

Background: Elevated levels of both tumor suppressor PDCD4 and arginine methyltransferase PRMT5 correspond to aggressive tumor growth.
Results: Nutrient deprivation increases methylated PDCD4, enhancing its interaction with eIF4A and promoting cell viability.
Conclusion: Methylation of PDCD4 is responsive to the cellular metabolic state and promotes cell survival.
Significance: PRMT5 controls PDCD4 in a context-dependent fashion to enhance tumor cell viability.
Keywords: Molecular Cell Biology, Oncogene, Protein Methylation, Translation Initiation Factor, Tumor Cell Biology, Tumor Suppressor Gene, PDCD4, PRMT5, eIF4A, Nutrient Deprivation
Abstract
The role of programmed cell death 4 (PDCD4) in tumor biology is context-dependent. PDCD4 is described as a tumor suppressor, but its coexpression with protein arginine methyltransferase 5 (PRMT5) promotes accelerated tumor growth. Here, we report that PDCD4 is methylated during nutrient deprivation. Methylation occurs because of increased stability of PDCD4 protein as well as increased activity of PRMT5 toward PDCD4. During nutrient deprivation, levels of methylated PDCD4 promote cell viability, which is dependent on an enhanced interaction with eIF4A. Upon recovery from nutrient deprivation, levels of methylated PDCD4 are regulated by phosphorylation, which controls both the localization and stability of methylated PDCD4. This study reveals that, in response to particular environmental cues, the role of PDCD4 is up-regulated and is advantageous for cell viability. These findings suggest that the methylated form of PDCD4 promotes tumor viability during nutrient deprivation, ultimately allowing the tumor to grow more aggressively.
Introduction
Programmed cell death 4 (PDCD4) expression is often lost or decreased in tumors, and, in many instances, loss of PDCD4 expression is found to correlate with poor patient outcome (1–5). Consistent with this, PDCD4 suppresses transformation and displays characteristics of a tumor suppressor in tissue culture-based assays for invasion, migration, and anchorage-independent cell growth (6–11). Moreover, PDCD4 knockout mice develop B cell lymphoma, and epidermis-specific PDCD4 overexpression in mice prevents carcinogen-induced tumor development (12, 13).
Mechanistically, the function of PDCD4 as a tumor suppressor is attributed to its inhibition of the RNA helicase eIF4A, a component of the eIF4F complex involved in removing the secondary structure in 5′ UTRs for efficient translation initiation. PDCD4 contains two well defined MA3 domains, a motif characterized to bind eIF4A (14). Structural studies using the MA3 domains separately or in tandem have revealed that PDCD4 engages eIF4A with both MA3 domains (15–20).
PDCD4 is highly modified by phosphorylation, which dictates both protein localization and stability. Phosphorylation by several kinases, including S6K, Akt, and PKC δ and ϵ, at serine 67 targets PDCD4 for proteasomal degradation (21–23), and, because overactivation of the PI3K-mTOR5 signaling cascade is a common occurrence in tumors (24, 25), this mechanism is thought to be the cause of down-regulation of PDCD4 in many tumors. PDCD4 levels have also been reported to decrease during the mitotic phase of the cell cycle, concomitant with other changes in the status of translational machinery (26). Additionally, phosphorylation of PDCD4 at serine 457 by Akt drives nuclear localization (27). This is important because the function of PDCD4 as a tumor suppressor is attributed to the cytoplasmic role of eIF4A inhibition.
We reported previously that the ability of PDCD4 to act as a tumor suppressor is context-dependent. Specifically, when PDCD4 was coexpressed with protein arginine methyltransferase 5 (PRMT5) in an orthotopic xenograft model of breast cancer, we observed accelerated tumor growth (5). PRMT5 symmetrically dimethylates arginine residues, adding bulkiness to cause changes in the interactions, localization, and stability of target proteins (28). We found that PDCD4 is targeted by PRMT5 at arginine 110 and that accelerated tumor growth in the xenograft mouse model was dependent on both catalytically active PRMT5 and a methyl competent site at Arg-110 of PDCD4. Analysis of breast cancer patient data for both PDCD4 and PRMT5 reinforced the notion that this interaction has oncogenic potential (5). These data suggest that PDCD4 function should be considered in a context-dependent manner and prompted us to define conditions that drive PDCD4 methylation and the function of methylated PDCD4.
Here we find that PDCD4 is methylated and promotes cell viability during nutrient-limiting conditions. Mechanistically, methylation of PDCD4-Arg-110 enhances the interaction with eIF4A, a partnership required for effects on cell viability. Upon recovery from starvation, methylated PDCD4 is phosphorylated rapidly and shuttled to the nucleus. Its levels decrease quickly but with somewhat slower kinetics. These results shed light on how posttranslational modifications of PDCD4 integrate cues from the cellular environment and provide a framework for understanding the context-dependent nature of PDCD4 function.
EXPERIMENTAL PROCEDURES
Constructs
eIF4A1 was received from a human open reading frame library (29), and a stop codon was added. eIF4A2 was cloned from a human cDNA library into pDONR221. Both eIF4A1 and eIF4A2 were recombined, using Gateway technology (Invitrogen), into the pDEST53 and pGEX4T vectors. pGEX4T-PDCD4 was generated as described previously (5). PDCD4 mutations D253A, D418A, and R110K were generated by site-directed mutation of the pDONR221 vector and then moved into the pDEST53 vector and the pcDNA3.1-nV5 Gateway vector. Catalytically dead PRMT5 was generated by site-directed mutagenesis of G367A and R368A as described previously (5).
Cell Culture and Treatments
MCF7 cells were cultured in DMEM:F12 (Hyclone), 10% FBS, 1× ITS (Invitrogen), and human EGF(10 ng/ml, BIOSOURCE) and HEK293 cells in DMEM (Invitrogen) with 10% FBS. Stable MCF7 cells were generated as described previously using a MCF7 subline, designated MCF7e, that stably expresses the mouse ecotropic receptor allowing for infection with murine-specific retroviruses (5). Stable HEK293 lines were generated by Lipofectamine LTX (Invitrogen) transfection of pEXP53-PDCD4 (wild-type or mutants) (N-terminal GFP tag) or pcDNA3.1-V5-PDCD4 (wild-type or R110K) (N-terminal V5 tag) according to the recommendations of the manufacturer and selected with G418 (Sigma) at 1 mg/ml and maintained in 0.5 mg/ml. Single clones were selected on the basis of GFP or V5 expression. DMEM without glucose (Invitrogen), HBSS (Corning, catalog no. 21-023), insulin (Sigma), adenosine-2,3-dialdehyde in dimethyl sulfoxide (AdOx, Sigma), and MG132 in dimethyl sulfoxide (Calbiochem) were used to treat cells, as indicated. For cell cycle synchronization experiments, nocodazole (Sigma) was added to medium at 100 ng/ml for 18 h, or thymidine (Sigma) at a 2.5 mm concentration was included for 16 h, followed by a 12-h release and an additional 16-h treatment.
In Vitro Methylation Reaction
Cells were lysed by Dounce homogenization in methyl buffer (1× PBS, 0.25% TritonX-100, 1 mm DTT, and 5% glycerol; when used with cells or cell lysate, this was supplemented with 2× complete protease inhibitors (Roche)). 2 μg of GST-PDCD4 was incubated with 600 μg of HEK293 cell lysate, plus or minus 100 μm S-adenosylmethionine (New England Biolabs) in a 300-μl final volume of methyl buffer. For radiolabeled methylation reactions, 1 μg of GST-PDCD4 or GST-PDCD4R110K was incubated with 20 μg of cell lysate and 1 μCi of [3H]AdoMet (PerkinElmer Life Sciences) in a 35-μl final volume. Samples for immunoblot analyses were taken prior to addition of [3H]AdoMet. The remainder of the reaction was incubated at 30 °C for 1 h, and SDS loading buffer was added. Following SDS-PAGE and transfer of samples to PVDF, the membrane was sprayed with EN3HANCE (PerkinElmer Life Sciences) according to the recommendation of the manufacturer and exposed to film.
Recombinant Protein Production
pGEX4T-PDCD4, pGEX4T-eIF4A1, and pGEX4T-eIF4A2 were transduced into BL21/RIL cells grown to 0.6 A600 and induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 1–3 h. Cells were pelleted, lysed by sonication in PBS with 0.126% deoxycholate, 500 μm PMSF, 5 μg/ml aprotinin, and 5 μg/ml leupeptin. Glutathione resin (GE) was used to purify GST proteins following a standard protocol.
Cell Harvest and Immunoblot Analyses
Cells were washed twice with cold PBS, scraped off the plate with Nonidet P-40 buffer (50 mm HEPES (pH 7.6), 2 mm EDTA, 5% glycerol, 0.5% Nonidet P-40, and 150 mm NaCl), incubated 30 min at 4 °C while rotating end-over-end, and spun down at 12,000 × g for 10 min at 4 °C. The supernatant (lysate) was then transferred to a fresh tube. SDS loading buffer was added to lysates, heated at 95 °C for 5 min, and subjected to SDS-PAGE. For Phos-tag gels, lysates were generated with modified Nonidet P-40 buffer (lacking EDTA and with 1.84 mm β-glycerophosphate, 100 μm sodium vanadate, and protease inhibitor mixture III EDTA-free (Millipore)), and 83 μm MnCl2 and 42 μm Phos-tag (Wako Pure Chemical Industries) were added to 8% SDS-PAGE gels. Gels were transferred to PVDF. Blots were completed according to LI-COR recommendations. Briefly, they were blocked for 1 h with LI-COR blocking buffer, primary antibody diluted in LI-COR blocking buffer + 0.2% Tween 20 for 1 h at room temperature or overnight at 4 °C, washed four times with PBS plus 0.1% Tween 20 (PBS-T) or Tris-buffered saline with 0.1% Tween 20 (TBS-T), secondary antibodies diluted in LI-COR blocking buffer with 0.2% Tween 20 and 0.02% SDS at 1:4000 for 1 h at room temperature, washed four times with PBS-T or TBS-T, and scanned using LI-COR Odyssey. Quantifications were done using the Odyssey application software, comparing integrated intensity.
GST Pulldown
Recombinantly produced and purified GST protein (2–5 μg) was incubated with glutathione resin (GE) for 1 h at room temperature. Beads were washed and incubated with 250–400 μg of cell lysate (harvested as for the in vitro methylation reaction) for 1 h at room temperature in methyl buffer. Beads were washed three times with methyl buffer and resuspended in 1× SDS loading buffer.
GFP Pulldown
Lysate (250 μg in 200 μl of Nonidet P-40 buffer, lysed as described for immunoblot analyses), was added to 8 μl of GFP-Trap A beads (ChromoTek) and rotated for 1 h at 4 °C. Beads were washed three times with 250 μl of Nonidet P-40 buffer and resuspended in 1× SDS loading buffer.
Antibodies
The following antibodies were used: PDCD4 (Abcam, catalog no. 51495), PDCD4 (Cell Signaling Technology, catalog no. 9535), PRMT5 (Abcam, catalog no. 31751), methylated PDCD4 purified from hybridomas 1A8 and 3E7 and eIF4A1 (Cell Signaling Technology, catalog no. 2490), actin (Sigma, catalog no. A2228), GFP (Abcam, catalog no. 290), V5 (Invitrogen, catalog no. 46-0705), phospho-PDCD4-Ser-457 (Abcam, catalog no. 74141), S6 kinase (Cell Signaling Technology, catalog no. 9202), phospho-S6 kinase Thr-389 (Cell Signaling Technology, catalog no. 9205), ribosomal protein S6 (Cell Signaling Technology, catalog no. 2317), phospho-ribosomal protein S6 Ser-235/236 (Cell Signaling Technology, catalog no. 4857), mTOR (Cell Signaling Technology, catalog no. 2972), and phospho-mTOR Ser-2448 (Millipore, catalog no. 09-213SP).
Immunofluorescence
MCF7 cells were grown on autoclaved glass coverslips. Cells were fixed with methanol for 5 min at −20 °C, blocked for 30 min with 3% BSA in PBS, and incubated with primary antibodies overnight at 4 °C. Coverslips were washed in PBS. Alexa Fluor secondary antibodies were diluted in blocking buffer at 1:1000, incubated for 1 h at room temperature, washed as stated previously, and mounted with ProLong Gold + DAPI (Invitrogen) or the DNA was counterstained with Hoechst 33258 and mounted with ProLong Gold (Invitrogen). Images were acquired with Zeiss AxioVision using a ×63 objective. Counts were performed on two experiments, each of which included at least two coverslips and a total of more than 300 cells.
FACS Analysis
Nocodazole-treated cells were harvested by gentle pipetting. For all cell other treatments, PBS with 2.5 mm EDTA was used to dislodge cells from the plate. Cells were washed in PBS, vortexed, and spun several times. The final pellet was resuspended in 200 μl of PBS, fixed with 800 μl of 100% ethanol, and stored for ∼1 day at −20 °C. Cells were stained with 500 μl of staining solution (50 μg/ml propidium iodide, 0.1% Triton X-100, and 0.2 mg/ml RNase A in PBS), incubated for 20 min at 37 °C, and stored at 4 °C until analysis on the flow cytometer (FACScan, BD Biosciences). Cells were analyzed using FlowJo (Tree Star Inc.). Gates were selected so that doublets or debris was removed to ensure that single cells were analyzed, and propidium iodide staining was quantified. Cells displaying 2N (diploid chromosomal content) through 4N DNA content were analyzed and graphed in FACS distribution plots.
Viability Assay
Cells (625 total) were plated per well of a 96-well dish (Nunc, catalog no. 165306). The medium was exchanged with HBSS after cell attachment (24 h) and assayed using CellTiter Glo (Promega) following the suggested protocol of the manufacturer. Day 0 readings were done in regular growth medium, and day 6 readings were done in HBSS. Luminescence readings were done using a BioTek Synergy HT plate reader. Controls of medium/HBSS with reagent, medium, and cells without reagent were negligible. Graphs, standard deviation, and p values were generated using GraphPad Prism6.
RESULTS
Generation of a Methyl-specific PDCD4 Antibody
To understand the context in which PDCD4 is arginine-methylated and begin to elucidate how methylation influences its function, we generated monoclonal antibodies directed against methylated PDCD4. A symmetrically dimethylated peptide derived from PDCD4, encompassing the methylation site identified previously at arginine 110 (5), was used as the immunogen (Fig. 1A). To establish the specificity of individual hybridoma clones, we compared the reactivity with bacterially produced GST-PDCD4 before or after this recombinant protein was incubated with HEK293 cell lysate as a source of methyltransferases, either in the absence or presence of supplemental AdoMet, which serves as the methyl donor. Levels of recombinant PDCD4 were shown to be loaded equally for each condition on the immunoblot using a commercial antibody that recognizes PDCD4 independently of methylation (Fig. 1B). Two independent monoclonal antibodies showed little to no reactivity with GST-PDCD4 alone (Fig. 1B, lanes 1 and 4), as expected, because bacteria lack arginine methyltransferases (30). Indeed, only when GST-PDCD4 was preincubated with both lysate and AdoMet is reactivity robust with the monoclonal antibodies (Fig. 1B, lanes 3 and 6). This pattern confirms that these are methyl-specific reagents, to which we refer as msPDCD4 (methyl-specific PDCD4) antibody. To ascertain whether dual detection of methylated PDCD4 and total PDCD4 using antibodies generated in separate species can be performed without cross-interference using different fluorescently labeled secondary antibodies, we probed similar samples either independently or simultaneously. Comparable signals were obtained, verifying that dual detection of methylated and total PDCD4 on the same immunoblot is accurate (Fig. 1C).
FIGURE 1.
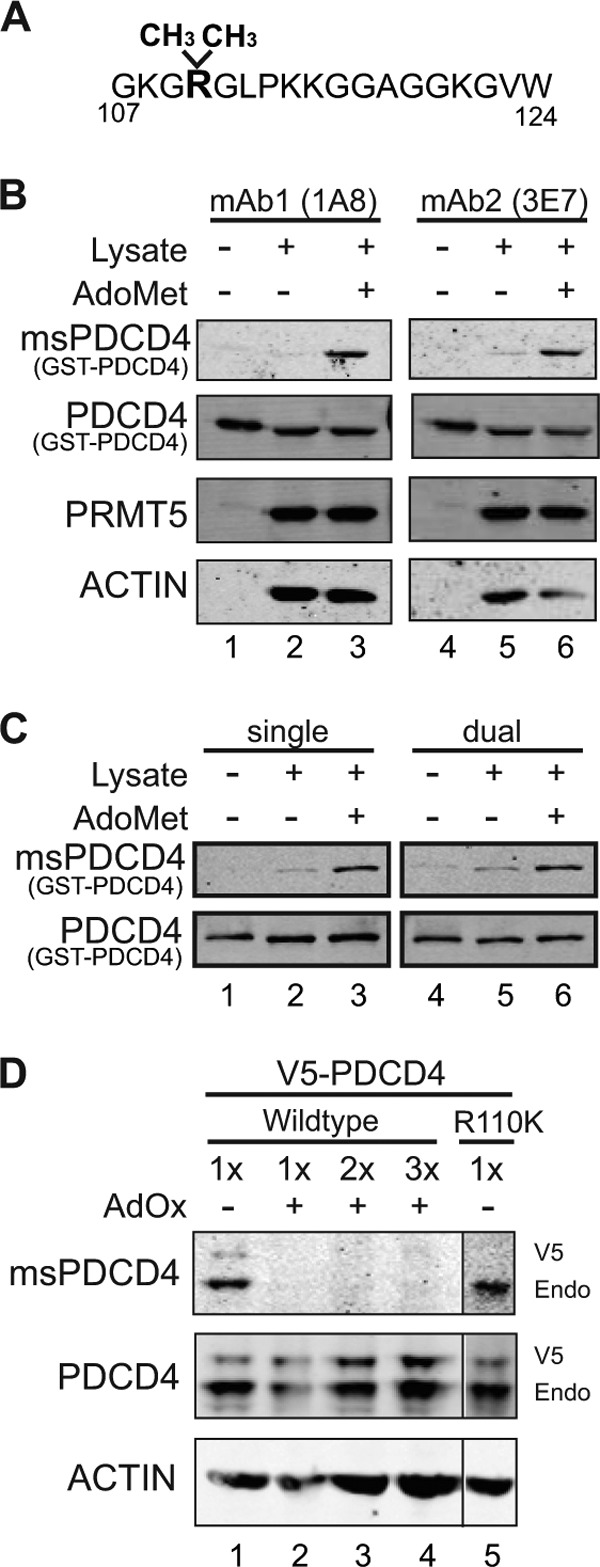
msPDCD4 antibody recognizes methylated PDCD4. A, sequence of the peptide used to develop methyl-PDCD4-specific antibodies. A cysteine residue was added to the C terminus for conjugation purposes. B, immunoblot analyses of GST-PDCD4 (lanes 1 and 4), in vitro methylation reactions where GST-PDCD4 was incubated with lysate (lane 2 and 5), or lysate and AdoMet (lanes 3 and 6) probed with antibodies as indicated. Either hybridoma line 1A8 or 3E7 was used for msPDCD4 detection. C, reactions prepared as described in B were probed either separately for total PDCD4 and methylated PDCD4 levels (single detection, left column) or simultaneously with both antibodies (dual detection, right column). In all blots, each primary antibody is tracked separately using species-specific secondary antibodies conjugated to different fluorophores. D, immunoblot analyses of HEK293 cells stably expressing V5-PDCD4 either untreated (lane 1) or treated with AdOx (20 μm for 20 h) (lanes 2–4; lane 2 loaded equal to lane 1, lane 3 twice the amount of protein as lane 1, and lane 4 three times the amount of protein as lane 1) or HEK293 cells stably expressing V5-PDCD4R110K (lane 5; loaded equal to lane 1 but normalized for endogenous (Endo) PDCD4 levels) and probed with antibodies as indicated.
To further confirm that this antibody is specific to methylated PDCD4, HEK293 cells expressing V5-tagged PDCD4 were treated with AdOx, which depletes cellular levels of AdoMet, generating a hypomethylated state. Cell lysate from this sample was then subjected to immunoblot analysis. In contrast to an antibody directed generally against PDCD4 that reacts with PDCD4 in all samples, the msPDCD4 antibody recognized V5-PDCD4 and endogenous PDCD4 only in untreated lysates. Reactivity was undetectable with protein from AdOx-treated lysate, even when loaded at increasing levels (Fig. 1D). Using lysate from HEK293 cells expressing V5-tagged PDCD4R110K, a mutation that retains charge but blocks arginine methylation (5), msPDCD4 antibody recognizes only the endogenous form of PDCD4 (Fig. 1D, lane 5). This data validates that msPDCD4 antibody reacts specifically with methylated PDCD4.
Nutrient Deprivation Enhances Methylation of PDCD4 to Promote Cell Viability
Although the above results confirm the specificity of this new reagent and indicate that the methylated form of PDCD4 is, in fact, present in cells, we were initially surprised by results when we used immunofluorescence to detect methylated PDCD4 in a panel of cell lines developed previously (5). This panel of MCF7-derived cell lines has stably integrated constructs: either expression cassettes for PRMT5 and PDCD4, PRMT5 and PDCD4R110K, or empty vectors. We found previously that, when both PDCD4 and PRMT5 are expressed in wild-type form, these cells grow more aggressively as orthotopic xenografts (5). Yet, probing this panel by immunofluorescence using msPDCD4 antibody revealed only sporadic reactivity, although it was selective for the PDCD4-PRMT5 line (data not shown). In the course of these experiments, however, it was noted that reactivity appeared stronger when cells were growing more densely, so we pursued a systematic investigation to test whether confluency alters the levels of methylated PDCD4. Immunoblot analysis clearly revealed that both methylated PDCD4 and total PDCD4 levels increase as cells become more confluent, whereas levels of PRMT5 protein remain unchanged (Fig. 2A). The increase in methylated PDCD4 is most robust in cells coexpressing elevated levels of both PDCD4 and PRMT5, suggesting that both methyl-competent PDCD4 and elevated PRMT5 levels are necessary for enhanced methylation. Cells coexpressing PDCD4R110K with PRMT5 displayed increased levels of total PDCD4 but not methylated PDCD4, indicating that methylation at Arg-110 does not directly influence PDCD4 levels. These data reveal that, under confluent conditions in tissue culture, PDCD4 levels are increased and PDCD4 methylation is increased.
FIGURE 2.

Methylated PDCD4 is increased upon nutrient deprivation and enhances cell viability during starvation. A, immunoblot analysis of MCF7 cells stably expressing PDCD4 and PRMT5 (lanes 4–6), PDCD4R110K and PRMT5 (lanes 7–9), or vector control (lanes 1–3) grown at increasing confluency and probed with antibodies as indicated. B, immunoblot analysis of MCF7 (PDCD4-PRMT5) stable cells grown at >100% confluency where the medium was not changed (lane 1) or exchanged for 3 h with fresh medium (lane 2), DMEM:F12 (lanes 3–5) supplemented with ITS (lane 4) or insulin (lane 5), or DMEM lacking glucose (lanes 6–8) and supplemented with ITS (lane 7) or insulin (lane 8). C, immunoblot analysis of MCF7 (PDCD4-PRMT5) cells grown under subconfluent conditions and then switched to DMEM:F12 (without growth factors) for the indicated times. D, immunoblot analysis of MCF7 (PDCD4-PRMT5) cells grown at subconfluent conditions in HBSS for the indicated times. E, immunoblot of MCF7 (PDCD4-PRMT5) cells grown under normal conditions (lane 1), in DMEM:F12 for 24 h (lane 2), or in HBSS for 24 h (lane 3). F, FACS analysis of DNA content, as detected by propidium iodide (PI) staining, of MCF7 (PDCD4-PRMT5) cells under normal growth conditions (−), after 24 h of growth in HBSS, after double-thymidine treatment (Thy), and after nocodazole treatment (Noc). G, immunoblot analysis of cells treated as described in F. H, MCF7 cells stably expressing vector controls, PDCD4 with PRMT5, or PDCD4R110K with PRMT5 were plated in 96-well dishes with regular medium and allowed to adhere overnight. The medium was then replaced with HBSS, and cell viability was measured with the Cell Titer Glo assay. Values are normalized to day 0 and reported as percentages. *, p = 0.0003; **, p = 0.0008.
Reasoning that nutrients and growth factors could be limiting in the medium of confluent cells, we replaced the medium in a confluent dish of MCF7(PDCD4-PRMT5) cells with fresh growth medium (DMEM:F12, 10% FBS, 1× ITS, and 10 ng/ml human EGF) for 3 h. Addition of this medium was sufficient to decrease both total and methylated PDCD4 levels (Fig. 2B, lanes 1 and 2), suggesting that, in confluent cells, metabolic limitations drive increases in PDCD4 levels and methylation. We then tested which components of the medium trigger the decrease in PDCD4 methylation and levels. DMEM:F12 supplemented with ITS or with insulin alone was sufficient to decrease methylated PDCD4 and total PDCD4 (Fig. 2B, lanes 3–5). Further, when medium lacking glucose but supplemented with ITS or insulin was transferred to confluent cells, methylated PDCD4/PDCD4 levels did not decrease to the same extent as when DMEM:F12, which contains glucose, was used. These results are consistent with both insulin and glucose being required to decrease methylated and total PDCD4 (Fig. 2B, lanes 3–8).
We next wanted to determine whether nutrient limitation is sufficient to drive the increased levels of methylated and total PDCD4 observed during confluency or whether other factors, such as increased cell-cell contacts, are required. PDCD4 levels are known to stabilize upon serum deprivation and decline upon serum stimulation (21), and, therefore, subconfluent MCF7 cells coexpressing PDCD4 and PRMT5 were serum-deprived over a 40-h time course and harvested every 8 h. Immunoblot analyses of lysates from these cells indicated that levels of total PDCD4 increased over time with serum deprivation, yet robust induction of methylated PDCD4 was not observed (Fig. 2C). We next tested a more stringent nutrient and growth factor deprivation using HBSS, which lacks amino acids and growth factors but contains essential salts and a low concentration of glucose (1g/liter). Immunoblot analysis of this experiment showed that methylated PDCD4 is elevated after 8 h, peaks at 24 h, and remains high throughout the remainder of the time course. PDCD4 levels follow a similar trend, whereas levels of PRMT5 do not change (Fig. 2D). To monitor signaling pathways that are modulated under these conditions, we tracked the mTOR pathway (24, 31). HBSS treatment led to a more pronounced modulation of mTOR signaling, as indicated by the lower levels of p-mTOR-Ser-2448 and pp70 S6 kinase-Ser-235/236 (Fig. 2E). These results indicate that PDCD4 levels and methylation are increased concomitantly with a threshold in repression of mTOR signaling and independently of changes specific to high confluency, such as increased cell-cell contacts. Because nutrient deprivation leads to changes in cell cycle distribution and levels of PDCD4 have been shown to decrease during mitosis (26), we next addressed whether cell cycle-driven changes in PDCD4 levels and methylation could account for the pattern observed during nutrient deprivation. To this end, we subjected MCF7 (PDCD4-PRMT5) cells either to a double thymidine treatment to arrest them in G1 or to nocodazole to block cells in mitosis (Fig. 2F) and compared these conditions directly to 24-h incubation in HBSS. Although HBSS treatment results in a significant drop in cells that are in S phase (27% in control versus 9% in HBSS) and a corresponding increase in cells with 2N DNA content (G1), this similarity to thymidine-arrested cells does not account for the observed increases in total and methylated PDCD4 (Fig. 2G). Following nocodazole treatment, we observed a robust enrichment in mitosis (78% versus 28% of the cells had 4N DNA content, Fig. 2F). However, this did not result in a change of PDCD4 levels or methylation status (Fig. 2G). This result differs from a recent report where mitotically arrested cells were observed to have lower levels of PDCD4 (26). The difference in results could be due to the different cell lines used and will be of interest to better understand. Here, however, we can conclude that a shift in cell cycle distribution does not account for changes in PDCD4 levels and methylation observed in MCF7 cells. Rather, signals unique to nutrient deprivation drive the activation of this pathway.
Tumors often have nutrient-deprived, poorly vascularized regions (32). Together with our previous observation of accelerated tumor growth when xenograft cells coexpress PDCD4 and PRMT5 (5), this suggests that methylation of PDCD4 may influence viability under nutrient deprivation. To address this possibility, a panel of MCF7 cells was cultured for a longer term (6 days) in HBSS and assessed for viability using the Cell Titer Glo assay. This revealed a statistically significant enhancement in the viability of cells with elevated PDCD4 and PRMT5 compared with cells expressing PDCD4R110K with PRMT5 (Fig. 2H). Vector control cells displayed a range of viability phenotypes, which may reflect the ability to turn on the PDCD4-PRMT5 pathway with lower efficiency. This panel of cell lines displays no differences in cell proliferation or viability under normal cell culture conditions (Ref. 5 and data not shown).
Nutrient Deprivation Changes Methylated PDCD4 Localization
Because the role of PDCD4 can be influenced by its subcellular distribution, which varies depending on the cell line and conditions of culture (27, 33, 34), we next wished to determine the localization of methylated PDCD4. Using the MCF7 cells expressing elevated PDCD4 and PRMT5, we tracked methylated PDCD4 levels and localization under conditions of normal cell culture, high confluency, and starvation. We observed an increase in the percentage of cells positive for methylated PDCD4 under both starvation and high confluency and a shift in subcellular localization from the nucleus to cytoplasm (Fig. 3, A–C). The shift to the cytoplasm is more dramatic under HBSS treatment, possibly because of the severity of the nutrient deprivation. We did not observe changes in PRMT5 localization or levels. We also observed that levels and localization of total PDCD4 increase and shift to the cytoplasm upon HBSS treatment (Fig. 3D).
FIGURE 3.

Localization of methylated PDCD4 changes upon nutrient deprivation. A, immunofluorescence of MCF7 (PDCD4-PRMT5) cells cultured under normal growth conditions (top row), confluent conditions (center row) or in HBSS for 24 h (bottom row), probing with antibodies against methylated PDCD4 (left column), PRMT5 (center column), and merged with DAPI to detect DNA (right column). B, counts of cells positive for methylated PDCD4 displayed as percentage of total cells with standard deviation. C, localization of methylated PDCD4 counted by localization of the predominant msPDCD4 signal, nuclear/cytoplasmic (Nuc/Cyto) cells had equal amounts of nuclear and cytoplasmic msPDCD4. Data are displayed as a percentage of total msPDCD4-positive cells with standard deviation. D and E, immunofluorescence of MCF7 (PDCD4-PRMT5) cells cultured in HBSS for 24 h and probed with antibodies against methylated PDCD4 (left panels), PDCD4 (D, center panel), eIF4A1 (E, center panel), or merged with DNA detected by Hoechst 33258 (right panels). Scale bars = 10 μm.
Methylated PDCD4 Enhances the Interaction with eIF4A and Requires This Interaction to Confer Enhanced Viability
The effects of PDCD4 when in the cytoplasm are attributed to its ability to bind eIF4A, an RNA helicase involved in the unwinding of the secondary structure to facilitate cap-dependent translation initiation (33). Probing for eIF4A1, we found it localized to the cytoplasm, as expected, in HBSS-treated samples (Fig. 3E). Given the cytoplasmic localization of methylated PDCD4 upon starvation, we next assessed whether methylation of PDCD4 modulates its interaction with eIF4A. GST-tagged eIF4A1 or eIF4A2 was used to retrieve binding partners from HEK293 lysate, and immunoblot analyses of these pulldowns were then probed with PDCD4 and msPDCD4 antibodies. Methylated PDCD4 is more robustly enriched than total PDCD4. When the same amount of input was compared for each antibody, methylated PDCD4 displayed an increased recovery with GST-eIF4A1 or GST-eIF4A2 versus total PDCD4 (Fig. 4A), indicating that methylation of PDCD4 promotes its interaction with eIF4A1 and eIF4A2.
FIGURE 4.

Methylated PDCD4 interacts more robustly with eIF4A and requires this interaction for enhanced viability during starvation. A, immunoblot analysis probed for PDCD4 and methylated PDCD4 (merged in first panel, single detection in second and third panels), showing recovery with recombinant GST-eIF4A1 (lane 1), GST-eIF4A2 (lane 2), or GST (lane 3) from HEK293 cell lysates. A titration of input material is loaded in lanes 4–6 (27, 17, and 7%, respectively). Coomassie stain confirms equal GST protein recovery. B, GFP pulldowns or input samples using HEK293 cells transiently transfected with GFP-eIF4A1 (lanes 1, 3, 5, and 7) or GFP (lanes 2, 4, 6, and 8). Cells were either switched into HBSS for 24 h (lanes 3, 4, 7, and 8) or fresh medium 2 h prior to harvesting for cell lysate (lanes 1, 2, 5, and 6). An immunoblot analysis was probed for methylated PDCD4 and total PDCD4 (merged, top panel; individual, bottom panel). The percentage input in the top panel was 20%. The immunoblot analyses shown in the bottom panel were loaded at equal input and pulldown and probed with eIF4A1 or GFP, as indicated, to confirm recovery as well as relative levels of GFP-eIF4A1 and endogenous (Endo) eIF4A1. The asterisk indicates a nonspecific band. C, quantification of experiments performed as in A to track methylated and total PDCD4 recovery with GST-eIF4A1 from MCF7 (PDCD4-PRMT5) cell lysate. ***, p = 0.0009. D, GST-eIF4A1 pulldowns from MCF7 cells expressing empty vector, PDCD4-PRMT5, or PDCD4D253A/D418A-PRMT5 grown under normal culture conditions (lanes 1–3 and 7–9) or in HBSS for 24 h (lanes 4–6 and 10–12). The equivalent of 15% input was loaded (lanes 7–12). E, immunoprecipitation of V5-tagged PDCD4 from lysates of MCF7 cells coexpressing PDCD4-PRMT5 (lane 1), PDCD4R110K-PRMT5 (lane 2), or PDCD4D253A/D418A-PRMT5 (lane 3) cultured in HBSS for 24 h. Immunoprecipitated (IP) material was probed for methylated PDCD4 (top) and total PDCD4 (bottom). The asterisk indicates the IgG heavy chain. F, MCF7 (PDCD4-PRMT5) and MCF7 (PDCD4D253A/D418A-PRMT5) cells cultured in HBSS for 6 days and measured for viability by Cell Titer Glo assay. Values were normalized to day 0 and expressed as percentages. **, p = 0.0002.
To test whether methylated PDCD4 is the preferential binding partner in a more physiological context, HEK293 cells were transiently transfected with GFP-eIF4A1 and then cultured in HBSS to maximize PDCD4 methylation. Material was recovered under these conditions or from cells that were cultured further in complete medium for 2 h to reduce methylated PDCD4 prior to lysis. GFP proteins, along with associated proteins, were then captured on an affinity matrix and subjected to immunoblot analysis. Again, relative to levels detected in the input, methylated PDCD4 was recovered with much greater efficiency than total PDCD4 (Fig. 4B). To quantify this effect, lysates from MCF7 (PDCD4-PRMT5) cells cultured in HBSS were used in a pulldown assay with GST-eIF4A1, and recovery was measured in four repeats of this experiment (Fig. 4C). We found consistently that methylated PDCD4 was recovered ∼3-fold more efficiently. Note that this is an underestimate of the difference between methylated and unmethylated PDCD4 because we detected both populations when tracking total levels. Thus, these data support the conclusion that methylation of PDCD4 promotes a more robust interaction with eIF4A1. PDCD4 contains two MA3 domains (amino acids 164–275 and 327–440) well characterized to interact with eIF4A (14–18, 35). Methylation occurs at Arg-110 in the N-terminal domain (5), which is thought to be an unstructured domain and is dispensable for binding to eIF4A. Because methylation of PDCD4 modulates its interaction with eIF4A (Fig. 4, A–C), we wanted to address whether methylation enhances the MA3 domain interaction with eIF4A or generates a new PDCD4-eIF4A interface independent of the MA3 domains. Point mutations D253A/D418A in the MA3 domains were generated in the context of V5-tagged PDCD4 and confirmed to eliminate binding to eIF4A under normal culture conditions, as reported previously (14) (Fig. 4D, lanes 1–3). Under starvation conditions, when PDCD4 methylation is increased, the PDCD4D253A/D418A mutant is still not recovered with GST-eIF4A1 (Fig. 4D, lane 6, V5 (PDCD4)). Mutation of the MA3 domains (PDCD4D253A/D418A) does not alter the ability to be methylated because an increase in methylation is observed upon expression of the PDCD4D253A/D418A mutant compared with vector control cells (Fig. 4D, compare lanes 10–12) and because immunoprecipitation of V5-tagged PDCD4 out of HBSS-treated lysates shows that both PDCD4 and PDCD4D253A/D418A are methylated to the same extent (Fig. 4E, compare lanes 1 and 3). This demonstrates that arginine methylation of PDCD4 works in concert with the MA3 domains to promote the interaction with eIF4A.
To test whether the heightened interaction between methylated PDCD4 and eIF4A contributes to enhanced viability conferred by PDCD4-PRMT5 expression during nutrient deprivation, MCF7 cells expressing PDCD4D253A/D418A with PRMT5 or PDCD4 with PRMT5 were cultured in HBSS and tracked for viability. Cells coexpressing PDCD4D253A/D418A and PRMT5 displayed decreased viability compared with cells with elevated PDCD4-PRMT5, indicating a requirement for the MA3-mediated interaction with eIF4A (Fig. 4F). No differences in growth under normal culture conditions were observed (data not shown). Together, these data suggest that PDCD4 requires the robust interaction with eIF4A mediated through both methylation at Arg-110 and the MA3 domains to enhance viability during starvation. Although the result that a feature of PDCD4 associated with its tumor suppressive activity is both elevated and required under conditions where PDCD4 confers enhanced viability appears paradoxical, these observations highlight the importance of cellular context in terms of PDCD4 function (discussed further below).
Upon Recovery from Starvation, Methylated PDCD4 Is Eliminated Quickly
In the tumor context, signals that stimulate neovasculature can restore nutrient levels, and tumor growth depends on responding dynamically to this changing situation (36). Given that nutrient deprivation increases PDCD4 methylation to modulate its interaction with eIF4A, it is equally important to understand how this pathway is reversed. To assess recovery from nutrient deprivation, MCF7 cells expressing elevated PDCD4 and PRMT5 were treated with HBSS for 24 h, placed in complete growth medium, and observed over a 2-h time course. Immunoblot analysis of these samples revealed that both methylated and total levels of PDCD4 are down-regulated quickly and return to steady-state levels of non-detectable methylated PDCD4 and low levels of total PDCD4 after 120 min (Fig. 5A). One signaling pathway that is activated upon the switch to optimal growth conditions is the Akt pathway, a kinase that has been found previously to regulate PDCD4. To determine whether the same signals converge to regulate methylated PDCD4, we first tracked phosphorylation of serine 457, known to be targeted by Akt (27). As expected, PDCD4-Ser-457 levels increase following a shift from HBSS to complete medium. Following a peak of abundance at 15–30 min, this phosphorylated form decreases in levels (Fig. 5B). To specifically probe whether methylated PDCD4 is itself phosphorylated, samples were subjected to SDS-gel electrophoresis using a gel supplemented with Phos-tag reagent, which enhances the separation of phosphorylated proteins by slowing their migration (37). This immunoblot analysis allowed us to track the phospho-isoforms of PDCD4, which rapidly accumulated by 15 min (Fig. 5C). This analysis also showed that methylated PDCD4 was part of this population, phosphorylated in a quantitative fashion with the same kinetics. To put the dynamics of PDCD4 regulation into context with mTOR signaling, we tracked key markers of this network during recovery from HBSS treatment. Under the conditions used here, downstream effectors of this pathway are active by 15 min of recovery because phosphorylated forms of both p70-S6 kinase and the downstream target, ribosomal protein S6, are detected robustly (Fig. 5D). The kinetics of S6 kinase activation are consistent with its role in driving proteasome-mediated turnover of PDCD4 (Ref. 21 and Fig. 6).
FIGURE 5.

Methylated PDCD4 is subject to phosphorylation and is dynamic in localization. MCF7 (PDCD4-PRMT5) cells cultured in HBSS for 24 h and then switched into complete medium for the indicated time. A, levels of methylated PDCD4 (msPDCD4) and PDCD4 were compared by immunoblot analysis (merged, top panel). B, levels of methylated PDCD4 (msPDCD4) and PDCD4 phosphorylated at S457 were tracked simultaneously by immunoblot (merged, top panel). C, methylated PDCD4 and total PDCD4 were tracked following separation on a gel containing Phos-tag reagent to enhance the resolution of phosphorylated proteins. D, total and phosphorylated (pT389) S6 Kinase (open arrowheads, p70; closed arrowheads, p85), along with a downstream target of this pathway, ribosomal protein S6, were tracked by immunoblot analysis. E, immunofluorescence detection of methylated PDCD4 (top row), PDCD4 (center row), and merged with DNA stained by Hoechst 33258 (bottom row). Scale bar = 10 μm.
FIGURE 6.
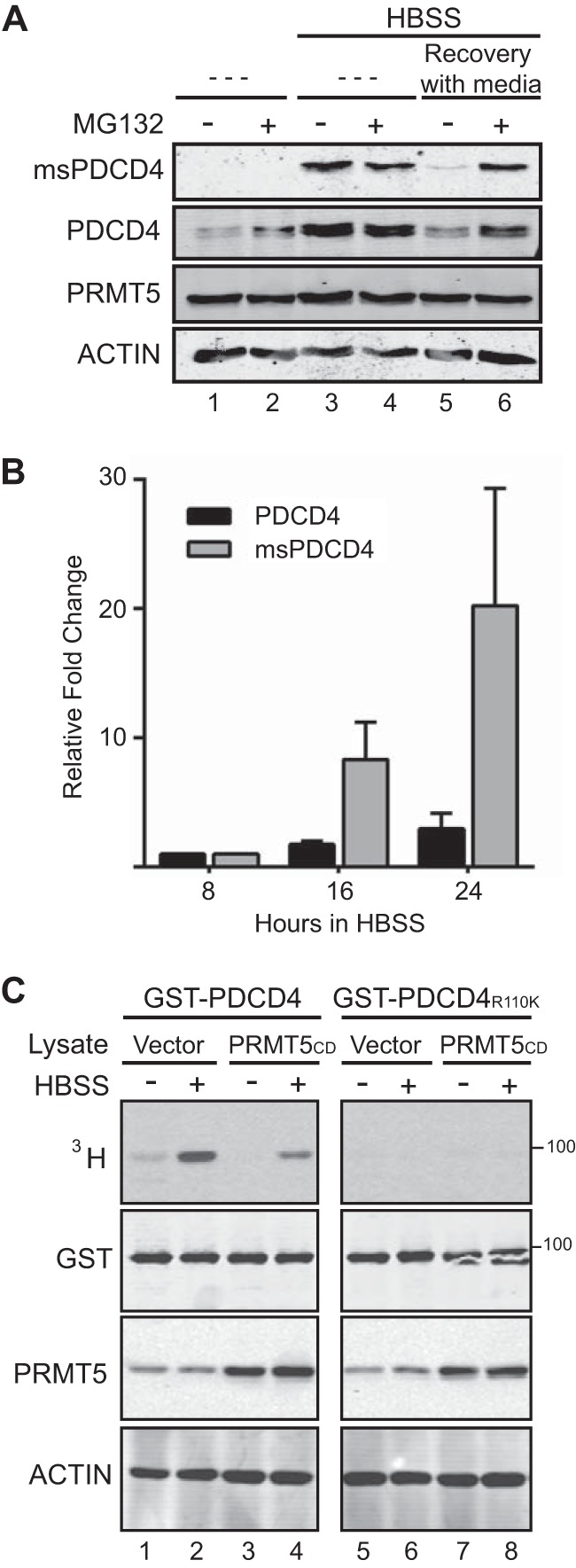
During nutrient deprivation, PDCD4 stabilization and increased PRMT5 activity converge to increase methylated PDCD4 levels. A, MCF7 (PDCD4-PRMT5) cells were treated with (lanes 2, 4, and 6) or without (lanes 1, 3, and 5) MG132 (10 μm, 1 h) under normal growth conditions (lanes 1 and 2) following culture in HBSS for 24 h (lanes 3 and 4) or following culture in HBSS for 24 h and then a period of recovery with media for 1 h (MG132 was added during recovery) (lanes 5 and 6). Immunoblot analyses of these cell lysates were probed with antibodies as indicated. B, quantification of methylated and total PDCD4 from three experiments (performed as in Fig. 2D), graphed as values relative to the 8-h time point. C, autoradiograph detecting tritiated proteins from in vitro methylation reactions where lysates from MCF7 expressing catalytically dead PRMT5 (mutation G367A/R368A) (lanes 3, 4, 7, and 8) or empty vector (lanes 1, 2, 5, and 6) were incubated with recombinant GST-PDCD4 (lanes 1–4) or GST-PDCD4R110K (lanes 5–8) and [3H]AdoMet. Immunoblot analyses were probed with antibodies as indicated.
Because PDCD4 subcellular localization is regulated by Akt phosphorylation at Ser-457 (27), we next tracked localization by immunofluorescence. Both total and methylated PDCD4 are shifted swiftly to the nucleus during recovery in complete medium, becoming almost completely nuclear by 30 min and returning to non-starved levels and localization after 120 min of recovery (Fig. 5E). These data suggest that methylated PDCD4 is subject to the same regulation shown previously for PDCD4 (21, 27). The quick shift in methylated PDCD4 levels and localization upon recovery from starvation suggests that methylated PDCD4 is highly regulated and that its function in the cytoplasm is reversed rapidly when cells recover from metabolically limiting conditions in synchrony with the global response to nutrient restoration. Notably, however, both phosphorylation at Ser-457 (Fig. 5B) and nuclear localization (Fig. 5E) are observable as soon as 5 min after nutrient addition, indicating that PDCD4 is highly sensitive to nutrient status and is engaged very rapidly in the cellular response to nutrient availability.
Enhanced PDCD4 Protein Stability and Increased PRMT5 Activity Converge to Increase Methylated PDCD4 during Nutrient Deprivation
Several kinases, including S6K, Akt, and the PKC δ and ϵ isoforms, have been shown to phosphorylate PDCD4 S67, targeting it for polyubiquitination and proteasomal degradation (21–23). To test whether methylated PDCD4 is subject to proteasomal degradation, MCF7 cells coexpressing PDCD4 and PRMT5 were treated with and without MG132 to block proteasomal degradation under normal growth, during HBSS treatment, and during recovery. During recovery from nutrient deprivation, treatment with MG132 was sufficient to sustain the levels of methylated and total PDCD4 (Fig. 6A, lanes 5 and 6), suggesting that proteasomal degradation is responsible for the decreases in methylated and total PDCD4. Treatment of nutrient-deprived cells with MG132 showed no change in the levels of total or methylated PDCD4 (Fig. 6A, lanes 3 and 4), suggesting that PDCD4 is not subject to proteasomal turnover during nutrient-limiting conditions. Lastly, treatment of cells under normal growth conditions showed stabilization of PDCD4 protein levels but not methylated PDCD4 (Fig. 6A, lanes 1 and 2).
To address whether the increase in methylated PDCD4 is due solely to increases in total PDCD4, we quantified the signal detected by immunoblot analysis of cells cultured in HBSS in three independent experiments (as in Fig. 2D). We found that the relative increase in total PDCD4 does not account for the appearance of methylated PDCD4 because the methylated PDCD4 signal increases at a faster rate (Fig. 6B). This suggests that the increase in methylated PDCD4 signal cannot be explained solely by increases in total PDCD4 and that additional factors are affecting methylated PDCD4 levels.
To test whether PRMT5 activity during starvation contributes to increased PDCD4 methylation independently of PDCD4 levels, in vitro methylation reactions were performed with [3H]AdoMet to track differences in methylation activity toward PDCD4. Lysates from MCF7 cells, cultured in HBSS for 24 h or left untreated, were incubated with GST-PDCD4 or GST-PDCD4R110K and [3H]AdoMet. We observed an increase in methylation of GST-PDCD4 in the HBSS-treated lysates compared with non-treated counterparts (Fig. 6C, lane 2 versus lane 1). Methylation was dependent on the presence of Arg-110 (Fig. 6C, lanes 5 and 6). In addition, when MCF7 cells expressing PRMT5CD were used (in which exogenous PRMT5 is catalytically dead (5)), levels of GST-PDCD4 methylation were reduced (Fig. 6C, lane 4), confirming the dependence on PRMT5 itself. These data indicate that PRMT5 has increased activity toward PDCD4 during starvation.
DISCUSSION
Many modes of regulation, including transcriptional, posttranscriptional, and posttranslational (for example, Refs. 2, 21, 23, 27, 38–43), impinge on the abundance and activity of PDCD4, underscoring a pivotal role for PDCD4. Although one important facet of PDCD4 function is its tumor-suppressive activity, previous work from our group revealed that the role of PDCD4 in cancer biology is context-dependent. When PDCD4 is coexpressed with PRMT5, it promotes tumor growth. Underscoring the context-dependent nature of PDCD4 and highlighting an additional mode of regulation, here we report that nutrient deprivation triggers methylation of PDCD4 that enhances the ability of PDCD4 to interact with eIF4A and promote cell viability. This leads to the model that promoting PDCD4 methylation at a certain time and place, with respect to tumorigenesis, confers an advantage on tumor growth.
PDCD4 has been connected previously to nutrient levels because PDCD4 protein is stabilized (21, 44) and changes subcellular localization (27) following growth factor removal. More recently, PDCD4 has been shown to inhibit autophagy by controlling the levels of ATG5, a component involved in autophagosome formation (45). We also observed that PDCD4 levels are induced upon serum deprivation and, to a larger extent, with HBSS treatment, yet we did not observe changes in ATG5 levels or LC3 puncta when comparing a vector control and PDCD4-PRMT5-expressing cells (data not shown). This could be due to differences in cell lines or PDCD4 expression levels.
Increased methylation of PDCD4 during nutrient deprivation is due, at least in part, to increases in PRMT5 activity toward PDCD4. Interestingly, PRMT5 activity toward histone H3 Arg-2 (H3R2) has been found recently to increase in liver cells during fasting, and this activity is required to promote transcription of genes required for gluconeogenesis (46). Together with our data, this raises the question of whether PRMT5 may play a key role in adapting to changing nutrient conditions via both a global transcriptional effect involving H3R2 methylation (46) and a translational effect mediated by methylation of PDCD4.
We found further that methylated PDCD4 is turned over by proteasomal degradation, occurring promptly during recovery from nutrient stress. This suggests that PDCD4 methyl marks are removed en masse by proteasomal degradation and not subject to demethylation in the circumstances we examined. This turnover allows the role of methylated PDCD4 to occur specifically during metabolic stress.
We describe that PDCD4-R110 methylation stabilizes the interaction with eIF4A. Previous studies defined that the MA3 domains mediate the interaction with eIF4A and suggest that the less structured N-terminal domain is dispensable for binding to eIF4A (14), and this has been found to be the case in structural studies (15–20). Our data suggest that the N-terminal domain, and, more specifically, arginine methylation of Arg-110, either reinforces the interaction between the MA3 domains and eIF4A to form a more robust interaction or makes the PDCD4 MA3 domains more accessible for interaction with eIF4A. Understanding this regulatory mechanism will be an interesting avenue to pursue.
The interaction between PDCD4 and eIF4A is also likely to be influenced by subcellular compartmentalization. Under nutrient deprivation, PDCD4 has been observed to shift to the cytoplasm (34), and we found this pattern to be true for methylated PDCD4 as well. Upon recovery, PDCD4, including the methylated population, is quickly localized to the nucleus prior to an overall decrease in levels. In a simple model, nuclear localization before and after nutrient deprivation sequesters PDCD4 away from eIF4A. Given previously characterized nuclear roles for PDCD4 in transcriptional regulation and DNA damage response (10, 47–49), however, it will be of interest to probe whether methylation of PDCD4 influences these roles. Nuclear compartmentalization of PDCD4 during recovery from nutrient deprivation may serve not just to terminate a cytoplasmic role but to promote specific roles in the nucleus as well.
Cells expressing elevated levels of PDCD4 and PRMT5 show enhanced viability when subjected to long-term culture in HBSS, and this phenotype requires that PDCD4 is both methyl-competent and able to bind eIF4A via the two MA3 domains. Solid tumors are characterized by lack of or incorrectly formed blood vessels, leaving regions hypoxic and nutrient-deprived (32). It is within these regions that we expect methylated PDCD4 to play a role, allowing the tumor to continue to expand by promoting viability and providing time for blood vessel recruitment. A role for methylated PDCD4 in nutrient-deprived regions could explain the results from our previous xenograft experiment where PDCD4-PRMT5 tumors grew larger (5). Similarly, a kinase that controls translation elongation, eEF2K, plays a role in regulating translation during nutrient deprivation and is thought to promote tumorigenesis by suppressing translation during nutrient deprivation (50). For both methylated PDCD4 and eEF2K, it is unclear whether enhanced viability is due to changes in global translation or whether specific mRNA transcripts are targeted.
Metabolic stress occurs later in tumorigenesis when the tumor has formed nutrient-deprived regions (51). Because methylation of PDCD4 occurs during metabolic stress, we speculate that methylated PDCD4 has a role later in tumor development. This could explain the paradoxical role of PDCD4 in tumorigenesis. PDCD4 could function as a tumor suppressor early during tumorigenesis, at a time when inhibition of translation prevents initiating events and early growth, whereas methylation of PDCD4 could occur during metabolic stress to acutely inhibit, or otherwise modulate, translation at a time when this promotes viability, ultimately allowing tumor expansion (51).
Acknowledgments
We thank past and present members of the Ullman and Rosenblatt laboratories for discussions. We also thank Andy Weyrich, Julie Hollien, Alana Welm, Don Ayer, and Markus Babst for discussions and feedback on the manuscript; Mohan Kaadige and Don Ayer for reagents; and Atakan Ekiz for help with FACS analysis.
This work was supported, in whole or in part, by NCI, National Institutes of Health Grant R21 CA173064 and by shared resources supported in part by National Institutes of Health Grant P30 CA042014 through the Huntsman Cancer Institute. This work was also supported by the Huntsman Cancer Foundation.
- mTOR
- mammalian target of rapamycin
- ITS
- insulin-transferrin-selenium
- HBSS
- Hanks' balanced salt solution
- ms
- methyl-specific
- AdOx
- adenosine-2,3-dialdehyde
- AdoMet
- S-adenosylmethionine.
REFERENCES
- 1. Mudduluru G., Medved F., Grobholz R., Jost C., Gruber A., Leupold J. H., Post S., Jansen A., Colburn N. H., Allgayer H. (2007) Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer 110, 1697–1707 [DOI] [PubMed] [Google Scholar]
- 2. Gao F., Wang X., Zhu F., Wang Q., Zhang X., Guo C., Zhou C., Ma C., Sun W., Zhang Y., Chen Y. H., Zhang L. (2009) PDCD4 gene silencing in gliomas is associated with 5′CpG island methylation and unfavourable prognosis. J. Cell. Mol. Med. 13, 4257–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang X., Wei Z., Gao F., Zhang X., Zhou C., Zhu F., Wang Q., Gao Q., Ma C., Sun W., Kong B., Zhang L. (2008) Expression and prognostic significance of PDCD4 in human epithelial ovarian carcinoma. Anticancer Res. 28, 2991–2996 [PubMed] [Google Scholar]
- 4. Fassan M., Cagol M., Pennelli G., Rizzetto C., Giacomelli L., Battaglia G., Zaninotto G., Ancona E., Ruol A., Rugge M. (2010) Programmed cell death 4 protein in esophageal cancer. Oncol. Rep. 24, 135–139 [DOI] [PubMed] [Google Scholar]
- 5. Powers M. A., Fay M. M., Factor R. E., Welm A. L., Ullman K. S. (2011) Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 71, 5579–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang H. S., Matthews C. P., Clair T., Wang Q., Baker A. R., Li C. C., Tan T. H., Colburn N. H. (2006) Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol. Cell. Biol. 26, 1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Santhanam A. N., Baker A. R., Hegamyer G., Kirschmann D. A., Colburn N. H. (2010) Pdcd4 repression of lysyl oxidase inhibits hypoxia-induced breast cancer cell invasion. Oncogene 29, 3921–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Q., Sun Z., Yang H. S. (2008) Downregulation of tumor suppressor Pdcd4 promotes invasion and activates both β-catenin/Tcf and AP-1-dependent transcription in colon carcinoma cells. Oncogene 27, 1527–1535 [DOI] [PubMed] [Google Scholar]
- 9. Leupold J. H., Yang H. S., Colburn N. H., Asangani I., Post S., Allgayer H. (2007) Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene 26, 4550–4562 [DOI] [PubMed] [Google Scholar]
- 10. Yang H. S., Knies J. L., Stark C., Colburn N. H. (2003) Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 22, 3712–3720 [DOI] [PubMed] [Google Scholar]
- 11. Cmarik J. L., Min H., Hegamyer G., Zhan S., Kulesz-Martin M., Yoshinaga H., Matsuhashi S., Colburn N. H. (1999) Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc. Natl. Acad. Sci. U.S.A. 96, 14037–14042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hilliard A., Hilliard B., Zheng S. J., Sun H., Miwa T., Song W., Göke R., Chen Y. H. (2006) Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J. Immunol. 177, 8095–8102 [DOI] [PubMed] [Google Scholar]
- 13. Jansen A. P., Camalier C. E., Colburn N. H. (2005) Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 65, 6034–6041 [DOI] [PubMed] [Google Scholar]
- 14. Yang H. S., Cho M. H., Zakowicz H., Hegamyer G., Sonenberg N., Colburn N. H. (2004) A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol. Cell. Biol. 24, 3894–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waters L. C., Veverka V., Böhm M., Schmedt T., Choong P. T., Muskett F. W., Klempnauer K. H., Carr M. D. (2007) Structure of the C-terminal MA-3 domain of the tumour suppressor protein Pdcd4 and characterization of its interaction with eIF4A. Oncogene 26, 4941–4950 [DOI] [PubMed] [Google Scholar]
- 16. Waters L. C., Strong S. L., Ferlemann E., Oka O., Muskett F. W., Veverka V., Banerjee S., Schmedt T., Henry A. J., Klempnauer K. H., Carr M. D. (2011) Structure of the tandem MA-3 region of Pdcd4 protein and characterization of its interactions with eIF4A and eIF4G: molecular mechanisms of a tumor suppressor. J. Biol. Chem. 286, 17270–17280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loh P. G., Yang H. S., Walsh M. A., Wang Q., Wang X., Cheng Z., Liu D., Song H. (2009) Structural basis for translational inhibition by the tumour suppressor Pdcd4. EMBO J. 28, 274–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. LaRonde-LeBlanc N., Santhanam A. N., Baker A. R., Wlodawer A., Colburn N. H. (2007) Structural basis for inhibition of translation by the tumor suppressor Pdcd4. Mol. Cell. Biol. 27, 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzuki C., Garces R. G., Edmonds K. A., Hiller S., Hyberts S. G., Marintchev A., Wagner G. (2008) PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc. Natl. Acad. Sci. U.S.A. 105, 3274–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang J. H., Cho Y. H., Sohn S. Y., Choi J. M., Kim A., Kim Y. C., Jang S. K., Cho Y. (2009) Crystal structure of the eIF4A-PDCD4 complex. Proc. Natl. Acad. Sci. U.S.A. 106, 3148–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dorrello N. V., Peschiaroli A., Guardavaccaro D., Colburn N. H., Sherman N. E., Pagano M. (2006) S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 [DOI] [PubMed] [Google Scholar]
- 22. Nakashima M., Hamajima H., Xia J., Iwane S., Kwaguchi Y., Eguchi Y., Mizuta T., Fujimoto K., Ozaki I., Matsuhashi S. (2010) Regulation of tumor suppressor PDCD4 by novel protein kinase C isoforms. Biochim. Biophys. Acta 1803, 1020–1027 [DOI] [PubMed] [Google Scholar]
- 23. Schmid T., Jansen A. P., Baker A. R., Hegamyer G., Hagan J. P., Colburn N. H. (2008) Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res. 68, 1254–1260 [DOI] [PubMed] [Google Scholar]
- 24. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silvera D., Formenti S. C., Schneider R. J. (2010) Translational control in cancer. Nat. Rev. Cancer 10, 254–266 [DOI] [PubMed] [Google Scholar]
- 26. Ramírez-Valle F., Badura M. L., Braunstein S., Narasimhan M., Schneider R. J. (2010) Mitotic raptor promotes mTORC1 activity, G2/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol. Cell. Biol. 30, 3151–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palamarchuk A., Efanov A., Maximov V., Aqeilan R. I., Croce C. M., Pekarsky Y. (2005) Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res. 65, 11282–11286 [DOI] [PubMed] [Google Scholar]
- 28. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rual J. F., Hirozane-Kishikawa T., Hao T., Bertin N., Li S., Dricot A., Li N., Rosenberg J., Lamesch P., Vidalain P. O., Clingingsmith T. R., Hartley J. L., Esposito D., Cheo D., Moore T., Simmons B., Sequerra R., Bosak S., Doucette-Stamm L., Le Peuch C., Vandenhaute J., Cusick M. E., Albala J. S., Hill D. E., Vidal M. (2004) Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res. 14, 2128–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bachand F. (2007) Protein arginine methyltransferases: from unicellular eukaryotes to humans. Eukaryot. Cell 6, 889–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hay N., Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 [DOI] [PubMed] [Google Scholar]
- 32. Vaupel P. (2004) Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 14, 198–206 [DOI] [PubMed] [Google Scholar]
- 33. Yang H. S., Jansen A. P., Komar A. A., Zheng X., Merrick W. C., Costes S., Lockett S. J., Sonenberg N., Colburn N. H. (2003) The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell. Biol. 23, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Böhm M., Sawicka K., Siebrasse J. P., Brehmer-Fastnacht A., Peters R., Klempnauer K. H. (2003) The transformation suppressor protein Pdcd4 shuttles between nucleus and cytoplasm and binds RNA. Oncogene 22, 4905–4910 [DOI] [PubMed] [Google Scholar]
- 35. Waters L. C., Oka O., Muskett F. W., Strong S. L., Schmedt T., Klempnauer K. H., Carr M. D. (2010) Resonance assignment and secondary structure of the middle MA-3 domain and complete tandem MA-3 region of the tumour suppressor protein Pdcd4. Biomol. NMR Assign. 4, 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hanahan D., Folkman J. (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 [DOI] [PubMed] [Google Scholar]
- 37. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 38. Asangani I. A., Rasheed S. A., Nikolova D. A., Leupold J. H., Colburn N. H., Post S., Allgayer H. (2008) MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27, 2128–2136 [DOI] [PubMed] [Google Scholar]
- 39. Lu Z., Liu M., Stribinskis V., Klinge C. M., Ramos K. S., Colburn N. H., Li Y. (2008) MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 27, 4373–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frankel L. B., Christoffersen N. R., Jacobsen A., Lindow M., Krogh A., Lund A. H. (2008) Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 283, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 41. Zhu S., Wu H., Wu F., Nie D., Sheng S., Mo Y. Y. (2008) Micro RNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 18, 350–359 [DOI] [PubMed] [Google Scholar]
- 42. Göke R., Barth P., Schmidt A., Samans B., Lankat-Buttgereit B. (2004) Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1). Am. J. Physiol. Cell Physiol. 287, C1541–C1546 [DOI] [PubMed] [Google Scholar]
- 43. Olson C. M., Donovan M. R., Spellberg M. J., Marr M. T., 2nd (2013) The insulin receptor cellular IRES confers resistance to eIF4A inhibition. eLife 2, e00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kakade D., Islam N., Maeda N., Adegoke O. A. (2014) Differential effects of PDCD4 depletion on protein synthesis in myoblast and myotubes. BMC Cell Biol. 15, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song X., Zhang X., Wang X., Zhu F., Guo C., Wang Q., Shi Y., Wang J., Chen Y., Zhang L. (2013) Tumor suppressor gene PDCD4 negatively regulates autophagy by inhibiting the expression of autophagy-related gene ATG5. Autophagy 9, 743–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsai W. W., Niessen S., Goebel N., Yates J. R., 3rd, Guccione E., Montminy M. (2013) PRMT5 modulates the metabolic response to fasting signals. Proc. Natl. Acad. Sci. U.S.A. 110, 8870–8875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kumar N., Wethkamp N., Waters L. C., Carr M. D., Klempnauer K. H. (2013) Tumor suppressor protein Pdcd4 interacts with Daxx and modulates the stability of Daxx and the Hipk2-dependent phosphorylation of p53 at serine 46. Oncogenesis 2, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shiota M., Izumi H., Tanimoto A., Takahashi M., Miyamoto N., Kashiwagi E., Kidani A., Hirano G., Masubuchi D., Fukunaka Y., Yasuniwa Y., Naito S., Nishizawa S., Sasaguri Y., Kohno K. (2009) Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res. 69, 3148–3156 [DOI] [PubMed] [Google Scholar]
- 49. Bitomsky N., Böhm M., Klempnauer K. H. (2004) Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun. Oncogene 23, 7484–7493 [DOI] [PubMed] [Google Scholar]
- 50. Leprivier G., Remke M., Rotblat B., Dubuc A., Mateo A. R., Kool M., Agnihotri S., El-Naggar A., Yu B., Somasekharan S. P., Faubert B., Bridon G., Tognon C. E., Mathers J., Thomas R., Li A., Barokas A., Kwok B., Bowden M., Smith S., Wu X., Korshunov A., Hielscher T., Northcott P. A., Galpin J. D., Ahern C. A., Wang Y., McCabe M. G., Collins V. P., Jones R. G., Pollak M., Delattre O., Gleave M. E., Jan E., Pfister S. M., Proud C. G., Derry W. B., Taylor M. D., Sorensen P. H. (2013) The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153, 1064–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones R. G., Thompson C. B. (2009) Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 23, 537–548 [DOI] [PMC free article] [PubMed] [Google Scholar]