

Background: Na+ interactions with the alanine serine cysteine transporters (ASCT) have not been well established.
Results: Identified residues involved in coordinating three Na+ ions.
Conclusion: ASCT1-mediated transport requires Na+ bound to only two of three Na+ sites; activation of the anion conductance requires only one Na+ bound.
Significance: ASCT1 has three Na+ sites, however, full occupancy is not required for function.
Keywords: Amino Acid Transport, Chloride Channel, Electrophysiology, Membrane Transport, Site-directed Mutagenesis, ASCT, EAAT, GltPh, Na+ Coupling
Abstract
The alanine, serine, cysteine transporters (ASCTs) belong to the solute carrier family 1A (SLC1A), which also includes the excitatory amino acid transporters (EAATs) and the prokaryotic aspartate transporter GltPh. Acidic amino acid transport by the EAATs is coupled to the co-transport of three Na+ ions and one proton, and the counter-transport of one K+ ion. In contrast, neutral amino acid exchange by the ASCTs does not require protons or the counter-transport of K+ ions and the number of Na+ ions required is not well established. One property common to SLC1A family members is a substrate-activated anion conductance. We have investigated the number and location of Na+ ions required by ASCT1 by mutating residues in ASCT1 that correspond to residues in the EAATs and GltPh that are involved in Na+ binding. Mutations to all three proposed Na+ sites influence the binding of substrate and/or Na+, or the rate of substrate exchange. A G422S mutation near the Na2 site reduced Na+ affinity, without affecting the rate of exchange. D467T and D467A mutations in the Na1 site reduce Na+ and substrate affinity and also the rate of substrate exchange. T124A and D380A mutations in the Na3 site selectively reduce the affinity for Na+ and the rate of substrate exchange without affecting substrate affinity. In many of the mutants that reduce the rate of substrate transport the amplitudes of the substrate-activated anion conductances are not substantially affected indicating altered ion dependence for channel activation compared with substrate exchange.
Introduction
The alanine, serine, cysteine transport (ASCT)4 system regulates the cellular uptake of small neutral amino acids (1, 2). Two isoforms have been identified, ASCT1 and ASCT2, which are expressed in various tissues and display distinct substrate selectivity profiles (3–6). ASCTs belong to the solute carrier family 1A (SLC1A) (3–5), along with the human excitatory amino acid transporters (EAATs) and the prokaryotic homolog GltPh (3, 7–9). ASCTs share ∼23% amino acid sequence identity with GltPh and 40% amino acid sequence identity with the EAATs (Fig. 1D) (9). Although all of these transporters belong to the same gene family, there are some striking differences in their transport mechanisms. ASCTs exchange small neutral amino acids in a K+-independent manner, whereas the EAATs transport acidic amino acids, with the counter-transport of K+, generating a net flux of charge during transport (10, 11). The EAATs and ASCTs both possess a thermodynamically uncoupled anion conductance, which is responsible for the current observed during ASCT1-mediated transport (12, 13). Like the EAATs and GltPh, ASCT-mediated transport and activation of the anion conductance is Na+-dependent (5, 13, 14), but the number of Na+ ions required for transport and the location of the Na+ binding sites are not established.
FIGURE 1.

The structure of a GltPh protomer, and sequence alignments of ASCTs, EAATs, and GltPh. A, GltPh protomer (Protein Data Bank code 2NWX) shown in the plane of the membrane, with the trimerization domain (TM1, -2, -4, and -5) in gray and the transport domain: TM3 (light brown), TM6 (blue), TM7 (orange), TM8 (purple), HP1 (yellow), and HP2 (red). l-Aspartate is shown in stick representation, and two bound Na+ ions are shown as light blue spheres. Close up view of the Na1 and Na2 binding sites (B), and proposed Na3 site (C) are shown, with residues targeted for mutation in this study shown in stick representation and labeled with GltPh numbering. The protein has been rotated for ease of visualization of each of the Na+ binding sites. Images were made using PyMol (43). D, sequence alignment of parts of TM3, HP2, TM7, and TM8 in EAAT1–3, ASCT1–2, and GltPh, where conserved residues are highlighted in black, and mutated residues are highlighted by yellow boxes.
GltPh is an aspartate transporter from Pyrococcus horikoshii that was first crystallized in 2004 by Yernool et al. (9) revealing the complex structure of this transporter family (Fig. 1A). GltPh exists as a homotrimer with each protomer containing 8 transmembrane domains (TM1–8) and 2 hairpin loops (HP1 and -2). A later crystal structure of GltPh revealed two Na+-selective cation binding sites, termed Na1 and Na2 (Fig. 1A) (15). Na1 is buried below bound substrate, formed by backbone carbonyls of residues in TM7 and TM8 along with a carboxyl group from Asp-405 in TM8 (Fig. 1B), which is equivalent to Asp-467 in ASCT1. This aspartate residue has been extensively studied in the EAATs, where it has been mutated to a range of alternate residues (16–18). Mutating this aspartate residue in Na1 of EAAT3 to an asparagine generates a transporter with unaltered substrate and Na+ binding capacity. However, this mutant transporter displayed impaired K+ coupling and thereby was locked in exchange mode. Mutating the equivalent residue in GltPh (D405N) decreased Na+ affinity (15), suggesting some variation in the Na1 site between EAATs and GltPh. More drastic mutations, for example, aspartate to alanine, result in an impaired transporter that is able to bind substrate but cannot translocate it. The second cation binding site observed in GltPh, Na2, is located above the binding site and is formed by backbone carbonyls from HP2 and TM7 (Fig. 1B) (15). A glycine residue in HP2 of EAAT1, and the equivalent serine residue in EAAT2, have been shown to influence the differences in cation selectivity between EAAT1 and EAAT2 (19, 20). This residue is in close proximity to the Na2 site (Fig. 1B), and therefore can be used as a probe for the Na2 site in ASCT1. The third Na+ binding site, Na3, was not observed in the crystal structure of GltPh. Mutagenesis and molecular dynamics (MD) studies in both GltPh and the EAATs have been used to propose the location and coordinating residues of the Na3 site (17, 21, 22). One proposed third Na+ site is coordinated by the side chains of Thr-92, Ser-93, Asn-310, Asp-312, and the backbone of Tyr-89 in GltPh. Fig. 1C depicts the cavity of the proposed Na3 site in GltPh, highlighting some of the residues involved in coordinating the Na+ ion. Although the mutations N310A and D312A in GltPh generated non-functional transporters, functional analysis of T92A and S93A, and the equivalent residues in EAAT1, confirmed their involvement in Na+ coordination (22).
The residues involved in coordinating the three established Na+ sites in the EAATs and GltPh are conserved in ASCTs (Fig. 1D). However, no mutational studies have investigated Na+ coupling in the neutral amino acid transporter, ASCT1. Despite the conservation of the Na+ coordinating residues at all three sites, models of 1:1 coupling stoichiometry of Na+ and amino acid exchange have been proposed (14, 23). More recently, Zander et al. (24) demonstrated that Na+ dependence of ASCT2 anion currents is biphasic, suggesting that ASCT2 is coupled to at least two Na+ ions. MD simulations in this study similarly suggest the presence of at least two, and possibly three, Na+ binding sites in ASCT2 (24). To probe Na+ binding sites in ASCTs we mutated coordinating residues within each of the sites proposed for the EAATs and GltPh. Mutations at all three proposed Na+ sites affected either the binding of substrate and/or Na+, or the rate of substrate exchange. Our results suggest that Na+ ions can bind to each of the proposed Na+ sites. However, binding of Na+ to either Na1 or Na3 is required for exchange of substrate, whereas the anion conductance can be activated in the absence of Na+ at Na1 or Na3.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
ASCT1 was subcloned into the plasmid oocyte transcription vector. Site-directed mutagenesis was performed using the Q5® Site-directed Mutagenesis Kit (New England BioLabs Inc.). Primers were designed using NEBaseChanger (New England BioLabs Inc.) and synthesized by Sigma Genosys (Sydney, Australia). DNA sequences of all mutations were confirmed by the Australian Genome Research Facility (Sydney, Australia). DNA was prepared using the PureLinkTM Quick Plasmid Miniprep Kit (Invitrogen), cDNA was linearized with SpeI (Promega) and mRNA was transcribed with T7 polymerase using the mMESSAGE mMACHINE kit (Ambion).
Electrophysiology
All chemicals were obtained from Sigma unless otherwise stated. Stage V oocytes were harvested from Xenopus laevis as described previously (25), and all surgical procedures followed a protocol approved under the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. 20 ng of cRNA was injected into oocytes and incubated in Cl− containing buffer (96 mm NaCl, 2 mm KCl, 1 Mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES, pH7.5) supplemented with 50 μg/ml of gentamycin, 2.5 mm sodium pyruvate, and 0.5 mm theophylline at 16–18 °C.
Two to 4 days after microinjection, current recordings were made using the two-electrode voltage clamp technique with a Geneclamp 500 amplifier (Axon Instruments, Foster City, CA) interfaced with a MacLab 2e chart recorder (ADI Instruments, Sydney, Australia) using the chart software, and a Digidata 1322A (Axon Instruments) controlled by an IBM-compatible computer using pClamp software (version 10, Molecular Devices, Union City, CA). The current-voltage relationships for substrate-elicited conductances were obtained by subjecting cells to 200-ms voltage pulses between −100 and +60 mV in 10-mV steps. Current-voltage relationships were calculated by subtracting steady state current measurements in the absence of substrate from the corresponding current measurements in the presence of substrate.
Recording solution for all experiments (except where otherwise stated) was normal frog Ringer's solution (Cl− containing buffer) with complete NO3− substitution for Cl−. For Na+ titrations, NMDG+ was used as the substitute cation, and total cation concentration was 150 mm. The pH of recording solutions was adjusted using HNO3 and NaOH or KOH. Recordings were made with the bath grounded via a 3 m KCl/agar bridge connected to a 3 m KCl reservoir to minimize offset potentials. Cells were washed with Cl− containing buffer between substrate applications to ensure that NO3 loading of the cell was not significant. Current (I) as a function of substrate concentration was fitted by least-squares analysis to a derivation of the Michaelis-Menten equation, I = Imax · [substrate]/([substrate] + EC50), where Imax is the maximum current generated and EC50 is the substrate concentration, which generates a half-maximal response. Na+ concentration responses were fit to the Hill equation, I/Imax = [substrate]n/([substrate]n + (EC50)n), where n is the Hill coefficient and all other terms are as described above.
Radiolabeled Uptake Experiments
Uptake of l-[3H]serine (PerkinElmer Life Sciences) was measured in oocytes expressing wild type and mutant ASCT1, and uninjected oocytes. Five oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]serine at room temperature. After 10 min, uptake was terminated by three rapid washes in ice-cold Cl− containing buffer followed by lysis in 50 mm NaOH and 1% SDS. l-[3H]Serine was measured by scintillation counting using a Trilux β counter (PerkinElmer Life Sciences).
Homology Model and MD Simulations
The ASCT1 homology model investigated in this study was created using the program MODELLER (26), using the crystal structure of GltPh with aspartate and Na+ bound at Na1 and Na2 as a template (PDB code 2NWX) (22). We replaced the aspartate substrate with serine and placed a Na+ at the Na3 site described by Bastug et al. (22). We have created the models including the ligands in the alignment to obtain the ASCT1 structure in the fully bound state. Once the model is ready, we constructed a trimer by superposing the ASCT1 monomer to chains A, B, and C of GltPh. The simulation system is prepared using the VMD software (27). We embedded the ASCT1 trimer in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine phospholipid bilayer and then solvated the protein-membrane complex in a box of water molecules with 45 Na+ ions and 48 Cl− ions. The extra Cl− ions are required to keep the system neutral, and their number changes depending on the ligands bound to the transporter. There are a total of 247 lipid molecules and 15,975 water molecules, adding up to ∼100,000 atoms in the simulation box. The system was equilibrated by gradually releasing the applied restraints as described elsewhere (28). All MD simulations are performed using the NAMD package (version 2.9) (29) with the CHARMM36 force field (30), using the same parameters as described previously (28). The mutations are performed by alchemically transforming the chosen residue(s) during 5 ns of simulations, and then slowly releasing the restraints applied in the substrate during further 5-ns simulations. After that the equilibrium simulations for a given system are performed.
RESULTS
Characterization of Wild Type ASCT
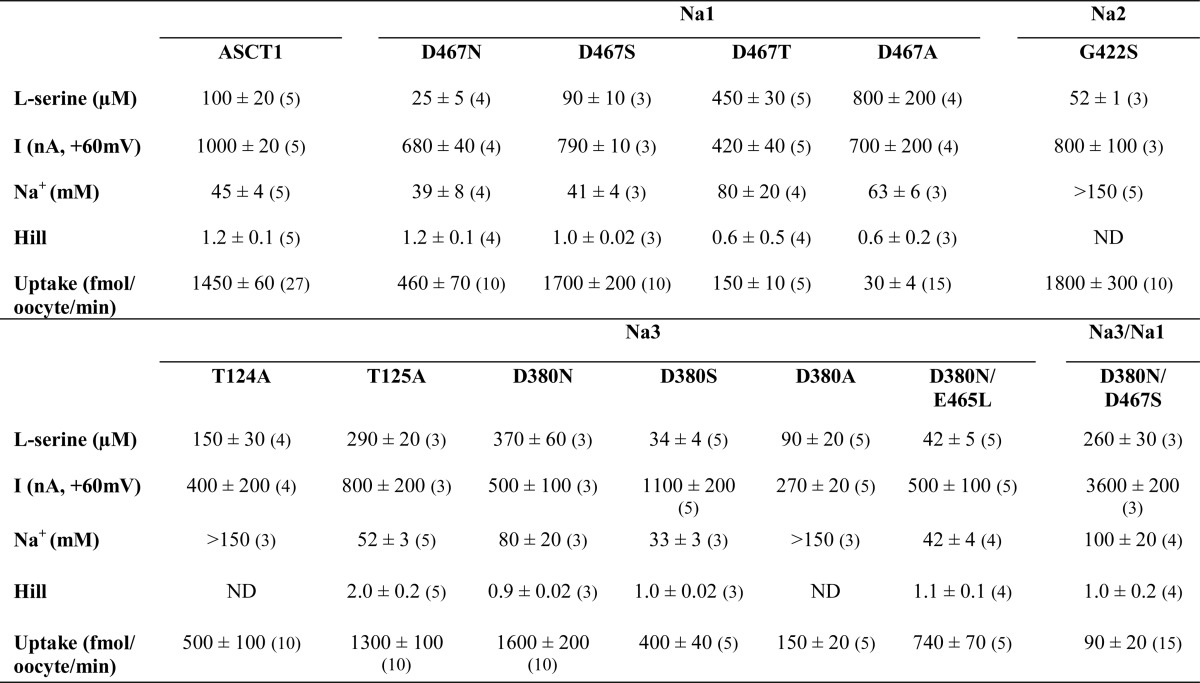
In standard Cl− based recording buffer, application of l-serine to oocytes expressing wild type ASCT1 generates small currents (e.g. 300 μm l-serine generates 17 ± 2 nA at +60 mV; Fig. 2A), which reverse direction at approximately −20 mV. This is indicative of activation of the uncoupled Cl− conductance of ASCT1 (13), which is activated by the binding of substrate and Na+. As exchange of neutral amino acids by ASCT1 is electroneutral, the only current observed is due to this uncoupled anion conductance that has a permeability sequence where some anions are more permeable than others (SCN− > NO3− > I− > Cl−) (13). Therefore, to enhance the amplitude and reliability of current measurements mediated by ASCT1, Cl− in the recording buffer was replaced with NO3−. Using this NO3− containing buffer, the application of l-serine to oocytes expressing ASCT1 at pH 7.5 generates large outward currents (e.g. 1 mm l-serine generates 1000 ± 20 nA at +60 mV; Fig. 2, A and B, Table 1). The EC50 for l-serine activation of the anion current at +60 mV is 100 ± 20 μm (Fig. 2B, Table 1), and the Na+ EC50 is 45 ± 4 mm with a Hill coefficient of 1.2 ± 0.1 (Fig. 2C, Table 1). It should be noted that saturation of Na+ was not achieved in the Na+ concentration-response assays due to limitations in the osmolarity for recording from the oocytes. Therefore the fit and subsequent parameters (Imax, EC50, and Hill coefficient) are only an estimate. However, the relative changes between wild type ASCT1 and mutant transporters provide useful insight into the actions of Na+. ASCT1 also supports significant levels of l-[3H]serine uptake (1450 ± 60 fmol/oocyte/min; Fig. 2D, Table 1).
FIGURE 2.

Mutations of Asp-467 in the proposed Na1 site affect substrate binding and transport. A, current-voltage relationship elicited by 300 μm l-serine in Cl− containing buffer (open squares) and NO3− containing buffer (closed squares), at pH 7.5 in wild type ASCT1. Sample currents in response to 100-ms voltage jumps from −30 to +60 mV (top panel depicts protocol) at 1 mm l-serine and 96 mm NaNO3 are shown for ASCT1 (B) and D467T (C) (lower panels). Imemb refers to the membrane potential, and V refers to the applied voltage. Concentration-response curves are shown for l-serine (D) and Na+ (E) in ASCT1 (closed triangles), D467N (circles), D467S (closed squares), D467T (diamonds), and D467A (open squares) at +60 mV. l-Serine concentrations were varied in a NO3− based buffer with 96 mm NaNO3. Na+ titrations were performed with 1 mm l-serine and NMDG+ as the substitute cation and leak currents were subtracted from baseline measurements. F, l-[3H]serine uptake into oocytes expressing wild type and mutant ASCT1 transporters. Oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]serine at room temperature, pH 7.5, for 10 min. Values presented are mean ± S.E., see Table 1 for n values.
TABLE 1.
Effect of Na+-site mutations on ASCT1 transport properties
l-Serine was applied at pH 7.5 in a NO3− containing buffer at 1 mm, and the current (I) at +60 mV was measured. Maximal concentrations of l-serine were used to establish a Na+ EC50 and Hill coefficient, where NMDG+ was used as the replacement cation. EC50 values are shown in μm for l-serine and mm for Na+. Oocytes expressing wild type and mutant transporters were incubated for 10 min in 10 μm l-[3H]serine (D467T and D467A were incubated in 50 μm), and uptake was measured. All values represent mean ± S.E. Sample sizes are shown in parentheses, where values represent the number of complete experiments. ND, not determined, in these cases the Na+ concentration response did not saturate and it was not possible to obtain an accurate estimate of the Hill coefficient.
The aim of this study was to investigate the number of Na+ ions required by ASCT1, and their location in comparison with corresponding sites in the EAATs. We generated multiple mutations in ASCT1 that correspond to the three known Na+ binding sites in the EAATs, to determine the Na+ coupling mechanism of ASCT1.
Na1 Site
The Na1 site in GltPh is coordinated by multiple backbone interactions, along with a carboxyl group of Asp-405 in TM8 (Fig. 1B). This aspartate residue has been widely studied in the EAATs, where it has been mutated to a range of alternate residues (16–18). To thoroughly characterize the Na1 site in ASCT1 we mutated the equivalent aspartate residue (Asp-467) to asparagine, serine, threonine, and alanine, and expressed the mutant transporters in X. laevis oocytes.
D467S mutant transporters expressed in oocytes display a phenotype very similar to that of wild type ASCT1 (Fig. 2, Table 1). The D467N mutant ASCT1 transporter has an apparent Na+ affinity and Hill coefficient that are similar to that of wild type ASCT1 (39 ± 8 mm and 1.2 ± 0.1; Fig. 2C, Table 1). These results are consistent with that observed from the equivalent mutation in the EAATs where Na+ binding is unaffected (16–18). However, serine is 4-fold more potent for D467N and the rate of l-[3H]serine exchange is reduced 3-fold compared with that of wild type ASCT (Fig. 2, Table 1). To further probe the existence of the Na1 site in ASCT1 we generated D467T and D467A mutants. In contrast to similar mutants in the EAATs (16), the mutants generated functional transporters, albeit with reduced serine and Na+ affinity (EC50 values for serine of 450 ± 30 and 800 ± 200 μm, respectively, and EC50 values for Na+ of 80 ± 20 and 63 ± 6 mm, respectively; Fig. 2, Table 1) and an ∼2-fold reduction of the amplitude of serine-activated currents for D467T (420 ± 40 nA; Fig. 2C, Table 1). The Hill coefficients for both D467T and D467A also appeared slightly shifted from that of wild type, although within error (0.6 ± 0.5 and 0.6 ± 0.2, respectively; Table 1). Despite the large outward currents mediated by D467T and D467A, the rates of [3H]serine uptake are not significantly above background (Fig. 2D). This suggests that perturbation of the Na1 site diminishes the ability of ASCT1 to translocate substrate, but does not largely affect the substrate-activated anion conductance. It is remarkable that the addition of a single methyl group, from serine (D467S) to threonine (D467T), can abolish the exchange of substrate while maintaining sufficient binding of substrate to activate the anion conductance.
Na2 Site
The Na2 site in GltPh is coordinated exclusively by backbone interactions (15), making it difficult to characterize using mutagenesis. However, a glycine residue in close proximity to the Na2 site (Fig. 1B) of EAAT1 can influence cation coupling of glutamate transporters (19, 20). Mutating this glycine residue in EAAT1 to serine diminishes the capacity of Li+ to support transport. This glycine residue is conserved in ASCT1 (Fig. 1D) and to investigate binding of Na+ at the proposed Na2 site in ASCT1 we mutated this glycine in HP2 to serine (G422S).
Oocytes expressing G422S displayed an EC50 for Na+ that was significantly greater than for wild type ASCT1 (>150 mm; Fig. 3B), whereas l-serine activation of the anion conductance (e.g. 1 mm l-serine generates 800 ± 100 nA at +60 mV; Fig. 3C, Table 1) estimates of the EC50 for l-serine (52 ± 1 μm; Fig. 3A), and rates of [3H]serine uptake (1800 ± 300 fmol/oocyte/min; Fig. 3C) are comparable to wild type ASCT1. These results suggest that introduction of a serine at position 422 in HP2 of ASCT1 impairs the binding of Na+, presumably at the Na2 site, without affecting substrate binding or translocation.
FIGURE 3.

Mutation of Gly-422 in the proposed Na2 site alters Na+ binding. Concentration-response curves are shown for l-serine (A) and Na+ (B) in wild type ASCT1 (triangles) and G422S (open triangles). l-Serine concentrations were varied in a NO3− based buffer with 96 mm NaNO3. Na+ titrations were performed with 1 mm l-serine and NMDG+ as the substitute cation. C, a sample current in response to 100 ms voltage jumps from −30 to +60 mV (top panel depicts protocol) at 1 mm l-serine and 96 mm NaNO3 is shown for G422S (lower panel). Imemb refers to the membrane potential, and V refers to the applied voltage. D, l-[3H]serine uptake into oocytes expressing wild type and G422S mutant ASCT1 transporters. Oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]serine at room temperature, pH 7.5, for 10 min. Values presented are mean ± S.E., see Table 1 for n values.
Na3 Site
A third Na+ ion was not identified in the crystal structure of GltPh. However, multiple studies of both GltPh and the EAATs have proposed a Na3 site (17, 21, 22). Na3 is believed to be coordinated by Thr-92 and Asp-312 (GltPh numbering) (17, 21, 22), and more recently Ser-93, Asn-310, and Tyr-89 have also been shown to be involved (22). In this study, we investigated Asp-380, Thr-124, and Thr-125 in ASCT1 (which correspond to Asp-312, Thr-92, and Ser-93 in GltPh; Fig. 1C).
Application of l-serine to oocytes expressing T124A or T125A transporters generates outward currents with small changes in EC50 values for serine (Table 1), but in the case of T124A, there is a significant reduction in Na+ affinity (EC50 > 150 mm; Fig. 4B) and a 3-fold reduction of uptake levels of l-[3H]serine (500 ± 100 fmol/oocyte/min; Fig. 4G). T125A displayed a similar affinity for Na+ compared with wild type ASCT1 (52 ± 3 mm), but the Hill coefficient shifted from 1.2 ± 0.1 in wild type ASCT1 to 2.0 ± 0.2 in T125A (Fig. 4B, Table 1), which suggests that Na+ interactions with the transporter have been altered by the mutation. Thus, Thr-124 and Thr-125 both appear to contribute to Na+ binding to ASCT1.
FIGURE 4.

Mutations in the proposed third Na+ site affect Na+ and substrate binding, and translocation. l-Serine concentration-response curves are shown for ASCT1 (A, C, and E; closed triangles), T124A (A; open circles), T125A (A; open diamonds), D380N (C; closed circles), D380S (C; closed triangles), D380A (C; open squares), D380N/D467S (E; diamonds), and D380N/E465L (E; closed circles). l-Serine concentrations were varied in a NO3− based buffer with 96 mm NaNO3. Na+ concentration-response curves are shown for ASCT1 (B, D, and F; closed squares), T124A (B; open circles), T125A (B; open diamonds), D380N (D; closed circles), D380S (D; closed triangles), D380A (D; open squares), D380N/D467S (F; diamonds), and D380N/E465L (F; closed circles). Na+ titrations were performed with 1 mm l-serine and NMDG+ as the substitute cation. G, a sample current in response to 100-ms voltage jumps from −30 to +60 mV (top panel depicts protocol) at 1 mm l-serine and 96 mm NaNO3 is shown for D380N/D467S (lower panel). Imemb refers to the membrane potential, and V refers to the applied voltage. H, l-[3H]serine uptake into oocytes expressing wild type and mutant ASCT1 transporters. Oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]serine at room temperature, pH 7.5, for 10 min. Values presented are mean ± S.E., see Table 1 for n values.
Aspartate 312 of GltPh is located in the highly conserved NMDGT motif in TM7 that is known to play crucial roles in substrate binding and coordination of Na3 (15, 17, 21, 31, 32). In the EAATs and GltPh, neutralizing this aspartate residue generates a non-functional transporter (17, 22). To investigate the role of the equivalent aspartate residue in coordinating the binding of a third Na+ ion to ASCT1, we generated D380N, D380S, and D380A mutations. Surprisingly, and in contrast to the EAATs, D380N generated outward currents in response to application of l-serine, albeit with a decreased affinity compared with that of wild type ASCT1 (370 ± 60 μm; Fig. 4C) but with EC50 values for Na+ (80 ± 20 mm; Fig. 4D), and uptake levels of l-[3H]serine (1600 ± 200 fmol/oocyte/min; Fig. 4G) that are comparable to wild type ASCT1. There are two possible explanations for these results: Na+ can still bind to the proposed Na3 site of the D380N mutant transporter and the transporter is functioning like wild type ASCT1; or Na+ is not bound in the Na3 site of D380N and the transporter does not require this aspartate residue for function. The D380S mutant displayed an apparent affinity for Na+ that is comparable to wild type ASCT1 (33 ± 3 mm, respectively; Fig. 4, C and D) but the EC50 for serine and uptake levels of l-[3H]serine were reduced 3- and 3.5-fold, respectively (34 ± 4 μm and 400 ± 40 fmol/oocyte/min; Fig. 4G). D380A also displayed an apparent affinity for l-serine that was similar to wild type ASCT1 (90 ± 20 μm; Fig. 4C) but the apparent Na+ affinity of D380A was reduced (>150 mm; Fig. 4D). Similarly, the uptake levels of l-[3H]serine into oocytes expressing D380A were reduced by 9.6-fold (150 ± 20 fmol/oocyte/min; Fig. 4G). The impaired ASCT1-mediated [3H]serine exchange observed with the D380A mutation suggests that disruption of the proposed Na3 site has caused a conformational change that no longer allows efficient movement of the transport domain or unbinding of substrate. However, despite the reduced rates of transport, the amplitudes of the substrate-activated anion conductances of all the Asp-380 mutant transporters are comparable to wild type ASCT1 (Table 1), which suggests that the remaining Na+ and substrate binding sites remain intact and that they are sufficient for activation of the anion channel.
It is surprising that neutralizing the proposed coordinating aspartates in either Na1 or Na3 can generate fully functional transporters that closely resemble wild type ASCT1, such as with D380N and D467S (Figs. 2 and 4). However, it is unclear from these results whether a Na+ ion is still bound at Na1 and Na3 in the D467S and D380N mutant transporters. In the event that Na+ can bind effectively to Na1 and Na3 in D467S and D380N mutant transporters, combining the two mutations should generate a transporter that continues to reflect wild type ASCT1. We therefore generated the double mutant transporter, D380N/D467S, to determine if Na1 and Na3 remain intact in both of the single mutants. Oocytes expressing the D380N/D467S double mutant transporter generate outward currents in response to application of l-serine (e.g. 1 mm l-serine generates 3600 ± 200 nA at +60 mV; Fig. 4G, Table 1), with a decreased affinity compared with wild type ASCT1 (260 ± 30 μm; Fig. 4E). The apparent Na+ affinity of D380N/D467S was reduced by 2-fold compared with that of wild type ASCT1 (100 ± 20 mm; Fig. 4F), and the levels of l-[3H]serine uptake were not significantly greater than background (Fig. 4G). Furthermore, the amplitude of the serine-activated anion conductance was increased by 3.8-fold (Table 1), which suggests that despite the slight impairment of Na+ and substrate binding, the lack of substrate exchange allows Na+ and serine to remain bound and generate greater anion current activation.
Molecular Dynamic Simulations
Our results suggest that either Asp-380 or Asp-467 may not be essential for ASCT1-mediated transport, and that substitution with other small polar residues such as asparagine or serine may suffice to maintain substrate and Na+ binding and exchange. The contrasting role of Asp-380 in ASCT1 compared with the equivalent residue in the EAATs led us to investigate the molecular details of substrate binding in the presence and absence of these two aspartate residues in an ASCT1 homology model. We have simulated the wild type ASCT1 model, as well as mutations of the Asp-380 and Asp-467 residues, with different combinations of ligands bound to the transporter (Table 2). In simulations of Na+ binding to GltPh, it is useful to introduce a Na1′ site that overlaps with both Na1 and Na3 (22). Na1′ is a Na+ site obtained by equilibrating the system for 10 ns in the absence of Na3, so that the Na1 ion becomes coordinated by both the Asp-380 and Asp-467 side chains in all monomers. We use this site because it overlaps with both the Na1 and Na3 sites allowing a quick transition to either one of them depending on the mutations introduced. The systems were simulated for 20 ns and the results are summarized in Table 2. Even though the three ASCT1 monomers exhibit slightly different behavior in various cases (see Table 2), we can draw some important conclusions based on our simulations. The fully bound system is very stable, with all the ligands (Na1, Na2, Na3, and serine) bound to the transporter during the 20 ns of simulations (Fig. 5A). In wild type ASCT1, there is a strong negative electric potential in the Na1/Na3 region caused by the presence of the Asp-380 and Asp-467 side chains. This is evidenced by the displacement of Na+ from Na2 to this location when either Na3, or both Na1 and Na3, are absent (Fig. 5B). Therefore, at least one Na+, most likely two, must bind to this region in the wild type ASCT1. This is supported by the observation that serine is unstable in two of the three monomers when both Na1 and Na3 are absent. Na3 always remains bound when we neutralize Asp-380, revealing that the negative charge of Asp-380 might not be essential for the binding of this Na+ in ASCT1. In the D380A mutation we see the displacement of the Glu-465 (TM8) side chain toward Na3 in one of the monomers (Fig. 5C).
TABLE 2.
MD simulations performed in wild type and mutant ASCT1 transporters
Various combinations of Na+ sites occupied with bound substrate were simulated. The length of each simulation is 20 ns, with a total of 240 ns for all systems.
| Ligands bound | Result from 20 ns of simulations |
|---|---|
| ASCT1-WT | |
| Na1, Na2, Na3, serine | All ligands remain bound to the transporter at their respective binding sites (Fig. 1A). |
| Na1′, Na2, serine | In monomers A and B, Na2 leaves its binding site and the two Na+ are now coordinated by the Asp-80 and Asp-467 side chains (Fig. 1B). In monomer C, Na2 remains bound, and so does Na1′. Serine remains bound in all subunits. |
| Na2, serine | Na2 moves to the Na1′ site in monomer A, to the Na1 site in monomer B, and remains bound in monomer C. Serine is unstable in the binding site and loses contacts in subunits B and C. |
| ASCT1-D380A | |
| Na1, Na2, Na3, serine | Na1 remains bound and Na2 is released to the solvent in all chains. Na3 remains bound in all subunits, being coordinated by Glu-465 in monomer A (Fig. 1C). Serine remains bound in all monomers. |
| Na1′, Na2, serine | Na1′ moves to the Na1 site in all chains. Na2 remains bound at subunits B and C. Serine remains bound in all monomers. |
| ASCT1-D380N | |
| Na1, Na2, Na3, Serine | Na1, Na3, and serine remain bound in all monomers. Na2 is released to the solvent in monomers A and C. |
| Na1′, Na2, serine | Na1′ moves to the Na1 site in all chains. Na2 is released to the solvent in monomers A and C. Serine remains bound in all monomers. |
| ASCT1-D467S | |
| Na1, Na2, Na3, serine | Na1 is released in all subunits (Fig. 5E). Na2 is released in monomers A and C. Na3 and Serine remain bound in all monomers. |
| Na1′, Na2, serine | Na1′ is shifted close to the Na3 site, coordinated by the Asp-380 side chain in all subunits (Fig. 1D). Na2 remains bound in monomers A and B. Serine remains bound in all monomers. |
| ASCT1-D380N/D467S | |
| Na1, Na2, Na3, serine | In all monomers both Na1 and Na2 are released, with Na3 and Serine remaining bound. |
| Na1′, Na2, serine | Na1′ and serine remain bound, and Na2 is released in all subunits. |
| Na2, serine | Na2 is released in chains B and C. Serine is unstable in the binding site and loses contacts in monomer B. |
FIGURE 5.

MD simulations of an ASCT1 homology model with mutations to the Na1 and Na3 sites. A, the ASCT1 model after 20 ns of simulations, showing the l-serine substrate (green sticks), as well Na+ (cyan sphere) bound at Na1, Na2, and Na3 sites. B, the Na1′ and Na2 sites occupied in wild type ASCT1, in the absence of a Na+ at Na3, after 20 ns of simulations. The Na+ bound at Na2 moves closer to Na1′ and are now coordinated by the Asp-380 and Asp-467 side chains. C, the new coordination of Na+ at Na3 in the D380A mutant transporter, in which we see the flipping of the Glu-465 side chain to coordinate a Na+ at Na3 in one of the chains of ASCT1. D, the new binding site of the Na+ bound at Na1′ in the D467S mutant transporter without a Na+ bound at Na3, after 20 ns of simulations. This Na+ is now coordinated by Ser-467 and Asp-380 side chains. E, the time evolution of the distance between the Ser-467 side chain oxygen and the Na1 ion in the D467S transporter with all ligands bound. Black represents chain A, red chain B, and green chain C. In all cases the Na+ bound at Na1 leaves its binding site.
The Na1 site displays contrasting behavior. The bound Na+ is always released when the Asp-467 side chain is neutralized and Na3 is bound to the transporter (Fig. 5E). In the absence of Na3, and with Asp-467 neutralized, a Na+ remains bound at Na1′, stabilized by the Asp-380 side chain (Fig. 5D). This suggests that neutralization of Asp-467 reduces the number of Na+ ions bound to ASCT1. The bound substrate remains stable in the binding site during most of the simulations, even when both Na1 and Na3 sites are perturbed with the double D380N/D467S mutant transporter (Table 2). This may explain why the substrate-activated anion conductance observed in the mutant transporters is largely unaffected by these mutations. The Na2 ion appears very sensitive to changes in the transporter or the bound ligands, since this Na+ is released in most of the simulations (Table 2). In electrophysiological experiments most mutations to the Na1 or Na3 sites did not appear to affect Na2 binding, as the apparent Na+ affinity remained similar to that of wild type ASCT1. One possible explanation is that the Na2 site identified in GltPh might be in a slightly different location in ASCT1.
As discussed above, the side chain of a glutamate residue in TM8 (Glu-465 in ASCT1) was observed to flip 180° toward the Na3 site, and coordinate the bound Na+. This glutamate residue is a leucine in the EAATs (Fig. 1D), and therefore may explain the opposing results observed in mutating Asp-380 in ASCT1 and the corresponding residues in the EAATs. To investigate the role of this glutamate residue (Glu-465) in coordinating Na3 in ASCT1 we mutated Glu-465 to the EAAT counterpart, in combination with a neutralized aspartate residue in Na3. Oocytes expressing D380N/E465L mutant transporters generated outward currents in response to application of l-serine, with a slightly reduced EC50 compared with wild type ASCT1 (42 ± 5 μm; Fig. 4E). The apparent Na+ affinity of D380N/E465L closely resembled that of wild type ASCT1 (42 ± 4 mm; Fig. 4F) and levels of l-[3H]serine uptake were significantly above background, however, only reached ∼50% of wild type ASCT1 (740 ± 70 fmol/oocyte/min; Fig. 4G). This suggests that Na3 is not required for the binding and transport of substrate by ASCT1, which is in stark contrast to the EAATs.
DISCUSSION
Three Na+ binding sites have been identified in both EAATs and GltPh (15, 17, 19–22, 28, 33–35). However, in the closely related neutral amino acid transporters, ASCT1 and -2, Na+ interactions have not been well characterized. In this study, we utilize mutagenesis and functional analysis in combination with MD simulations to investigate the three possible Na+ sites in ASCT1. Asp-467 in Na1 of ASCT1 can be replaced by amino acids such as asparagine or serine while maintaining effective substrate and Na+ binding, and activation of the anion conductance (Fig. 2). However, the introduction of more hydrophobic residues such as alanine or threonine at position 467 diminishes substrate exchange. Similarly in the Na3 site, the D380N mutation does not affect substrate binding or transport, whereas D380A affects the apparent Na+ affinity and substrate exchange. This suggests that the coordinating residues in Na1 and Na3 of ASCT1 must remain hydrophilic for translocation to occur. MD simulations performed by Mwaura et al. (18) show that a fully hydrophobic environment is created around the coordinating aspartate of Na1 when it is mutated to alanine (D454A in EAAT3, equivalent to D467A in ASCT1), collapsing the aqueous access pathway to the binding site. The structural changes associated with collapsing Na+ sites may be interrupting the movement of the transport domain during translocation, or the unbinding of substrate to allow exchange. However, in our MD simulations and also those by Mwaura et al. (18), a Na+ remains bound at Na3, despite the variations observed in physical experiments. The disparity between the physical and computational experiments investigating Na3 is yet to be understood.
Interestingly, combining mutations in Na1 and Na3 of ASCT1 that individually resemble wild type (D467S and D380N, respectively) generate a transporter with diminished transport rates. The substrate affinity of D467S/D380N remains unchanged from wild type ASCT1, and our MD simulations show that the substrate is stable within the binding pocket. This indicates that neutralizing the coordinating aspartate residue in either Na1 or Na3 subtly alters the binding of Na+ at each respective site, without affecting the transporter phenotype. However, when Na+ binding is altered at both sites simultaneously, substrate can bind but cannot be effectively exchanged, as seen with hydrophobic mutations to the Na+ sites (D467A, D380A). This indicates that effective binding of at least one Na+ at either Na1 or Na3 is required for substrate translocation by ASCT1. This is in contrast to the EAATs, where substrate binding is directly correlated with Na+ binding at the Na3 site (17, 22).
In the EAATs similar results were seen with mutations at the Na1 site, where neutralization of the coordinating aspartate residue does not alter substrate or Na+ affinities (16–18). However, there is a striking contrast between the EAATs and ASCT1 in respect to mutations of the Na3 site. In the EAATs, mutations of the coordinating aspartate (D367N in EAAT3, D398C in EAAT2) markedly reduce the Na+ affinity, generating an ineffective transporter (17, 32). In contrast, mutating D380N in ASCT1 generates a transporter with similar phenotype to wild type ASCT1. Our MD simulations revealed a glutamate residue (Glu-465) that is unique to ASCTs and may coordinate the Na+ at Na3 when Asp-380 is neutralized. However, when both of the coordinating aspartate and glutamate residues were neutralized in ASCT1 (D380N/E465L), the transport characteristics remained unaltered from wild type, reinforcing our hypothesis that only one of Na1 or Na3 is required to be occupied for substrate translocation by ASCT1.
Previous studies have reported a much higher Na+ affinity for ASCT2 in the absence of substrate (0.25–2.0 mm) (23, 24) compared with the EAATs (EAAT3 is 100 mm (17)). This raises the question: is Na+ unbinding during ASCT-mediated exchange, or does Na+ function as an allosteric modulator at some of the Na+ sites? Zander et al. (24) propose that Na+ acts as allosteric modulators at Na1 and Na3 due to the reported high affinity, an unlikely event of unbinding. Our results demonstrate the modulatory role Na+ plays in both transport and anion conductance of ASCT1, however, as Na+ dissociation could not been investigated in this study, further research is required to fully answer this question.
Tao et al. (17) demonstrated that the D367N mutation in Na3 of EAAT3 dramatically reduced Na+ affinity, and in turn reduced the substrate affinity by 500-fold. From this it was determined that substrate binding was directly related to Na+ binding at Na3. In our study, the equivalent mutation D380A in Na3 of ASCT1 reduced the Na+ affinity, but did not affect substrate binding. The study by Tao et al. (17) also demonstrated that reducing the affinity of Na+ at the Na3 site of ASCT2 reduced substrate affinity by only 4-fold, compared with a 500-fold reduction in EAAT3. This leads us to conclude that substrate binding in ASCT1 is not critically dependent on Na+ binding at Na3. The disparity between the effects of Na3 mutations in EAAT3 and ASCT1 could be explained by the charge of the substrates. EAATs transport negatively charged acidic amino acids, whereas ASCT1 (and ASCT2) transports neutral amino acids. Two Na+ ions bound below the substrate binding site, as in Na1 and Na3, would most likely assist in the coordination of a negatively charged substrate, such as glutamate. On the other hand, neutral amino acids are unlikely to require a charged binding partner. They do, however, require the neutralization of the strong negative electric potential generated by the side chains of Asp-467 and Asp-380, as seen in our MD simulations. Thus, neutral amino acid exchange by ASCT1 does not require Na+ to be bound at Na1 and Na3 for efficient substrate binding and activation of the anion conductance. However, efficient translocation of substrate still requires the binding of at least one Na+ to Na1 or Na3.
Despite disruptions in the ability to exchange substrate, effective activation of the anion conductance is maintained in all of the mutant transporters described in this study. Although the amplitude of some substrate-activated currents are reduced compared with wild type (e.g. D467T, Table 1), the current sizes are still relatively large indicating that anion conductance is not impaired. This is in contrast to the EAATs where there is no observable substrate-activated anion conductance in mutant transporters equivalent to D467T or D467A (16). The reductions observed in the amplitude of the serine-activated currents are often correlated with altered substrate or Na+ binding, however, a direct effect on anion conductance cannot be ruled out. In any case, changes in the amplitude of the serine-activated currents do not correlate with changes in the rates of transport, for example, the double mutant D380N/D467S displayed an exaggerated anion conductance, with minimal transport activity (Fig. 4, Table 1). This demonstrates the distinction between the processes of transport and activation of the anion conductance, which has also been demonstrated in the EAATs (36–38). Varying transport rates, from background levels to levels exceeding wild type ASCT1, exerted little effect on substrate affinity and activation of the anion conductance. This suggests that the structural changes necessary for opening the anion channel in ASCT1 do not require complete translocation of the substrate, as proposed in the EAATs and GltPh (34, 36, 38, 39). These results also provide some insight into the involvement of Na+ in the activation of the anion conductance. Although we know that Na+ alone binds (presumably) to Na1/Na3 and activates a leak conductance in ASCTs (23), this study demonstrates the lesser involvement of Na+ in activation of the substrate-activated anion conductance. The multiple perturbations introduced into Na1 and Na3 do not heavily influence the ability of substrate to activate the anion conductance. However, the activation of anion conductance remains Na+ dependent, which suggests that Na2 plays an important role in the activation of the anion conductance. This proposal is consistent with the lack of anion conductance observed when the non-transportable blocker threo-β-benzyloxyaspartate is bound to the EAATs or GltPh, where the Na2 site is disrupted by threo-β-benzyloxyaspartate propping HP2 open (15, 40–42). Similarly, benzylserine, an ASCT inhibitor structurally based on threo-β-benzyloxyaspartate, inhibits the anion conductance of ASCTs (23).
In conclusion, we propose that ASCT1 has three Na+ binding sites, consistent with those observed in the EAATs and GltPh. However, the three Na+ ions play a different role in ASCT1 compared with the EAATs. In the EAATs, Na+ bound at Na1 and Na3 is required for binding of the negatively charged substrate. In ASCT1 on the other hand, we propose that substrate and Na2 can bind and activate the anion conductance regardless of the occupancy state of Na1 and Na3, although for ASCT1-mediated substrate exchange, at least one of Na1 or Na3 must be occupied.
Acknowledgments
We thank Cheryl Handford for expert technical assistance and all who maintain the University of Sydney X. laevis colony. MD simulations were performed using the HPC facilities at the National Computational Infrastructure (Canberra), and the Victorian Life Sciences Computation Initiative (Melbourne).
This work was supported in part by National Health and Medical Research Council of Australia Project Grant APP1048784.
- ASCT1
- alanine, serine, cysteine transporter
- EAAT
- excitatory amino acid transporter
- SLC1
- solute carrier family 1
- TM
- transmembrane domain
- HP
- hairpin loop
- GltPh
- prokaryotic aspartate transporter
- MD
- molecular dynamics
- NMDG
- N-methyl-d-glucamine.
REFERENCES
- 1. Christensen H. N., Liang M., Archer E. G. (1967) A distinct Na+-requiring transport system for alanine, serine, cysteine, and similar amino acids. J. Biol. Chem. 242, 5237–5246 [PubMed] [Google Scholar]
- 2. Christensen H. (1990) Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev. 70, 43–77 [DOI] [PubMed] [Google Scholar]
- 3. Arriza J. L., Kavanaugh M. P., Fairman W. A., Wu Y. N., Murdoch G. H., North R. A., Amara S. G. (1993) Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J. Biol. Chem. 268, 15329–15332 [PubMed] [Google Scholar]
- 4. Shafqat S., Tamarappoo B. K., Kilberg M. S., Puranam R. S., McNamara J. O., Guadaño-Ferraz A., Fremeau R. T., Jr. (1993) Cloning and expression of a novel Na+-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J. Biol. Chem. 268, 15351–15355 [PubMed] [Google Scholar]
- 5. Utsunomiya-Tate N., Endou H., Kanai Y. (1996) Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J. Biol. Chem. 271, 14883–14890 [DOI] [PubMed] [Google Scholar]
- 6. Bröer A., Brookes N., Ganapathy V., Dimmer K. S., Wagner C. A., Lang F., Bröer S. (1999) The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 73, 2184–2194 [PubMed] [Google Scholar]
- 7. Fairman W. A., Vandenberg R. J., Arriza J. L., Kavanaugh M. P., Amara S. G. (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375, 599–603 [DOI] [PubMed] [Google Scholar]
- 8. Arriza J. L., Eliasof S., Kavanaugh M. P., Amara S. G. (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. U.S.A. 94, 4155–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yernool D., Boudker O., Jin Y., Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 [DOI] [PubMed] [Google Scholar]
- 10. Zerangue N., Kavanaugh M. (1996) Flux coupling in a neuronal glutamate transporter. Nature 383, 634–637 [DOI] [PubMed] [Google Scholar]
- 11. Ryan R. M., Kortt N. C., Sirivanta T., Vandenberg R. J. (2010) The position of an arginine residue influences substrate affinity and K+ coupling in the human glutamate transporter, EAAT1. J. Neurochem. 114, 565–575 [DOI] [PubMed] [Google Scholar]
- 12. Ryan R. M., Mitrovic A. D., Vandenberg R. J. (2004) The chloride permeation pathway of a glutamate transporter and its proximity to the glutamate translocation pathway. J. Biol. Chem. 279, 20742–20751 [DOI] [PubMed] [Google Scholar]
- 13. Zerangue N., Kavanaugh M. P. (1996) ASCT-1 is a neutral amino acid exchanger with chloride channel activity. J. Biol. Chem. 271, 27991–27994 [DOI] [PubMed] [Google Scholar]
- 14. Bröer A., Wagner C., Lang F., Bröer S. (2000) Neutral amino acid transporter ASCT2 displays substrate-induced Na+ exchange and a substrate-gated anion conductance. Biochem. J 346, 705–710 [PMC free article] [PubMed] [Google Scholar]
- 15. Boudker O., Ryan R. M., Yernool D., Shimamoto K., Gouaux E. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 [DOI] [PubMed] [Google Scholar]
- 16. Teichman S., Qu S., Kanner B. (2009) The equivalent of a thallium binding residue from an archeal homolog controls cation interactions in brain glutamate transporters. Proc. Natl. Acad. Sci. U.S.A. 106, 14297–14302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tao Z., Zhang Z., Grewer C. (2006) Neutralization of the aspartic acid residue Asp-367, but not Asp-454, inhibits binding of Na+ to the glutamate-free form and cycling of the glutamate transporter EAAC1. J. Biol. Chem. 281, 10263–10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mwaura J., Tao Z., James H., Albers T., Schwartz A., Grewer C. (2012) Protonation state of a conserved acidic amino acid involved in Na+ binding to the glutamate transporter EAAC1. ACS Chem. Neurosci. 3, 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y., Kanner B. I. (1999) Two serine residues of the glutamate transporter GLT-1 are crucial for coupling the fluxes of sodium and the neurotransmitter. Proc. Natl. Acad. Sci. U.S.A. 96, 1710–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang S., Ryan R. M., Vandenberg R. J. (2009) The role of cation binding in determining substrate selectivity of glutamate transporters. J. Biol. Chem. 284, 4510–4515 [DOI] [PubMed] [Google Scholar]
- 21. Tao Z., Rosental N., Kanner B. I., Gameiro A., Mwaura J., Grewer C. (2010) Mechanism of cation binding to the glutamate transporter EAAC1 probed with mutation of the conserved amino acid residue T101. J. Biol. Chem. 285, 17725–17733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bastug T., Heinzelmann G., Kuyucak S., Salim M., Vandenberg R. J., Ryan R. M. (2012) Position of the third Na+ site in the aspartate transporter GltPh and the human glutamate transporter, EAAT1. PLoS ONE 7, e33058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grewer C., Grabsch E. (2004) New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J. Physiol. 557, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zander C. B., Albers T., Grewer C. (2013) Voltage-dependent processes in the electroneutral amino acid exchanger ASCT2. J. Gen. Physiol. 141, 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poulsen M. V., Vandenberg R. J. (2001) Niflumic acid modulates uncoupled substrate-gated conductances in the human glutamate transporter EAAT4. J. Physiol. 534, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eswar N., Eramian D., Webb B., Shen M.-Y., Sali A. (2008) Protein structure modeling with MODELLER. in Structural Proteomics, pp. 145–159, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 27. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 28. Heinzelmann G., Baştuğ T., Kuyucak S. (2011) Free energy simulations of ligand binding to the aspartate transporter GltPh. Biophys. J. 101, 2380–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., Jr., Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seal R. P., Leighton B. H., Amara S. G. (2000) A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron 25, 695–706 [DOI] [PubMed] [Google Scholar]
- 32. Zarbiv R., Grunewald M., Kavanaugh M. P., Kanner B. I. (1998) Cysteine scanning of the surroundings of an alkali-ion binding site of the glutamate transporter GLT-1 reveals a conformationally sensitive residue. J. Biol. Chem. 273, 14231–14237 [DOI] [PubMed] [Google Scholar]
- 33. Huang Z., Tajkhorshid E. (2010) Identification of the third Na+ site and the sequence of extracellular binding events in the glutamate transporter. Biophys. J. 99, 1416–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Borre L., Kanner B. I. (2001) Coupled, but not uncoupled, fluxes in a neuronal glutamate transporter can be activated by lithium ions. J. Biol. Chem. 276, 40396–40401 [DOI] [PubMed] [Google Scholar]
- 35. Shrivastava I. H., Jiang J., Amara S. G., Bahar I. (2008) Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J. Biol. Chem. 283, 28680–28690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ryan R. M., Vandenberg R. J. (2002) Distinct conformational states mediate the transport and anion channel properties of the glutamate transporter EAAT-1. J. Biol. Chem. 277, 13494–13500 [DOI] [PubMed] [Google Scholar]
- 37. Borre L., Kavanaugh M. P., Kanner B. I. (2002) Dynamic equilibrium between coupled and uncoupled modes of a neuronal glutamate transporter. J. Biol. Chem. 277, 13501–13507 [DOI] [PubMed] [Google Scholar]
- 38. Seal R. P., Shigeri Y., Eliasof S., Leighton B. H., Amara S. G. (2001) Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc. Natl. Acad. Sci. U.S.A. 98, 15324–15329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shabaneh M., Rosental N., Kanner B. (2014) Disulfide cross-linking of transport and trimerization domains of a neuronal glutamate transporter restricts the role of the substrate to the gating of the anion conductance. J. Biol. Chem. 289, 11175–11182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shigeri Y., Shimamoto K., Yasuda-Kamatani Y., Seal R. P., Yumoto N., Nakajima T., Amara S. G. (2001) Effects of threo-β-hydroxyaspartate derivatives on excitatory amino acid transporters (EAAT4 and EAAT5). J. Neurochem. 79, 297–302 [DOI] [PubMed] [Google Scholar]
- 41. Shimamoto K., Lebrun B., Yasuda-Kamatani Y., Sakaitani M., Shigeri Y., Yumoto N., Nakajima T. (1998) DL-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol. Pharmacol. 53, 195–201 [DOI] [PubMed] [Google Scholar]
- 42. Ryan R. M., Mindell J. A. (2007) The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 14, 365–371 [DOI] [PubMed] [Google Scholar]
- 43. Schrodinger L. (2010) The PyMOL Molecular Graphics System, version 3.1, Schroedinger, LLC, New York [Google Scholar]