in the manufacturer's technical bulletin. Cyclophilin B (Cypb) expression was used as an internal control. The primer sequences of the Cypb gene were 5′-TGCCGGAGTCGACAATGAT-3′ (forward) and 5′-TGGAAGAGCACCAAGACAGACA-3′ (reverse). The primers to detect Mogat2 mRNAs were located on exon 1 and exon 2, respectively; the primer sequences were 5′-TGGGAGCGCAGGTTACAGA-3′ (forward) and 5′-CAGGTGGCATACAGGACAGA-3′ (reverse). The 2−ΔΔCt method was used to calculate the -fold change in gene expression (18).
In Vitro Monoacylglycerol O-Acyltransferase Assays
MGAT activity assays were performed with total tissue homogenates as described previously (11). Reactions were started by adding homogenates to the assay mixture and stopped after 5 min by adding chloroform:methanol (2:1, v/v). The lipids were extracted, dried, and separated by thin-layer chromatography (TLC) on silica gel G-60 plates with the solvent system hexane:diethyl ether:acetic acid (80:20:1, v/v/v). Lipid bands were visualized with iodine vapor, and products were identified by comparison with the migration of lipid standards. The incorporation of radioactive substrates into lipid products was also visualized by an imaging scanner (Typhoon FLA 7000, GE Healthcare) followed by scraping and counting in a scintillation counter (Packard Tri-Carb 2200 CA liquid scintillation counter analyzer).
Monoacylglycerol Uptake and Processing in the Small Intestine
To examine the uptake and processing of MAG, micelles containing radiolabeled tracers were injected into ligated intestine pouches created in anesthetized mice. Taurocholate micelles were prepared with 2-mono-[U-14C]oleoyl-racemic glycerol (∼15 Ci/mol; American Radiolabeled Chemicals, St. Louis, MO) and unlabeled oleate, and isolated pouches in both the proximal and distal intestine were created as described previously (16, 19). Two minutes after the micelle preparation was injected, the proximal and distal small intestinal pouches were excised. The luminal content was washed out with 10 mm taurocholate in PBS and collected. Lipids from the proximal small intestine and the distal small intestine were extracted with chloroform:methanol (2:1, v/v) and separated by TLC using a two-solvent system (first by chloroform:acetone:methanol:acetic acid:water (50:20:10:10:5, v/v/v/v/v) and then by hexane:diethyl ether:acetic acid (80:20:1, v/v/v)). The bands corresponding to TAG, DAG, and MAG were scraped after iodine staining and transferred into scintillation vials for counting of radioactivity.
Spatial and Temporal Distribution of Fat Absorption in the Intestine
To examine the anatomical distribution of lipid absorption, we intragastrically challenged mice with a radiolabeled lipid bolus and assessed the levels of radioactivity along the length of the small intestine. Briefly, mice were acclimated to 60 kcal % fat diet for at least 1 week, fasted for 4 h, and then gavaged with 2 μCi of tri-[carboxyl-14C]oleoylglycerol ([14C]TAG; American Radiolabeled Chemicals) in 200 μl of olive oil. After 2 h, the entire gastrointestinal tract was excised. The small intestine was rinsed and cut into 2-cm sections. Each section was digested overnight in 500 μl of 1 n NaOH at 65 °C. Radioactivity was counted after adding 4 ml of Optima Gold scintillation mixture (PerkinElmer Life Sciences) in a scintillation counter.
To assess the temporal distribution, the rate at which dietary fat entered circulation, mice acclimatized to high fat feeding for 1 week were fasted for 6 h and injected with 500 mg/kg surfactant Pluronic F127 NF Prill Poloxamer 407 (a gift from BASF Corp., Florham Park, NJ) to inhibit the clearance of plasma TAG. Ten minutes later, mice were intragastrically challenged with 200 μl of olive oil containing 2 μCi of [14C]TAG. Blood was collected before (time 0) and at the indicated times for TAG measurement and scintillation counting. Plasma TAG was measured by enzymatic assay (InfinityTM Triglycerides Lipid Stable Reagent, Thermo Fisher Scientific Inc., Middletown, VA).
Plasma Glucagon-like Peptide 1 (GLP-1) and Glucose-dependent Insulinotropic Peptide (GIP) Levels
To determine postprandial GLP-1 and GIP levels in circulation, mice acclimatized to high fat feeding for 1–2 weeks were fasted for 4 h and then challenged with an intragastric bolus of 300 μl of mixed meal (Ensure® supplemented with olive oil) containing 0.65 kcal and 42 mg of fat. Plasma was collected 2 h later in the presence of 100 μm dipeptidyl-peptidase IV inhibitor (EMD Millipore Corp., Billerica, MA). GLP-1 and GIP levels in plasma were assessed using total GLP-1 and GIP ELISA kits (Millipore).
Metabolic Phenotyping Studies
To assess phenotypes related to energy balance, mice were housed in a metabolic phenotyping system with housing and wood chip bedding similar to the home cage environment (LabMaster modular animal monitoring system; TSE Systems, Chesterfield, MO). Male mice (3 months old) were acclimated to individual housing and metabolic cages for 1 week before experiments and fed the indicated diets sequentially for 3 days each and high fat diet for up to 2 additional weeks. Data collection and analysis were performed as reported previously (15, 16). A subset of mice was housed continuously in the metabolic cages for 3 weeks to collect longitudinal food intake data in the metabolic chambers. Food intake after acclimation to high fat feeding was also examined in an independent cohort of mice in standard cages.
Body Weight Response to Chow or High Fat Feeding
To examine the effects of the interaction between diet and MGAT2 on long term energy balance, mice were fed a regular mixed meal chow at weaning (3 weeks), switched from chow to a 60 or 10 kcal % fat diet at 12 weeks of age, and weighed weekly for 10 weeks. Female mice were switched from chow to high fat diet at 10 months of age and weighed weekly for 10 weeks. Following high fat feeding, tissues and plasma were harvested for weighing and biochemical assays.
Biochemical Assays
Hepatic and fecal lipids were extracted using the method of Folch et al. (20). Samples were homogenized in PBS and extracted in chloroform:methanol (2:1, v/v). After repeating the extraction, the organic phases were combined, dried, and weighed in a new glass vial. To determine the amount of TAG, free fatty acids, and cholesterol in these extracts, samples were resuspended in 1 ml of chloroform containing 1% Triton X-100, evaporated to dryness, resuspended in 1 ml of H2O, and assayed using enzymatic kits (Infinity Triglycerides Lipid Stable Reagent, Thermo Fisher Scientific Inc.; Cholesterol E, Non-Esterified Fatty Acid assay, Wako Diagnostics). Protein concentration was measured by the bicinchoninic acid assay (Thermo Fisher Scientific Inc.). Hepatic glycogen contents were determined as described (21). Fecal samples were collected from individually housed mice acclimatized to the 60 kcal % fat diet for 1 week.
Glucose Metabolism
To examine glucose metabolism prior to high fat feeding, chow-fed mice aged 3 months were bled after a 6-h fast for blood glucose and plasma insulin measurement. To examine glucose metabolism following high fat feeding, mice were fed a 60 kcal % fat diet for 8–11 weeks, and then a glucose tolerance test was performed. Briefly, male mice were fasted for 6 h beginning at 7 a.m. and then injected with glucose (1 g/kg of body weight, intraperitoneal, 10% glucose in PBS). Blood glucose was measured immediately before and at defined intervals after glucose injection using a hand-held glucometer (OneTouch Ultra, LifeScan, Inc., Milpitas, CA). Insulin was measured by ELISA (Crystal Chem, Inc., Downers Grove, IL).
Statistical Analyses
All data are presented as mean ± S.E. p < 0.05 was considered statistically significant. Each experiment was performed with independent samples at least twice to confirm reproducibility of the results. For comparisons between two groups, Student's t tests were used. For comparisons between four groups, a one-way analysis of variance followed by Tukey's multiple comparison test was used. Differences measured over time were compared using repeated measures two-way analysis of variance to determine the main effects of and interactions between time and genotype. Analyses were conducted using GraphPad Prism statistical analysis software (version 5.01; GraphPad Software, La Jolla, CA).
RESULTS
Generation of Intestine-specific MGAT2-deficient Mice
To examine the effects of inactivating MGAT2 specifically in the intestine, we introduced a Cre recombinase under the control of the intestine-specific villin promoter into Mogat2f/f mice, which carry two loxP sites flanking exon 2 of the Mogat2 gene (Fig. 1A), as described under “Experimental Procedures.” All of the offspring were born with floxed Mogat2 alleles, and half expressed the tissue-specific Cre enzyme. To confirm the efficiency and tissue specificity of MGAT2 ablation in the Cre-expressing floxed mice (Mogat2IKO), we measured the MGAT2 mRNA expression level and MGAT activity in several tissues, including the small intestine, kidney, and white and brown adipose tissue, where MGAT2 expression has been reported (11, 12) as well as liver where MGAT2 is not expressed in adult mice (11). We found that Mogat2f/f mice expressed MGAT2 mRNA at levels similar to that found in wild type mice. In both groups, MGAT2 was expressed predominantly in the intestine when mice were fed either a high fat (Fig. 1B) or a low fat (data not shown) diet, and the intestinal expression levels were up-regulated more than 5-fold upon high fat feeding. Mogat2−/− mice had no detectable MGAT2 mRNA in any tissue examined. In contrast, Mogat2IKO mice showed MGAT2 expression levels similar to those of Mogat2f/f littermates in examined tissues except in the small intestine where MGAT2 expression remained absent even after the continuous renewal of intestinal epithelia. Mice in all four groups had no detectable MGAT2 mRNA in liver (data not shown), indicating that lacking MGAT2 specifically in the intestine does not induce hepatic MGAT2 expression in adult mice.
The levels of MGAT activity correlated with the levels of MGAT2 mRNA detected. With the highest levels found in the intestine, Mogat2f/f mice showed MGAT activity similar to that of wild type mice. Ablation of MGAT2 message in the intestine of Mogat2IKO mice reduced intestinal MGAT activity by ∼70% as compared with Mogat2f/f mice (Fig. 1C). This difference in MGAT activity was similar to that seen between Mogat2−/− and wild type mice. The levels of MGAT activity were low in all other tissues examined. In these tissues, the differences between wild type and Mogat2−/− mice were statistically significant, whereas the differences between Mogat2f/f and Mogat2IKO mice were not. These results indicated that Mogat2IKO mice are deficient in MGAT2 specifically in the intestine.
Deletion of MGAT2 in the Intestine Reduces MAG Uptake/Esterification and Delays Fat Absorption
Mogat2−/− mice exhibit reduced MAG uptake and esterification in the intestine (16). To confirm that deletion of MGAT2 in the intestine impairs MAG uptake and esterification, we infused taurocholate micelles containing fatty acids and 2-mono-[U-14C]oleoyl-racemic glycerol directly into ligated pouches created in both the proximal and distal intestine. After a 2-min incubation, ∼45% of the label injected was taken up and accumulated in the proximal intestine segment of wild type mice (Fig. 2A), whereas less than 1% of label appeared in the circulation with the rest still in the lumen (data not shown). A similar level of accumulation was found in Mogat2f/f mice. In contrast, less than 10% of MAG tracer was taken up in the proximal intestine of Mogat2−/− mice and likewise in Mogat2IKO mice. In wild type and Mogat2f/f mice, uptake was greater in the proximal intestine than in the distal intestine. In Mogat2−/− and Mogat2IKOmice, the uptakes were low compared with that in respective controls, and there was no difference between the proximal and the distal intestine. In general, the levels of MAG uptake were associated with the production of DAG and TAG. In wild type and Mogat2f/f mice, 90% of the tracer was recovered as TAG in the proximal intestine. In Mogat2−/− and Mogat2IKO mice, the proportion of MAG incorporated into DAG and TAG was greatly reduced compared with that in littermates, demonstrating the predominant role of intestinal MGAT2 in catalyzing the esterification process. Deletion of MGAT2 in the intestine inhibited both uptake and esterification of MAG.
FIGURE 2.

Intestine-specific inactivation of MGAT2 inhibits monoacylglycerol uptake and esterification, alters distribution of fat absorption, and delays appearance of TAG in the circulation. A, monoacylglycerol uptake and esterification in isolated intestinal pouches after injection of a micelle preparation containing fatty acids and 2-mono-[U-14C]oleoyl-racemic glycerol tracer. Bars represent accumulation of MAG and incorporation of tracers into DAG and TAG in each intestinal segment of WT, Mogat2−/− (KO), Mogat2f/f (M2f/f), and Mogat2IKO (M2IKO) mice. n = 3–5/genotype. Differences between bars without the same letter are statistically significant. B, distribution of dietary TAG in 2-cm segments of the small intestine 2 h after an oral gavage of [14C]TAG in mice acclimatized to high fat feeding. n = 7–13/genotype. Blood radioactivity (C) and plasma TAG (TG) (D) in mice were determined after injection of the lipase inhibitor P407 and gavage with olive oil containing [14C]TAG. n = 5–8/genotype. Differences between curves without the same letter are statistically significant. Error bars represent S.E.
To assess the spatial distribution of fat absorption, we challenged mice with an intragastric bolus of oil containing [14C]TAG and measured radiolabeled lipid accumulated along the length of the small intestine. Consistent with the location of MAG uptake and esterification, a substantial amount of radiolabeled lipid accumulated in the proximal small intestine of wild type and Mogat2f/f mice. In Mogat2−/− and Mogat2IKO mice, there was significantly less accumulation of radiolabeled lipid in the proximal half of the intestine than in respective control mice (Fig. 2B). The pattern of radiolabeled lipids present in the small intestine of Mogat2IKO mice resembled that of Mogat2−/− mice.
To determine the rate of fat absorption, we treated mice with a lipoprotein lipase inhibitor and an intragastric bolus of [14C]TAG-containing oil. In wild type and Mogat2f/f mice, the labeled dietary and total fat increased in circulation at a similar rate; whereas in Mogat2−/− and Mogat2IKO mice, these increases were diminished to a similar degree (Fig. 2, C and D). In all four genotypes, the majority of radioactivity in blood was found as triacylglycerol (data not shown). These findings suggest that deletion of MGAT2 in the intestine was sufficient to not only alter the kinetics of lipid processing in the intestine but also modulate the delivery of dietary lipid into the circulation and other tissues.
Deletion of Intestinal MGAT2 Increases Postprandial Plasma GLP-1
To examine whether the change in fat absorption leads to a change in gut hormone secretion, we measured the levels of GLP-1 and GIP 2 h after an intragastric challenge with a high fat meal in mice that had been acclimatized to high fat feeding. Compared with wild type mice, Mogat2−/− mice showed an increased level of plasma GLP-1 (Fig. 3A). Likewise, Mogat2IKO mice had a higher level of GLP-1 than Mogat2f/f mice, linking the change in circulating GLP-1 level to the deficiency of MGAT2 in the intestine. In contrast, GIP levels were not significantly different across genotypes (Fig. 3B).
FIGURE 3.

Inactivation of intestinal MGAT2 increases postprandial plasma GLP-1. WT, Mogat2−/− (KO), Mogat2f/f (M2f/f), and Mogat2IKO (M2IKO) mice were challenged with a liquid meal, and plasma samples were collected 2 h later. Plasma GLP-1 (A) and GIP (B) were measured by ELISA. n = 7–10/genotype. Differences between bars without the same letter are statistically significant. Error bars represent S.E. ns, not statistically significant.
Deletion of Intestinal MGAT2 Protects Mice against Diet-induced Obesity
To determine whether intestinal MGAT2 modulates long term energy balance, we monitored body weight of mice over time. When mice were fed a standard mixed meal chow low in fat (accounting for ∼14 kcal %), deficiency of MGAT2 globally or specifically in the intestine did not alter long term energy balance as mice grew and gained weight similarly across genotypes (Fig. 4A). Conversely, when mice were fed a high fat diet, both wild type and Mogat2f/f male mice exhibited positive energy balance and gained weight rapidly, whereas Mogat2−/− and Mogat2IKO mice gained weight at a significantly lower rate than their littermate controls (Fig. 4B). When switched to a high fat diet at 3 months of age, wild type and Mogat2f/f mice gained 17.7 and 17.1 g after 10 weeks, respectively. In contrast, Mogat2−/− and Mogat2IKO mice gained only 6.0 and 10.7 g, resulting in body weights 28 and 15% lighter than their littermates, respectively. Thus, both Mogat2−/− and Mogat2IKO mice gained less weight than their littermates, whereas Mogat2IKO mice gained significantly more weight than Mogat2−/− mice.
FIGURE 4.

Intestine-specific inactivation of MGAT2 protects against excessive weight gain induced by diets. A, male mice fed chow from weaning through 23 weeks of age. n = 7–13/genotype. B, male mice switched to a high fat diet at 12 weeks of age. n = 17–28/genotype. C, tissue masses (WAT, inguinal fat; BAT, interscapular fat) of mice fed a high fat diet for ∼3 months. n = 13–15/genotype. D, females switched to high fat feeding at 10 months of age. n = 7–8/genotype. E, male mice switched to a low fat diet at 12 weeks of age. n = 14/genotype. WT, black squares; Mogat2−/− (KO), white circles; Mogat2f/f (M2f/f), dark gray diamonds; and Mogat2IKO (M2IKO), light gray triangles. Error bars represent S.E. Error bars not shown are smaller than the symbols. * or #, p < 0.05 versus littermate controls by t test or repeated measures analysis of variance, respectively. CHO, carbohydrate.
The differences in body weight were largely due to differences in fat mass. White and brown adipose tissues were significantly smaller in Mogat2−/− mice (63 and 45%, respectively) compared with wild type controls. In Mogat2IKO mice, white and brown adipose tissues were also smaller (38 and 36%, respectively) when compared with Mogat2f/f controls (Fig. 4C). Lean mass, represented by calf and heart, was not decreased in Mogat2−/− or Mogat2IKO mice. A small increase of heart mass in Mogat2−/− mice over controls reached statistical significance; whether the difference is biologically significant remains to be determined.
The effect of intestinal MGAT2 on weight gain was independent of age or sex as mature female Mogat2IKO mice were also protected from excess weight gain when they were switched to high fat feeding at 10 months of age (Fig. 4D). After 10 weeks of high fat feeding, female Mogat2IKO mice gained only 59% as much as Mogat2f/f controls (12.9 versus 21.9 g, respectively). In addition, Mogat2IKO mice gained less weight than Mogat2f/f controls (2.1 versus 3.8 g, respectively) after consuming a low fat diet rich in refined carbohydrate (Fig. 4E), suggesting that the protective effect is not limited to high fat feeding. Taken together, these data indicate that, like global deletion of MGAT2, deficiency of intestinal MGAT2 is sufficient to deter excess accumulation of body fat resulting from positive energy balance; however, the effect is to a lesser degree than that of deletion of MGAT2 in all tissues.
Deletion of Intestinal MGAT2 Modulates Short Term Energy Balance and Substrate Utilization
To explore the physiological mechanisms by which intestinal MGAT2 modulates energy balance, we examined energy intake and expenditure using indirect calorimetry when mice were fed the standard chow or a semipurified diet containing 10, 45, or 60% of calories from fat consecutively for 3 days on each diet. Mogat2−/− mice exhibited increased energy expenditure compared with wild type littermates as indicated by increased oxygen consumption (Fig. 5A). The increases, ranging from 11 to 15%, were significant under all diet conditions and were more pronounced when dietary fat was high. Similar, but more moderate, elevations in oxygen consumption were observed when comparing Mogat2IKO mice with their littermate controls (Fig. 5B). The increases ranged from 4 to 7% and were statistically significant except when mice were exposed to the 45 kcal % diet. Together with the long term weight gain, these data suggest that MGAT2 in the intestine contributes significantly to the regulation of energy expenditure, and MGAT2 in extraintestinal tissues could also play a role.
FIGURE 5.

Inactivation of intestinal MGAT2 alters energy balance. A and B, 3-month-old male mice (wild type, black; Mogat2−/−, red; Mogat2f/f, blue; and Mogat2IKO, green) were sequentially fed chow and then fed semipurified diets containing 10, 45, and 60% of calories from fat for 3 days each. Oxygen consumption adjusted for baseline body weight at the start of each diet (VO2), RER, and cumulative food intake were measured continuously. Data from each mouse were pooled from the same time of the day of the same diet treatment. n = 16–20/genotype. Graphs represent average days. Gray areas mark the dark phase of the light cycle (6 p.m. to 6 a.m.). C, short term body weight of mice during the 12-day metabolic phenotyping experiment. Differences between curves without the same letters are statistically significant. D, food intake of mice housed continuously in the metabolic cages for 21 days (n = 16–20/genotype, days 1–12; n = 4–9/genotype, days 13–21). The arrow indicates the start of high fat feeding. Fecal output (E) and lipid (F), free fatty acid (FFA) (G),and cholesterol (H) content were determined during the week following acclimatization to high fat feeding. Mogat2−/−, KO; Mogat2f/f, M2f/f; Mogat2IKO, M2IKO. Error bars represent S.E. ns, not statistically significant.
Energy expenditure is fueled by energy-yielding nutrients from the diet when intake is not limiting. In all four groups of mice, the respiratory exchange ratio (RER) of carbon dioxide production over oxygen consumption largely reflected the composition of nutrients in diets with lower RERs when the levels of dietary fat increased (Fig. 5, A and B, middle panels). Nonetheless, when fed diets rich in fat (45 or 60 kcal %), Mogat2−/− mice oxidized higher proportions of fatty acids than did wild type controls as indicated by 4% lower RERs. Mogat2IKO mice showed similar differences in RER when compared with Mogat2f/f controls fed the corresponding diets.
Food intake was greater in Mogat2−/− mice when fed chow or a low fat diet, compensating for increases in energy expenditure (Fig. 5A, lower panel) as reported previously (15). When they were switched onto high fat diets, which contain more metabolizable energy, Mogat2−/− mice decreased their food intake to a greater extent than did wild type littermates (Fig. 5A). Interestingly, these contrasting effects of diet on food intake were recapitulated between Mogat2IKO and Mogat2f/f mice, indicating an essential role of intestinal MGAT2 in modulating satiety or aversion induced by dietary fat (Fig. 5B).
During the 12-day experiment in metabolic chambers, wild type mice gained weight when fed the calorie-dense, semipurified diets (Fig. 5C). The weight gain was especially evident when fat content was high. In contrast, Mogat2−/− mice maintained their body weights throughout. These contrasting differences were also recapitulated between Mogat2f/f and Mogat2IKO mice (Fig. 5C).
The effect on caloric intake caused by MGAT2 deficiency during high fat feeding appears to be transient, whereas the effect on energy expenditure is persistent. Both Mogat2−/−and Mogat2IKO mice showed acute decreases in caloric intake when they were first exposed to a high fat diet (Fig. 5D, arrow). After housing continuously in the metabolic cages for 3 weeks, all four groups of mice consumed a similar level of calories (Fig. 5D). Similar food intake following high fat feeding was also confirmed in an independent experiment using regular cages (data not shown). After acclimatizing to high fat feeding, there was no difference in fat absorption quantitatively between Mogat2−/− and wild type mice or between Mogat2IKO and Mogat2f/f mice as indicated by total fecal output and residual lipids, free fatty acids, and cholesterol in the excrement (Fig. 5, E–H).
Deletion of Intestinal MGAT2 Protects Mice from Diet-induced Metabolic Disorders
Mogat2−/− mice are protected from several comorbidities of obesity (14). We next investigated whether intestine-specific ablation of MGAT2 can also protect mice from hepatic steatosis, hypercholesterolemia, and impaired glucose tolerance induced by high fat feeding. After long term high fat feeding, wild type mice developed hepatic steatosis, whereas Mogat2−/− mice were protected from increases in mass and TAG content of the liver (Fig. 6, A and B). To a similar degree, Mogat2IKO mice had significantly smaller livers and lower hepatic TAG content than their littermate controls (Fig. 6, A and B), whereas there was no difference in glycogen content between genotypes (Fig. 6C). In addition, Mogat2−/− and Mogat2IKO mice had lower plasma cholesterol than littermate controls (Fig. 6D). Their plasma TAG levels also trended lower than controls, but the differences did not reach statistical significance (Fig. 6E).
FIGURE 6.

Intestine-specific inactivation of MGAT2 protects mice against hepatic steatosis induced by high fat feeding. Male WT, Mogat2−/− (KO), Mogat2f/f (M2f/f), and Mogat2IKO (M2IKO) mice were fasted for 6 h after ∼3 months of high fat feeding. Liver mass (A), hepatic triacylglycerol (TG) (B) and glycogen (C), and plasma cholesterol (D) and triacylglycerol (TG) (E) were determined. n = 6–9/genotype. Differences between bars without the same letter are statistically significant. Error bars represent S.E. ns, not statistically significant.
Prior to high fat feeding, similar levels of fasting blood glucose and plasma insulin were found in mice across genotypes (Fig. 7, A and B). After 8 weeks of high fat feeding, levels of fasting blood glucose in Mogat2−/− and Mogat2IKO mice were 75% that of their littermates (Fig. 7C), whereas levels of fasting plasma insulin in these mice were approximately half that of their respective controls (Fig. 7D). A decrease in both fasting blood glucose and insulin suggests an enhancement in glucose tolerance. Indeed, when challenged with an intraperitoneal injection of glucose, both Mogat2−/− and Mogat2IKO mice disposed of glucose faster than did control littermates (Fig. 7, E and F). Following a glucose challenge, Mogat2−/− and Mogat2IKO mice had significantly reduced net increases in blood glucose (38 and 46% lower area under the curve, respectively).
FIGURE 7.

Intestine-specific inactivation of MGAT2 protects mice from impaired glucose metabolism following high fat feeding. Blood glucose and plasma insulin from 6-h-fasted WT, Mogat2−/− (KO), Mogat2f/f (M2f/f), and Mogat2IKO (M2IKO) mice fed a regular chow (A and B) or high fat diet (C and D) were measured. Differences between bars without the same letter are statistically significant. E and F, net blood glucose change following an intraperitoneal injection of glucose (1 mg/g of body weight, 10% glucose in PBS). n = 6–14/genotype. Error bars represent S.E. ns, not statistically significant. #, p < 0.05 versus area under the curve of littermate controls.
DISCUSSION
Mice without a functional MGAT2 enzyme absorb normal quantities of dietary fat but exhibit increased energy expenditure. As a result, they are protected from obesity and other metabolic disorders induced by the agouti mutation or by high fat feeding (14, 15). In this study, we examined whether intestinal MGAT2 is responsible for these phenotypes. We found that deletion of MGAT2 specifically in the intestine of mice altered intestinal TAG metabolism and delayed fat absorption. These Mogat2IKO mice had reduced weight gain and were protected against obesity-associated comorbidities induced by high fat feeding. Although the protection against weight gain was not to the same extent as that in the global deficiency of MGAT2, the partial reduction in weight gain was sufficient to normalize hepatic steatosis and glucose intolerance as seen in Mogat2−/− mice. These findings support the concept that intestinal lipid metabolism plays a crucial role in the regulation of systemic energy balance and suggest that targeting MGAT2 in the small intestine may be a feasible approach to reduce weight gain and avoid associated metabolic disorders. Complementing findings from gain-of-function studies using intestine-specific transgenic mice (16), these findings also implicate extraintestinal MGAT activity in the regulation of whole body energy balance.
MGAT2 is highly expressed in the small intestine where the role of MGAT activity in fat absorption is well established (4, 5). MGAT2 appears to be a major intestinal MGAT as Mogat2IKO mice, like Mogat2−/− mice, exhibited significantly reduced intestinal MGAT activity (Fig. 1C). The residual activity could come from other enzymes. For example, DGAT1, one of the two known DGAT enzymes (6), also exhibits MGAT activity in vitro and is expressed in the intestine (22–24); however, it does not appear to account for the activity as mice deficient in both MGAT2 and DGAT1 still exhibited similar levels of residual MGAT activity in the intestine.4 Lysophosphatidylglycerol acyltransferase 1 (LPGAT1) also exhibits MGAT activity (25). As LPGAT1 is expressed in the intestine, it could contribute to the residual MGAT activity. Like that of DGAT1, the expression of LPGAT1 is not altered by MGAT2 deficiency (data not shown).
Despite the presence of alternative enzymes, the biochemical and physiological functions of MGAT2 cannot be fully compensated for; the uptake and esterification of MAG were greatly reduced and entry of dietary fat into circulation was delayed in Mogat2IKO mice (Fig. 2). Diacylglycerol, the product of MGAT, can also come from the glycerol 3-phosphate pathway, which is predominant in most tissues (26, 27). The enzymes involved, glycerol-3-phosphate acyltransferase, 1-acylglycerol-3-phosphate acyltransferase, and phosphatidate phosphatase (lipin), are all expressed in the intestine, but the alternative pathway did not fully compensate for the MGAT pathway either. Consistent with our previous report (16), results from Mogat2IKO mice show that MGAT2 determines the levels of MAG uptake and esterification in the intestine not because of its systemic effects, such as gastric emptying, but in a cell-autonomous fashion. These findings support the model that lipid intermediates generated from different pathways have preferred destinations. MGAT2 likely mediates TAG synthesis coupled to chylomicron assembly because a lack of MGAT2 in the intestine altered the spatial distribution and temporal kinetics of fat absorption.
We postulated that MGAT2 modulates systemic energy balance most likely through its role in the intestine because MGAT2 is highly expressed only in the intestine in mice (11, 12). Intestine initiates hormonal and neural signals in response to the presence of substances in the lumen (1–3). A change in the spatial distribution of fat absorption in Mogat2−/− mice is associated with altered levels of gut hormones, including GLP-1 (14). In this study, we also found that Mogat2IKO mice had an increased GLP-1 level in response to a fatty meal (Fig. 3). In addition to its effects on glucose metabolism, GLP-1 is known to reduce food intake and contribute to weight loss (28). When exposed to a diet rich in fat (45 or 60 kcal %), Mogat2IKO mice exhibited a similar transient reduction in food intake as seen in Mogat2−/− mice (Fig. 5), indicating that changes in food intake are a result of altered intestinal lipid metabolism. Moreover, MGAT2 exhibits high activity toward MAGs with polyunsaturated fatty acyl groups (11), including sn-2-arachidonoylglycerol. sn-2-Arachidonoylglycerol is an endogenous ligand for the cannabinoid receptors. MGAT2 may thus modulate endocannabinoid signaling, which is known to regulate food intake and energy balance (29). It is not known whether MGAT2 is expressed in the taste buds, enteroendocrine cells, or the enteric nerve system. Interestingly, overexpression of MAG lipase, an enzyme competing with MGAT for MAG substrate, leads to decreases in sn-2-arachidonoylglycerol, increases in food intake, decreases in energy expenditure, and obesity in mice (30).
The reduction in food intake and the increases in energy expenditure may have both contributed to the decrease in weight gain of Mogat2IKO mice when fed a high fat diet. However, the reduction in food intake induced by high fat feeding was transient, whereas the increase in energy expenditure induced by the deficiency of intestinal MGAT2 was persistent. The differences in food intake disappeared after acclimatization to high fat feeding mainly because wild type and Mogat2f/f mice also reduced their food intake. This adaptation could be due to long term regulators, such as leptin secreted from their expanding adipose tissue (31). When Mogat2IKO mice first consumed a high fat diet, the greater reduction in food intake would have decreased diet-induced thermogenesis and thus blunted their increases in energy expenditure. As the dietary fat content increased to 60 kcal %, differences in energy expenditure were pronounced despite the reduced food intake. After acclimatization to the high fat feeding for 1 week, all groups of mice ate the same and absorbed the same amount of fat. However, the weight gain in Mogat2IKO mice remained much lower than that in controls (Fig. 4), consistent with an increase in energy expenditure. Compared with their respective littermate controls, Mogat2−/− and Mogat2IKO mice both had significant decreases in RER, suggesting a higher proportion of fatty acid oxidation upon high fat feeding. Throughout the 12-day metabolic chamber study, Mogat2−/− and Mogat2IKO mice maintained energy balance, matching their energy intake with expenditure. They likely oxidized most of the consumed dietary fat. In contrast, wild type and Mogat2f/f mice gained weight, likely storing some dietary fat. Moreover, when fed a diet low in fat (chow or 10 kcal % fat), Mogat2−/− mice ate more to compensate for increased energy expenditure, and they lost weight when food intake was limited to the levels of wild type littermates (15). Mogat2IKO mice also showed increases in food intake when fed chow or the low fat diet while gaining less weight than controls, indicating a persistent increase in energy expenditure regardless of dietary components.
The extent of energy expenditure increases in Mogat2IKO mice was not as great as that of Mogat2−/− mice. These findings are complementary to studies performed in Mogat2−/− mice with an intestine-specific MGAT2 transgene (16). Reintroducing MGAT2 in the intestine of Mogat2−/− mice reduces energy expenditure, enhancing metabolic efficiency and propensity to gain weight upon high fat feeding. However, the recovery is only partial as these mice do not gain as much weight as wild type mice. Taken together, these findings indicate that intestinal MGAT2 is necessary but not sufficient to maximize metabolic efficiency and suggest that MGAT2 in other tissues also regulates energy balance. The identity of these tissues remains to be determined. The low levels of MGAT2 expression and activity in the brown and white adipose tissues may have cell-autonomous effects, modulating the partitioning of substrates between use and storage. Several lines of mice lacking other enzymes involved in TAG metabolism in the adipose tissues also exhibit alterations in systemic energy balance (32–36). In addition, the involvement of adipose tissues can be indirect. For example, the sympathetic nervous system is a major regulator of brown adipose tissue, which is crucial for energy expenditure in rodents and may also play a role in humans (37). The level of MGAT2 expression in brain was not above the detection limit (data not shown). However, it is possible that MGAT2 may modulate the metabolism of the endogenous cannabinoid sn-2-arachidonoylglycerol in certain neurons in the central nervous system that regulate food intake and energy expenditure.
The phenotypes of Mogat2IKO mice recapitulated the protection against diet-induced metabolic disorders seen in Mogat2−/− mice, suggesting that inhibition of intestinal MGAT2 alone is sufficient to protect against hepatic steatosis and hyperglycemia associated with obesity. These aspects of MGAT2 functions resemble those of DGAT1. Informed by data from genetically engineered mice, several DGAT1 inhibitors have been developed and show promising effects in delaying fat absorption, reducing weight gain, alleviating hepatic steatosis, and enhancing insulin sensitivity in rodent models (38–40). However, for humans, their long term efficacy is unknown, and their safety could be a concern. Unlike mice, humans do not express DGAT2 in the intestine (41). Inhibiting the only DGAT enzyme in human intestine may block TAG synthesis completely and lead to side effects with gastrointestinal distress and malabsorption as observed in patients with a null mutation in the DGAT1 gene and in some clinical trials of DGAT1 inhibitors (42, 43). Conversely, inhibiting human MGAT2 is less likely to block TAG synthesis to the same extent as the alternative glycerol-3-phosphate acyltransferase pathway is intact in humans. In addition, MGAT3, a homolog of MGAT2, is also expressed in human intestine. As MGAT3 is not expressed in mice, whether MGAT3 could compensate fully for MGAT2 in humans remains to be determined. MGAT3 as well as MGAT2 is highly expressed in human subjects with non-alcoholic fatty liver disease, and its expression was reduced significantly after weight loss (44). Thus, inhibiting both MGAT2 and MGAT3 may present a therapeutic opportunity.
In summary, we generated and characterized mice lacking MGAT2 specifically in the intestine. Deficiency of intestinal MGAT2 was sufficient to deter excess fat accumulation resulting from high fat feeding, suggesting that MGAT2 in the intestine normally coordinates nutrient absorption and substrate utilization in the periphery. Although the protection is not to the same extent as with global deletion of MGAT2, these findings suggest that inhibiting intestinal MGAT2 alone is sufficient to blunt excessive weight gain and that the moderate decrease in weight gain is sufficient to protect against fatty liver and glucose intolerance associated with obesity. Our findings support the idea that MGAT2 in the intestine could be a feasible target to decrease the propensity of storing excess dietary calories.
Acknowledgments
We thank Taylor Bahn for technical assistance in PCR assays and mouse husbandry and Sherry Tanumihardjo for editorial input.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK088210 (to E. Y.) and T32DK007665 (to D. N.). This work was also supported by United States Department of Agriculture Grant WIS01442.
C.-L. E. Yen, unpublished results.
- MGAT
- acyl-CoA:monoacylglycerol acyltransferase
- DAG
- diacylglycerol
- DGAT
- acyl-CoA:diacylglycerol acyltransferase
- GIP
- glucose-dependent insulinotropic peptide
- GLP-1
- glucagon-like peptide 1
- LPGAT1
- lysophosphatidylglycerol acyltransferase 1
- MAG
- monoacylglycerol
- RER
- respiratory exchange ratio
- TAG
- triacylglycerol
- [14C]TAG
- tri-[carboxyl-14C]oleoylglycerol.
REFERENCES
- 1. Badman M. K., Flier J. S. (2005) The gut and energy balance: visceral allies in the obesity wars. Science 307, 1909–1914 [DOI] [PubMed] [Google Scholar]
- 2. Woods S. C., Seeley R. J., Porte D., Jr., Schwartz M. W. (1998) Signals that regulate food intake and energy homeostasis. Science 280, 1378–1383 [DOI] [PubMed] [Google Scholar]
- 3. Chambers A. P., Sandoval D. A., Seeley R. J. (2013) Integration of satiety signals by the central nervous system. Curr. Biol. 23, R379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kayden H. J., Senior J. R., Mattson F. H. (1967) The monoglyceride pathway of fat absorption in man. J. Clin. Investig. 46, 1695–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mansbach C. M., 2nd, Gorelick F. (2007) Development and physiological regulation of intestinal lipid absorption. II. Dietary lipid absorption, complex lipid synthesis, and the intracellular packaging and secretion of chylomicrons. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G645–G650 [DOI] [PubMed] [Google Scholar]
- 6. Yen C. L., Stone S. J., Koliwad S., Harris C., Farese R. V., Jr. (2008) Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49, 2283–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mansbach C. M., Siddiqi S. A. (2010) The biogenesis of chylomicrons. Annu. Rev. Physiol. 72, 315–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abumrad N. A., Davidson N. O. (2012) Role of the gut in lipid homeostasis. Physiol. Rev. 92, 1061–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kindel T., Lee D. M., Tso P. (2010) The mechanism of the formation and secretion of chylomicrons. Atheroscler. Suppl. 11, 11–16 [DOI] [PubMed] [Google Scholar]
- 10. Yen C. L., Stone S. J., Cases S., Zhou P., Farese R. V., Jr. (2002) Identification of a gene encoding MGAT1, a monoacylglycerol acyltransferase. Proc. Natl. Acad. Sci. U.S.A. 99, 8512–8517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yen C. L., Farese R. V., Jr. (2003) MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J. Biol. Chem. 278, 18532–18537 [DOI] [PubMed] [Google Scholar]
- 12. Cao J., Lockwood J., Burn P., Shi Y. (2003) Cloning and functional characterization of a mouse intestinal acyl-CoA:monoacylglycerol acyltransferase, MGAT2. J. Biol. Chem. 278, 13860–13866 [DOI] [PubMed] [Google Scholar]
- 13. Cheng D., Nelson T. C., Chen J., Walker S. G., Wardwell-Swanson J., Meegalla R., Taub R., Billheimer J. T., Ramaker M., Feder J. N. (2003) Identification of acyl coenzyme A:monoacylglycerol acyltransferase 3, an intestinal specific enzyme implicated in dietary fat absorption. J. Biol. Chem. 278, 13611–13614 [DOI] [PubMed] [Google Scholar]
- 14. Yen C. L., Cheong M. L., Grueter C., Zhou P., Moriwaki J., Wong J. S., Hubbard B., Marmor S., Farese R. V., Jr. (2009) Deficiency of the intestinal enzyme acyl CoA:monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nat. Med. 15, 442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelson D. W., Gao Y., Spencer N. M., Banh T., Yen C. L. (2011) Deficiency of MGAT2 increases energy expenditure without high-fat feeding and protects genetically obese mice from excessive weight gain. J. Lipid Res. 52, 1723–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao Y., Nelson D. W., Banh T., Yen M. I., Yen C. L. (2013) Intestine-specific expression of MOGAT2 partially restores metabolic efficiency in Mogat2-deficient mice. J. Lipid Res. 54, 1644–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pinto D., Robine S., Jaisser F., El Marjou F. E., Louvard D. (1999) Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. J. Biol. Chem. 274, 6476–6482 [DOI] [PubMed] [Google Scholar]
- 18. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 19. Storch J., Zhou Y. X., Lagakos W. S. (2008) Metabolism of apical versus basolateral sn-2-monoacylglycerol and fatty acids in rodent small intestine. J. Lipid Res. 49, 1762–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 21. Doi R., Oishi K., Ishida N. (2010) CLOCK regulates circadian rhythms of hepatic glycogen synthesis through transcriptional activation of Gys2. J. Biol. Chem. 285, 22114–22121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yen C. L., Monetti M., Burri B. J., Farese R. V., Jr. (2005) The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J. Lipid Res. 46, 1502–1511 [DOI] [PubMed] [Google Scholar]
- 23. Buhman K. K., Smith S. J., Stone S. J., Repa J. J., Wong J. S., Knapp F. F., Jr., Burri B. J., Hamilton R. L., Abumrad N. A., Farese R. V., Jr. (2002) DGAT1 is not essential for intestinal triacylglycerol absorption or chylomicron synthesis. J. Biol. Chem. 277, 25474–25479 [DOI] [PubMed] [Google Scholar]
- 24. Cheng D., Iqbal J., Devenny J., Chu C. H., Chen L., Dong J., Seethala R., Keim W. J., Azzara A. V., Lawrence R. M., Pelleymounter M. A., Hussain M. M. (2008) Acylation of acylglycerols by acyl coenzyme A:diacylglycerol acyltransferase 1 (DGAT1). Functional importance of DGAT1 in the intestinal fat absorption. J. Biol. Chem. 283, 29802–29811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hiramine Y., Emoto H., Takasuga S., Hiramatsu R. (2010) Novel acyl-coenzyme A:monoacylglycerol acyltransferase plays an important role in hepatic triacylglycerol secretion. J. Lipid Res. 51, 1424–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coleman R. A., Mashek D. G. (2011) Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev. 111, 6359–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takeuchi K., Reue K. (2009) Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 296, E1195–E1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campbell J. E., Drucker D. J. (2013) Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837 [DOI] [PubMed] [Google Scholar]
- 29. Quarta C., Mazza R., Obici S., Pasquali R., Pagotto U. (2011) Energy balance regulation by endocannabinoids at central and peripheral levels. Trends Mol. Med. 17, 518–526 [DOI] [PubMed] [Google Scholar]
- 30. Chon S. H., Douglass J. D., Zhou Y. X., Malik N., Dixon J. L., Brinker A., Quadro L., Storch J. (2012) Over-expression of monoacylglycerol lipase (MGL) in small intestine alters endocannabinoid levels and whole body energy balance, resulting in obesity. PLoS One 7, e43962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryan K. K., Woods S. C., Seeley R. J. (2012) Central nervous system mechanisms linking the consumption of palatable high-fat diets to the defense of greater adiposity. Cell Metab. 15, 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., Kratky D., Wagner E. F., Klingenspor M., Hoefler G., Zechner R. (2006) Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 734–737 [DOI] [PubMed] [Google Scholar]
- 33. Ellis J. M., Li L. O., Wu P. C., Koves T. R., Ilkayeva O., Stevens R. D., Watkins S. M., Muoio D. M., Coleman R. A. (2010) Adipose acyl-CoA synthetase-1 directs fatty acids toward β-oxidation and is required for cold thermogenesis. Cell Metab. 12, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cortés V. A., Curtis D. E., Sukumaran S., Shao X., Parameswara V., Rashid S., Smith A. R., Ren J., Esser V., Hammer R. E., Agarwal A. K., Horton J. D., Garg A. (2009) Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 9, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Csaki L. S., Reue K. (2010) Lipins: multifunctional lipid metabolism proteins. Annu. Rev. Nutr. 30, 257–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitra M. S., Chen Z., Ren H., Harris T. E., Chambers K. T., Hall A. M., Nadra K., Klein S., Chrast R., Su X., Morris A. J., Finck B. N. (2013) Mice with an adipocyte-specific lipin 1 separation-of-function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 642–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harms M., Seale P. (2013) Brown and beige fat: development, function and therapeutic potential. Nat. Med. 19, 1252–1263 [DOI] [PubMed] [Google Scholar]
- 38. Birch A. M., Buckett L. K., Turnbull A. V. (2010) DGAT1 inhibitors as anti-obesity and anti-diabetic agents. Curr. Opin. Drug Discov. Devel. 13, 489–496 [PubMed] [Google Scholar]
- 39. Ables G. P., Yang K. J., Vogel S., Hernandez-Ono A., Yu S., Yuen J. J., Birtles S., Buckett L. K., Turnbull A. V., Goldberg I. J., Blaner W. S., Huang L. S., Ginsberg H. N. (2012) Intestinal DGAT1 deficiency reduces postprandial triglyceride and retinyl ester excursions by inhibiting chylomicron secretion and delaying gastric emptying. J. Lipid Res. 53, 2364–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cao J., Zhou Y., Peng H., Huang X., Stahler S., Suri V., Qadri A., Gareski T., Jones J., Hahm S., Perreault M., McKew J., Shi M., Xu X., Tobin J. F., Gimeno R. E. (2011) Targeting Acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J. Biol. Chem. 286, 41838–41851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cases S., Stone S. J., Zhou P., Yen E., Tow B., Lardizabal K. D., Voelker T., Farese R. V., Jr. (2001) Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 276, 38870–38876 [DOI] [PubMed] [Google Scholar]
- 42. Denison H., Nilsson C., Löfgren L., Himmelmann A., Mårtensson G., Knutsson M., Al-Shurbaji A., Tornqvist H., Eriksson J. W. (2014) Diacylglycerol acyltransferase 1 inhibition with AZD7687 alters lipid handling and hormone secretion in the gut with intolerable side effects: a randomized clinical trial. Diabetes Obes. Metab. 16, 334–343 [DOI] [PubMed] [Google Scholar]
- 43. Haas J. T., Winter H. S., Lim E., Kirby A., Blumenstiel B., DeFelice M., Gabriel S., Jalas C., Branski D., Grueter C. A., Toporovski M. S., Walther T. C., Daly M. J., Farese R. V., Jr. (2012) DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Investig. 122, 4680–4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hall A. M., Kou K., Chen Z., Pietka T. A., Kumar M., Korenblat K. M., Lee K., Ahn K., Fabbrini E., Klein S., Goodwin B., Finck B. N. (2012) Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. J. Lipid Res. 53, 990–999 [DOI] [PMC free article] [PubMed] [Google Scholar]