

Background: Signaling through the IL-22 axis is essential for immunity, yet there is little known about its regulation.
Results: The IL-22R is phosphorylated by GSK-3β that stabilizes the receptor from degradation to control cell repair.
Conclusion: Protein stability of IL-22R, modulated by GSK-3β, is a key mechanism impacting cell repair.
Significance: Levels of IL-22R protein controlled by GSK-3β may be important for intervention.
Keywords: Cytokine, Glycogen Synthase Kinase 3 (GSK-3), Inflammation, Lung Injury, Protein Degradation, Cytokine
Abstract
Signaling through the interleukin (IL)-22 cytokine axis provides essential immune protection in the setting of extracellular infection as part of type 17 immunity. Molecular regulation of IL-22 receptor (IL-22R) protein levels is unknown. In murine lung epithelia, IL-22R is a relatively short-lived protein (t½ ∼1.5 h) degraded by the ubiquitin proteasome under normal unstimulated conditions, but its degradation is accelerated by IL-22 treatment. Lys449 within the intracellular C-terminal domain of the IL-22R serves as a ubiquitin acceptor site as disruption of this site by deletion or site-directed mutagenesis creates an IL-22R variant that, when expressed in cells, is degradation-resistant and not ubiquitinated. Glycogen synthase kinase (GSK)-3β phosphorylates the IL-22R within a consensus phosphorylation signature at Ser410 and Ser414, and IL-22 treatment of cells triggers GSK-3β inactivation. GSK-3β overexpression results in accumulation of IL-22R protein, whereas GSK-3β depletion in cells reduces levels of the receptor. Mutagenesis of IL-22R at Ser410 and Ser414 results in receptor variants that display reduced phosphorylation levels and are more labile as compared with wild-type IL-22R when expressed in cells. Further, the cytoskeletal protein cortactin, which is important for epithelial spreading and barrier formation, is phosphorylated and activated at the epithelial cell leading edge after treatment with IL-22, but this effect is reduced after GSK-3β knockdown. These findings reveal the ability of GSK-3β to modulate IL-22R protein stability that might have significant implications for cytoprotective functions and therapeutic targeting of the IL-22 signaling axis.
Introduction
The “type 17” immune response is important for host defense against extracellular pathogens including bacteria and fungi and is active through a robust induction of cytotoxic and phagocytic cell types to eradicate infectious threats (1, 2). Interleukin (IL)-23 is the upstream cytokine in this system that causes expression of effector cytokines, IL-17 and IL-22, by diverse cells including T-helper 17 and innate lymphoid cells. These cytokines lead to induction of antimicrobial host defense factors including cytokines, chemokines, and antimicrobial proteins such as the regenerating islet-derived 3γ. At mucosal surfaces exposed to the external environment including skin, gut, and respiratory organ systems, the type 17 effector responses appear to be mediated, in part, through the activity of the effector cytokine IL-22, which is secreted in large quantities by Th17 lymphocytes (3) and innate lymphoid cells (4). The functional receptor for IL-22 signaling, IL-22R,3 is only expressed on epithelial cells of these organs. Importantly, the activity of IL-22 in the setting of infection in gastrointestinal and respiratory systems augments epithelial bacterial killing and stimulates epithelial proliferation (5). Blockade of IL-22 signaling in animal models of lung and liver bacterial infection disables robust mucosal immunity with disseminated infection and death (6, 7). IL-22 is also necessary for tissue repair after influenza infection with up-regulation in human bronchial epithelia of chronically infected hosts (8). Conversely, a hyperactive Th17 signal has been associated with the inflammatory disorders psoriasis and colitis via up-regulation of IL-23 and IL-22 signaling, and polymorphisms of IL-23R have been associated with inflammatory disease (9, 10). Despite these roles of IL-22 in innate immunity and tissue repair, very little is known about the molecular regulation of its cognate receptor, IL-22R.
The cellular regulation of cytokine receptors by their degradation through the proteasomal or lysosomal systems is an intense area of interest because changes in the cellular expression and recycling of these surface proteins determines their function in the setting of human disease. We have recently characterized the ubiquitin (Ub)-dependent degradation of the IL-33 receptor, ST2 (11), and the TNF receptor-associated factors (12). The IL-22R is a type 2 cytokine receptor whose family members include interferon receptors A and B, the IL-10R, and the IL-20R. Functional receptors are heterodimers with IL-22Ra and IL-10Rb binding IL-22 leading to signaling through activation of JAK-STAT phosphorylation and STAT3-dependent gene transcription (13). The activation of the IL-22R also mediates activation of the mitogen-activated protein kinase (MAPK), the extracellular signal-related kinase (ERK), and the c-Jun N-terminal kinase (JNK) (13). Hence, insights into modulation of the abundance and therefore signaling of this important receptor may enhance our understanding and treatment of infectious and inflammatory disorders. Although ligand-dependent ubiquitination and degradation have been described for the IL-10R (14) and the interferon-α receptor (15), specific mechanisms of cellular degradation of the other type 2 cytokine receptors have not, to our knowledge, been elucidated. Here we characterize post-translational modification and molecular regulation of the IL-22R in epithelial cells to determine the manner by which cells modulate the availability of this inflammatory signal.
EXPERIMENTAL PROCEDURES
Cell Cultures and Reagents
Murine lung epithelial (MLE-12) cells were maintained with HITES medium supplemented with 10% FBS in a 37 °C incubator and 5% CO2 as described previously (31). Anti-IL-22R antibody and recombinant mouse IL-22R were from R&D Systems, Inc. (Minneapolis, MN). V5 antibody, mammalian expression plasmid pcDNA3.1/HisV5-topo, and Escherichia coli Top10 competent cells were purchased from Invitrogen. Hyperactive glycogen synthase kinase (GSK)-3β plasmid was a generous gift from Dr. John Engelhardt (32). The HA-ubiquitin construct was a generous gift from Dr. Peter M. Snyder. Phospho-serine antibodies were from Cell Signaling (Danvers, MA). The GSK-3, mouse V5 monoclonal, and phospho-GSK-3 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Immobilized protein A/G beads were obtained from Pierce. Proteasome inhibitor MG132 and kinase inhibitor PP1 was from Calbiochem. HA antibody, leupeptin, cycloheximide, and phosphatase inhibitor mixtures were from Sigma. Gel extraction kits and QIAprep spin miniprep kits were from Qiagen (Valencia, CA). All materials in highest grades used in the experiments are commercially available.
Fluorescent Immunostaining
MLE cells at a concentration of 105 cells/ml were transiently transfected and inoculated into glass-bottomed 35-mm plates for 48 h. Cells were cultured with IL-22 (90 ng/ml) or PBS, washed with cold PBS twice, and fixed with 4% paraformaldehyde for 1 h, and then we incubated the fixed cells with staining solution (0.1% Triton X-100 in PBS with 1% goat serum) for 30 min. The cells were then incubated with anti-phospho-cortactin (Cell Signaling) or IL-22R (Millipore) antibody (1:100) in staining solution for 10 h. Plates were washed three times and incubated with fluorescence-conjugated goat anti-rabbit secondary antibodies for another 1 h. Plates were then washed three times for 10 min. Images were acquired by a combination laser-scanning microscope system (Nikon A1, Nikon (Mellville, NY)), and the results were analyzed through Nikon NIS-Elements software.
Immunoprecipitation and Immunoblotting
MLE cells during exponential growth were treated with 2 mm Ca2+ for 2 h, and the cells were lysed with lysis buffer (0.3% Triton X-100 (v/v) in PBS and 1:1000 protease inhibitor mixture). Lysates were sonicated and centrifuged at 13,000 rpm for 10 min. Cell lysates (containing 1 mg of protein) were incubated and rotated with 2 μg of anti-V5 or anti-phospho-serine at 4 °C for 4 h and then incubated with 30 μl of protein A/G-agarose beads for another 3 h, and the beads were spun down and washed with lysis buffer three times. The washed beads were mixed with SDS-PAGE loading dye prior to SDS-PAGE and immunoblot analysis. Immunoblotting was performed as described previously (31).
Cloning and Mutagenesis
Mouse IL-22R cDNA was purchased from Open Biosystems (Huntsville, AL), and all primers were from Integrated DNA Technologies (Coralville IA). The coding region of the gene was cloned into pcDNA 3.1 by using the following primers: forward (5′-ccacctgaagacactgac-3′) and reverse (5′-ggattcccactgcacagtcagg-3′). C-terminal truncations of IL-22R were generated by PCR using the forward primer and the following reverse primers: del449 (5′-ctgtagagaaaggtcccctgg-3′) and del423 (5′-gggagtggagaggatgcc-3′). IL-22R serine and lysine mutants were generated by site-directed mutagenesis (Stratagene, La Jolla, CA) with the following primers: S410A forward (5′-ctgtgtgtgtggaagacgctggcaaagctctacc-3′) and reverse (5′-ggtagagtctttgccagcgtcttccacacacacag-3′); S414A forward (5′-gactctggcaaagacgctaccccaggcatcc-3′) and reverse (5′-ggatgcctggggtaggcgtctttgccagagtc-3′); K426R forward (5′-cactcccaaatacctcaggacaaaaggtcagctcc-3′) and reverse (5′-ggagctgaccttttgtcctgaggtatttgggagtg-3′); K428R forward (5′-cccaaatacctcaagacaagaggtcagctccagga-3′) and reverse (5′-tcctggagctgacctcttgtcttgaggtatttggg-3′); K449R forward (5′-caggggacctttctctacagagagtcacctcct-3′) and reverse (5′-aggaggtgactctctgtagagaaaggtcccctg-3′); and K540R forward (5′-ctcccttgtgtgtccaagggatgagggtcc-3′) and reverse (5′-gagggaacacacaggttccctactcccagg-3′).
In Vitro GSK-3β Kinase Phosphorylation Assay
Recombinant purified mouse IL-22R (100 ng per reaction, R&D Systems) was used directly (see Fig. 4) or wild-type IL-22R, S410A, or S414A mutant IL-22Rs were immunoaffinity-purified for experiments (see Fig. 5). Constructs were expressed in cells and lysed in Buffer A (PBS with 0.5% Triton X-100 and 8 mg/ml protease inhibitors (Roche Applied Science)) with sonication. The cleared cell lysates were incubated with V5 antibody overnight and with protein A/G-agarose beads for 2 h with rotation at 4 °C. The beads were washed three times with IL-22R. In vitro phosphorylation reactions were conducted by combining either 40 μl of protein A/G-agarose bead-bound IL-22R and 10 μl of kinase assay buffer (25 mm MOPS, 12.5 mm β-glycerol phosphate, 25 mm MgCl2, 5 mm EGTA, 2 mm EDTA, 0.25 mm DTT, pH 7.2) or recombinant proteins into assay buffer for a final volume of 50 μl. All reactions contained 1 μCi of [γ-32P]ATP (PerkinElmer) and 0.1 μg of recombinant active kinase GSK-3β, protein kinase C (Life Technologies), ERK (Millipore), or heat-inactivated GSK-3β (see Fig. 5) per reaction for 1 h at room temperature. Active GSK-3β was denatured at 95 °C for 10 min as the kinase negative control. The reactions were terminated by the addition of SDS loading buffer, and the samples were separated by SDS-PAGE and transferred to nitrocellulose membranes. Phosphorylated IL-22R was visualized by autoradiography, and then immunoblotting for IL-22R (see Fig. 4) or V5 (see Fig. 5) was performed on the same membranes.
FIGURE 4.

Regulation of IL-22R protein stability by GSK-3β. A, primary sequence of IL-22R contains a putative GSK-3β phosphorylation domain. B, MLE cells were treated with 90 ng/ml IL-22, and lysates were probed for total GSK-3β and Ser21 phosphorylated GSK-3α (p-GSK-3α, upper band) or Ser9 phosphorylated GSK-3β (p-GSK-3β, lower band) by immunoblotting showing phosphorylation. C, in vitro phosphorylation of IL-22R was assessed by kinase assays with recombinant active kinases GSK-3β, protein kinase C, or ERK (2 μg/ml), with or without recombinant mouse IL-22R (Rec mIL-22R, 100 ng per reaction), and 32P-labeled (p32) ATP for 1 h followed by SDS-PAGE, immunoblotting, and autoradiography of blots. The bottom immunoblot was probed for mouse IL-22R (lower panel). D, cells exposed to IL-22 (90 ng/ml) and transfected with V5 IL-22R were subjected to phospho-serine immunoprecipitation (IP: p-Ser) followed by V5 immunoblotting (IB: V5) to determine levels of serine phosphorylation. E, modulation of IL-22R levels by GSK-3β was performed by transfection of cells with a plasmid encoding constitutively active enzyme (GSK-3β OE), or shRNA constructs; GSK-3β doublet indicates active isoform (upper band). F, co-transfection of GSK-3β and V5-IL-22R (3 μg each) shows effects of GSK-3β on receptor stability, with immunoblotting for IL-22R and GSK-3β. G, V5-IL-22R protein half-life with CHX in cells transfected with IL-22R and empty vector or the constitutively active enzyme (GSK-3β OE). Data represent n = 2 for kinase assays and immunoprecipitation experiments; n = 3 or more for all other panels. The bottom graphs in D and F show densitometric values after quantitation of individual bands on immunoblots. In G, the bottom graph shows the relative -fold change in V5 signal of IL-22R with time after normalizing to β-actin band intensities.
FIGURE 5.

GSK-3β phosphorylation sites impact IL-22R protein stability. A, MLE cells were transfected with the V5 WT IL-22R or Ser → Ala IL-22R mutants at residues Ser410 or Ser414 or an empty vector or GSK-3β plasmids for 48 h before immunoblotting. GSK-3β OE, constitutively active enzyme. B, V5 IL-22R constructs after cellular transfection were subjected to immunopurification (IP: V5) with beads coated with anti-V5 antibody and then assayed for phosphorylation by incubation with recombinant active or heat-inactivated GSK-3β and 32P-radiolabeled (p32) ATP followed by SDS-PAGE with autoradiography (upper panel) and immunoblotting (IB, lower panel). C, cellular degradation kinetics of wild-type (WT) and S410A IL-22R-V5 constructs exposed to CHX 48 h after cellular transfection with densitometry at 0 and 30 min CHX. D, graph shows relative densitometric values after normalizing to initial IL-22R levels by quantitation of individual bands on immunoblots to illustrate IL-22R Ser variant protein stability. Data are representative of at least two experiments.
RESULTS
The IL-22R Is Degraded by the Ubiquitin Proteasome
MLE cells express IL-22R as evidenced by immunoblotting with an IL-22R antibody. Immunoblot analysis of IL-22R expression in the presence of the protein synthesis inhibitor cycloheximide (CHX) reveals cellular degradation of the receptor over a period of ∼4 h (Fig. 1A), with a cellular half-life of ∼2 h (Fig. 1A, graph). Treatment with the proteasome inhibitor, MG132, prevents receptor degradation, but the lysosomal inhibitor, leupeptin, did not affect levels of IL-22R degradation. Stimulation of MLE cells with recombinant IL-22 cytokine but not heat-inactivated IL-22 accelerated depletion of the IL-22R with a half-life of ∼30 min after agonist treatment (Fig. 1B). IL-22R levels are restored 3 h after continuous IL-22 treatment in the absence of the protein synthesis inhibitor cycloheximide, suggesting synthesis of new IL-22R (data not shown).
FIGURE 1.
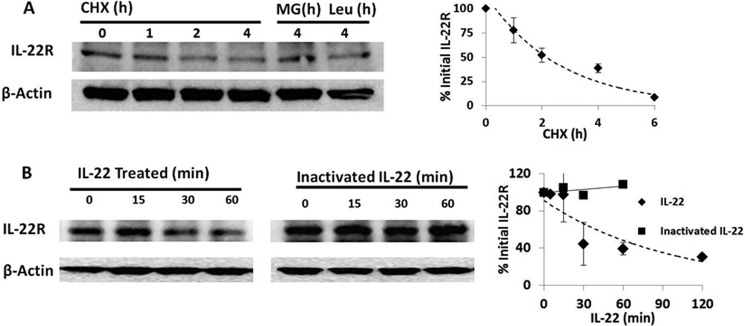
IL-22 receptor degradation under native and stimulated conditions. A, MLE cells were incubated with CHX (10 μg/ml) and MG132 (MG, 20 μg/ml) or leupeptin (Leu, 20 μg/ml), and IL-22R immunoblotting was performed with monoclonal anti-IL-22R (R&D Systems). Right, a densitometric plot of IL-22R signal decay versus time of CHX exposure with fit line; half-life, ∼90 min. B, IL-22R immunoblotting from MLE cells treated with 90 ng/ml recombinant mouse IL-22 cytokine or 90 ng/ml heat-inactivated (95 °C for 5 min) IL-22 and densitometry of immunoreactive IL-22R bands. Data are representative of three independent experiments averaged for graphical analysis and standardized to the initial IL-22R signal. Data represent mean ± S.E.
Lys449 Is a Ubiquitination Acceptor Site within IL-22R
To determine the specific molecular determinants of receptor ubiquitination, we cloned the mouse IL-22R cDNA into a pcDNA 3.1 v5 epitope-tagged expression plasmid and constructed C-terminal truncation mutants of the receptor (Fig. 2A). Ectopically expressed IL-22R in this vector was detected using anti-V5 antibody demonstrating reduction of immunoreactive IL-22R-V5 after treatment with IL-22 (Fig. 2B), consistent with results observed with endogenous IL-22R. MLE cells were transfected with truncation plasmids, and cells were exposed to medium alone or with MG132 to induce accumulation of mutant proteins, suggestive of accumulation of polyubiquitinated IL-22R. A construct lacking the ubiquitin acceptor site may not accumulate after MG132 exposure as it might be processed by alternative pathways for degradation. Fig. 2C shows that MLE cells transfected with full-length (584 amino acids) IL-22R-containing plasmid (IL-22RFL) or a C-terminal deletion construct (IL-22R449del) lacking 135 residues displayed low levels of expression without the addition of MG132, suggestive of proteins that are ubiquitinated and rapidly degraded. However, a mutant protein (IL-22R423del) lacking putative Ub acceptor sites was robustly expressed in cells without exposure to MG132, suggesting that the ubiquitin acceptor site resides between Lys423 and Lys449. Moreover, in the presence of MG132, we observed that both the IL-22RFL and the IL-22R449 constructs accumulated, suggesting the presence of accumulation of polyubiquitinated proteins. In contrast, levels of the IL-22R423 deletion variant comparatively decreased with MG132 treatment, suggesting an extraproteasomal degradation mechanism (e.g. lysosome (Fig. 2C)). To determine whether the stability of these proteins is indeed due to Ub-dependent degradation of IL-22R, we overexpressed each construct in the presence or absence of excess Ub plasmid with an HA epitope tag to increase Ub-dependent degradation. Fig. 2D shows that when correcting for loading, overexpression of Ub plasmid causes depletion of both the IL-22RFL and the IL-22R449 deletion proteins, but not the IL-22R423 deletion variant. The results indicate that ubiquitination of the IL-22R in the region between residues 424 and 449 is necessary and sufficient for receptor degradation. To confirm the specific Lys ubiquitination acceptor site within the IL-22R C terminus, we substituted Arg at each of the three Lys residues in the 424–449 region (K426R K428R, and K449R). We also analyzed the IL-22R with a Ub protein modification online database (16), predicting that either Lys449 or Lys540 might serve as Ub acceptor sites in this region. Thus, we also generated a K540R mutation within IL-22R. In Fig. 3, A and B, we show that transfection and expression of the IL-22R plasmid variants in cells with cycloheximide treatment results in protein degradation of full-length K426R, K428R, and K540R constructs, but not the K449R protein. Immunoprecipitation of HA-Ub and immunoblotting for IL-22R reveals modest polyubiquitination of full-length and K540R IL-22R, but reduced levels with the K449R mutant, implicating Lys449 as the likely Ub acceptor residue (Fig. 3C).
FIGURE 2.

Mapping of IL-22R regions regulated by the ubiquitin proteasome. A, full-length (FL) and C-terminal IL-22R deletion constructs were cloned into the pcDNA 3.1 TOPO mammalian expression vector (Life Technologies) with a V5 epitope tag. B, MLE cells were transiently transfected with the pcDNA-IL-22R full-length plasmid (4 μg) for 48 h, after which cells were treated with or without IL-22 treatment (90 ng/ml) for 30 min followed by immunoblotting with primary antibody targeting IL-22R, V5, or β-actin. C, transfected MLE cells with pcDNA-IL-22R constructs were incubated with or without MG132 for 4 h and then analyzed for accumulation by immunoblotting for V5. D, IL-22R423del is degradation-resistant. IL-22R constructs were co-transfected with an empty vector or HA-tagged ubiquitin (3 μg each plasmid) and evaluated 48 h later with immunoblotting to V5, HA, or β-actin; construct stability was assessed with densitometry of V5 signals relative to β-actin. Data are representative of three independent experiments or more for all blots shown. Band intensities in C and D were normalized to β-actin, and the data represent mean ± S.E.
FIGURE 3.

Lysine 449 is an IL-22R ubiquitin acceptor site. A, V5-IL-22R degradation kinetics in CHX-treated MLE cells transfected with wild-type IL-22R or the K449R mutant IL-22R plasmid (4 μg). B, densitometry kinetics of protein degradation for wild type (WT) or four IL-22R Lys → Arg mutants after expression in MLE cells. C, levels of polyubiquitination of V5-IL22R constructs assessed in transfected MLE cells after co-immunoprecipitation (Co IP) of the HA-Ub followed by V5 or HA immunoblotting to detect wild-type (WT), K449R, or K540R mutant proteins that are modified in cells. Data are representative of two independent experiments.
Regulation of the IL-22R Stability by Glycogen Synthase Kinase 3β
Phosphorylation of surface receptors and other membrane proteins modulates their vulnerability to ubiquitin-dependent degradation (17). To determine whether such post-translational modifications might impact the stability of the IL-22R, we scanned the primary sequence of the receptor for consensus phosphorylation domains and found that the C-terminal intracellular portion of the receptor contains the consensus domain for GSK-3β ((Ser/Thr)-X-X-X-(Ser/Thr)) (18) at residues 410–414 (Fig. 4A). Activity of this kinase has been implicated in both stabilizing and destabilizing a number of its protein substrates (19), and we have characterized GSK-3β as important for the Ub-dependent processing of the receptor for IL-33, ST2, as well as the acyl-CoA:lysophosphatidylcholine acyltransferase (11, 20). We therefore assessed whether IL-22 signaling affects GSK-3β kinase and whether the kinase governs IL-22R stability. First, we observed that IL-22 treatment of cells increased GSK-3β phosphorylation levels, indicating that the agonist reduces kinase activity (Fig. 4B).
To determine whether GSK-3β directly phosphorylates the IL-22R, we used an in vitro phosphorylation assay. Here reactions contained 32P-radiolabeled ATP with inclusion of recombinant mouse IL-22R and recombinant GSK-3β with appropriate controls. We observed that GSK-3β incubated with IL-22R results in a robust signal at the predicted IL-22R molecular weight (Fig. 4C). This signal was absent with GSK-3β alone in the absence of substrate or IL-22R with protein kinase C (PKC); IL-22R also appears to be phosphorylated by the MAP kinase ERK. IL-22R also possesses a putative ERK consensus phosphorylation sequence (Pro-X-(Ser/Thr)-Pro) around Thr488. However, pharmacologic inhibition of ERK signaling with the small molecule inhibitor PD98059 did not modulate IL-22R stability (data not shown). These observations suggest that IL-22R is prone to phosphorylation by GSK-3β in vitro with some degree of specificity.
To evaluate IL-22R regulation by GSK-3β in cells, we used a phospho-serine antibody to immunoprecipitate proteins from V5-IL-22R-transfected MLE cell lysates after treatment with IL-22. The immunoprecipitates were then subjected to V5 immunoblotting, demonstrating a rapid decrease in receptor phosphorylation after cytokine treatment (Fig. 4D). We next successfully manipulated GSK-3β levels in cells using ectopic overexpression plasmids or silencing methods (Fig. 4E). We then tested the effect of these approaches on IL-22R stability as GSK-3β phosphorylation can form a phospho-degron recognized by ubiquitin E3 ligases to mediate substrate degradation by the ubiquitin proteasome in some cases. However, Fig. 4F shows a direct correlation between levels of GSK-3β and immunoreactive IL-22R content in cells, indicating that increased GSK expression in cells stabilizes the receptor, whereas depletion of GSK-3β by shRNA depletes IL-22R protein. By analogy, the cellular protein half-life of IL-22R is prolonged with GSK-3β overexpression (Fig. 4G).
To investigate molecular signatures within IL-22R that might be regulated by the kinase, we expressed IL-22 variants harboring mutations at putative GSK-3β phosphorylation sites with or without ectopically expressed GSK-3β plasmid in cells (Fig. 5A). As described above, overexpression of constitutively active GSK-3β plasmid increased immunoreactive wild-type IL-22R; however, IL-22R variants harboring mutations at putative GSK-3β Ser phosphorylation sites are not stabilized with overexpression of GSK-3β plasmid (Fig. 5A). The phosphorylation of IL-22R by recombinant GSK-3β was markedly reduced in vitro using these Ser variants as substrates (Fig. 5B). In addition, the overall abundance and cellular life span of the IL-22R S410A construct in MLE cells are decreased as compared with the wild-type receptor (Fig. 5C). This was confirmed with densitometric analysis of immunoblots under identical exposure settings showing that both the S410A and the S414A constructs are present at only 58 and 46%, respectively, to that of the wild type (not shown). Likewise when we exposed transfected cells to cycloheximide, a loss of 30% of receptor from basal expression in S410A and S414A constructs as compared with loss of 10% of wild-type receptor at 30 min was observed (Fig. 5D).
Modulation of GSK-3β Changes IL-22R-dependent Cell Spreading
The activity of IL-22 signaling on epithelia is a mitogenic stimulus for cell spreading and barrier formation. To model this, we used a scratch assay in MLE cells where a confluent layer of cells is scratched with a pipette tip and photomicrographs of the cell layer are acquired immediately after scratch and 24 h later, with image analysis by ImageJ to quantitate the scratch area. We observed that treatment with IL-22 enhances MLE epithelial recovery, consistent with prior studies (6) on human bronchial epithelia (Fig. 6, A and B). We also treated cells with the Src kinase inhibitor PP1. Src kinase is active in the formation of focal adhesions and cell migration and phosphorylates the cortactin molecule among others. PP1 effectively blunts IL-22-induced epithelial cell recovery after scratch of cell monolayers (Fig. 6B). We next determined phosphorylation of the cortactin protein, a proximal step in actin polymerization required for cell spreading. We observed an increase in cortactin phosphorylation over time with IL-22 treatment without significant changes in total cortactin (Fig. 6C). We next investigated whether IL-22R modulation by GSK-3β changes cellular responses to IL-22. In fluorescent immunocytochemical analysis, we observed phosphorylated cortactin on the leading edges of cells transfected with empty vector and treated with IL-22 as compared with untreated cells, whereas the IL-22R staining intensity is reduced after cytokine treatment (Fig. 6D). The staining for both IL-22R and phospho-cortactin is also reduced with GSK-3β knockdown by shRNA with or without IL-22R treatment (Fig. 6E). Moreover, cellular expression of IL-22R was increased after expressing active GSK-3β plasmid in cells, and robust cortactin phosphorylation was also observed when we treated cells with IL-22 (Fig. 6F, quantitation in Fig. 6G). These data support a mechanistic model whereby the stabilizing effect of GSK-3β on IL-22R corresponds physiologically to IL-22-triggered phosphorylation of the cortactin molecule, which promotes actin polymerization and cell spreading that fortify the epithelial barrier in lung epithelial cells.
FIGURE 6.

IL-22 treatment enhances cellular spreading and triggers cortactin activation. A and B, MLE cells were grown to 95% confluence on polystyrene 6-well culture dishes that were then scratched with a pipette tip and treated with IL-22 (90 ng/ml) with vehicle (Veh, DMSO) or Src kinase inhibitor, PP1, at the concentrations shown. Photomicrographs were taken of the identical well coordinates of the scratched cells immediately and 24 h later. Scale bar = 200 μm. B, images were analyzed with ImageJ for scratch area, and the relative decrease in scratch area over time is shown as recovery (*, p < 0.05 by Student's t test, n = 12). Data represent mean ± S.E. C, MLE cells were treated with IL-22 (90 ng/ml) after a 15-min pretreatment with vehicle or 50 nm PP1 for indicated times, and lysates were immunoblotted for phospho-cortactin (P-Cortactin) (Tyr421) and β-actin. D–F, MLE cells were transfected with empty vector (D) or plasmids coding for GSK-3β shRNA (E) or active GSK-3β (F) and treated with vehicle (PBS) or IL-22 (90 ng/ml) prior to staining with IL-22R antibody (upper panels, green fluorescence) or phospho-cortactin antibody (lower panels, red fluorescence). Confocal fluorescent micrographs were acquired with identical exposure conditions. Arrowheads indicate likely focal adhesions on leading edges of cells. Scale bars = 10 μm. G, quantitation of phospho-cortactin intensity from at least four images from each condition displayed graphically as mean ± S.E. RLU, relative light units.
Modulation of an IL-22R Degradation Variant on Cell Spreading by GSK-3β
Because the K449R variant of the IL-22 receptor is degradation-resistant, we next tested whether the cellular responses to IL-22 stimulation were augmented after expression of this plasmid in epithelia. Cells transfected with IL-22RK449R plasmid display enhanced recovery in the scratch assay when treated with IL-22 (Fig. 7A), and also show increased cortactin phosphorylation (data not shown).
FIGURE 7.

Effect of a Lys449 IL-22R variant on cell spreading and regulation by GSK-3β. A, MLE cells were transfected with either WT or a K449R variant IL-22R protein and grown to 95% confluence before scratch assay without or with IL-22 treatment (*, p < 0.05 for n = 6). Data represent mean ± S.E. B, MLE transfected with IL-22R WT- or K449R-containing plasmids were co-transfected with vector control or the GSK-3β shRNA-containing plasmid. Cells were treated with cycloheximide, and lysates were analyzed by immunoblotting for the V5 signal. C, densitometric kinetics of V5 signals over time from immunoblots in B.
Because both site-specific ubiquitination and site-specific phosphorylation are important in the cellular regulation of IL-22R abundance, we next tested whether GSK-3β phosphorylation protects IL-22R from ubiquitination at the Lys449 site. We transfected plasmids encoding either WT or K449R IL-22R plasmid into cells, co-transfected empty vector or the GSK-3β shRNA plasmid, and measured protein half-life in the presence of cycloheximide. Fig. 7 (B and C) demonstrates that although IL-22RK449R is degradation-resistant as compared with WT IL-22R under native conditions without GSK-3β depletion, introduction of GSK-3β shRNA into these cells causes accelerated degradation of both the WT IL-22R and the K449R IL-22R variant. This indicates that regulatory control of IL-22R protein stability is independently mediated by site-specific ubiquitination and constitutive GSK-3β activity (Fig. 8).
FIGURE 8.

Schematic of IL-22 signaling and protein stability. Right, IL-22R is polyubiquitinated at Lys449, leading to degradation by the ubiquitin proteasome. Middle and left, IL-22 ligation of the IL-22R leads to phosphorylation (P) of cortactin, which promotes cell spreading and epithelial recovery from insult, and phosphorylation of GSK-3β, inactivating the kinase. Reduced GSK-3β activity limits its ability to phosphorylate and stabilize the IL-22R as this kinase normally increases the abundance of the receptor.
DISCUSSION
This study represents the first demonstration that the IL-22 receptor, which is critical for Th-17-mediated host defense but also implicated in inflammatory pathology, is regulated at the level of protein stability through the activity of the ubiquitin-proteasome system in lung epithelial cells. Lys449 is a putative ubiquitin acceptor site within the receptor. We also show that IL-22R availability is coordinately regulated by GSK-3β as this kinase constitutively phosphorylates and stabilizes the receptor via site-specific phosphorylation, whereas IL-22 signaling itself causes inactivation of GSK-3β in these cells. Another unique observation is demonstration that the IL-22 signaling axis triggers cortactin phosphorylation as a downstream action that regulates epithelial barrier integrity. Because of the stabilizing effects of GSK-3β activity on IL-22R abundance, we see that manipulation of GSK-3β levels in cells modulates the downstream phosphorylation of cortactin.
The mapping of the specific ubiquitin acceptor site to the intracellular Lys449 residue within IL-22R implies that the receptor can be targeted for degradation while on the cell surface without requiring internalization. In MLE during basal culture conditions, IL-22R has a short half-life (∼1.5 h) that is comparable with the stability of the IL-10R (14). After ligation of IL-22R by its natural agonist, however, receptor degradation is accelerated, possibly reflecting a combination of both proteasomal and nonproteasomal pathways for degradation, consistent with the processing of the related cytokine receptors.
Our observations that mutation of the Lys449 in IL-22R stabilizes the receptor in normal conditions but not in the setting of GSK-3β knockdown suggest that these regulatory mechanisms are distinct and possibly additive in control of receptor abundance. We have not excluded ubiquitination at an alternative residue or even multiubiquitination in the setting of IL-22 ligation as another mode of receptor regulation, but this was not explored for this study. The surface processing of IL-22R that we observe resembles molecular characterization of related type 2 cytokine receptors, the interferon-α receptor and IL-10 receptor, both of which are shown to be ubiquitinated and degraded by the lysosome after ligation through the action of the SCF E3 ubiquitin ligase β-TrCP (14, 15).
The phosphorylation of substrates by GSK-3β has pleiotropic effects on its targets and is directed, in part, by the minimal recognition motif Ser-X-X-X-Ser (18). In prior studies from our laboratory, we show that GSK-3β induced the phosphorylation of the cytokine receptor for IL-33, ST2, to facilitate FBXL19-mediated ubiquitination and degradation of ST2 (11); we have also described GSK-3β phosphorylation-dependent ubiquitin-proteasome degradation of the acyl-CoA lysophosphatidylcholine acyltransferase (LPCAT) via the action of β-TrCP (21). In this system, however, it appears that GSK-3β activity stabilizes the IL-22R by phosphorylation at the consensus sequence of residues 410–414. Although we have not conclusively demonstrated the ability of GSK-3β to modulate IL-22R levels in a site-specific manner in vivo, we present several lines of evidence implicating the kinase in stabilizing the receptor. GSK-3β phosphorylates IL-22R in vitro, and depletion of endogenous levels of the kinase destabilizes the receptor. Further, ectopic expression of highly active GSK-3β increases IL-22R protein stability. Last, we have identified sites within a consensus GSK-3β recognition signature in IL-22R that appear to impact receptor stability. The first residue, Ser410 of IL-22R, likely represents the bona fide phosphorylation acceptor site, whereas Ser414 may represent a priming or kinase dock site for GSK-3β recognition as mutagenesis of either site results in a protein with reduced phosphorylation in vitro that exhibits increased protein turnover when expressed in cells. Last, although GSK-3β has been shown to prime multiple substrates for degradation through the lysosome or ubiquitin proteasome, our results are consistent with other studies showing stabilization (22–24) or changes in cellular compartmentalization (25) of some GSK-3β substrates after phosphorylation. Indeed, the loss of IL-22R phosphorylation by GSK-3β may be induced by IL-22 cytokine signaling itself, and this event may be involved in rapid IL-22R degradation observed with cytokine treatment.
Biologically, there is a precedent for these observations of IL-22 signaling in innate immunity, cell injury, and repair. Infection containment by IL-22 signaling was previously demonstrated in vivo and implies that barrier function of the mucosal epithelia is augmented (6). Cortactin activation is important for epithelial integrity and host defense to infection as it orchestrates actin polymerization in the setting of infection (26, 27), and we have recently described activity of ERK (which is activated by IL-22 signaling (13)) in the modulation of cortactin stability (28). Regarding the effects of IL-22 signaling on GSK-3β, multiple growth factors including epidermal growth factor trigger inactivation of GSK-3β by phosphorylation at Ser9 (19, 29), so it appears biologically harmonious that IL-22, a proliferative factor, would exhibit a similar effect. Additionally, IL-22 and another proliferative cytokine IL-6 both activate STAT3, which has been shown to activate the AKT kinase (7, 30), whose targets include the GSK-3β Ser9.
These observations on IL-22 signaling need to be translated from studies on the mouse receptor to human systems, but there are important differences in the structure of IL-22R between species. First, the human IL-22R lacks the Lys449 residue we find necessary for mouse IL-22R proteasomal degradation. Thus, an analogous acceptor site within the human sequence with a juxtaposed recognition motif for an E3 ligase may exist. Another important observation within the primary sequences is that both species possess the GSK phosphorylation domain. In the mouse protein, this domain also contains the consensus sequence (DSGXXS) for the E3 ligase β-TrCP (FBXW1), whereas the human protein sequence is GSGKDS. The processes for ubiquitination and proteasomal degradation as well as the role of phosphorylation to coordinately dictate surface concentrations of IL-22R may thus differ between the two species, which will be an area of interest for additional investigation.
Acknowledgments
We acknowledge Dr. Jay Kolls and Dr. Derek Pociask for technical and intellectual suggestions.
This work was supported in part by the United States Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, and a Merit Review Award from the United States Department of Veterans Affairs (to R. K. M. and N. M. W.). This work was also supported, in part, by National Institutes of Health R01 Grants HL096376, HL097376, HL098174, HL081784, UH2 HL123502, and P01 HL114453 (to R. K. M.), HL116472 (to B. B. C.), and HL01916 (to Y. Z.), and an American Heart Association Award 12SDG9050005 (to J. Z.).
- IL-22R
- interleukin-22 receptor
- GSK
- glycogen synthase kinase
- Ub
- ubiquitin
- MLE
- murine lung epithelial
- CHX
- cycloheximide
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Bettelli E., Korn T., Oukka M., Kuchroo V. K. (2008) Induction and effector functions of TH17 cells. Nature 453, 1051–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aujla S. J., Kolls J. K. (2009) IL-22: a critical mediator in mucosal host defense. J. Mol. Med (Berl.) 87, 451–454 [DOI] [PubMed] [Google Scholar]
- 3. Khader S. A., Gaffen S. L., Kolls J. K. (2009) Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2, 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spits H., Artis D., Colonna M., Diefenbach A., Di Santo J. P., Eberl G., Koyasu S., Locksley R. M., McKenzie A. N., Mebius R. E., Powrie F., Vivier E. (2013) Innate lymphoid cells: a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 [DOI] [PubMed] [Google Scholar]
- 5. Liang S. C., Tan X. Y., Luxenberg D. P., Karim R., Dunussi-Joannopoulos K., Collins M., Fouser L. A. (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203, 2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aujla S. J., Chan Y. R., Zheng M., Fei M., Askew D. J., Pociask D. A., Reinhart T. A., McAllister F., Edeal J., Gaus K., Husain S., Kreindler J. L., Dubin P. J., Pilewski J. M., Myerburg M. M., Mason C. A., Iwakura Y., Kolls J. K. (2008) IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zenewicz L. A., Yancopoulos G. D., Valenzuela D. M., Murphy A. J., Karow M., Flavell R. A. (2007) Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27, 647–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pociask D. A., Scheller E. V., Mandalapu S., McHugh K. J., Enelow R. I., Fattman C. L., Kolls J. K., Alcorn J. F. (2013) IL-22 is essential for lung epithelial repair following influenza infection. Am. J. Pathol. 182, 1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eken A., Singh A. K., Treuting P. M., Oukka M. (2014) IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 7, 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zwiers A., Kraal L., van de Pouw Kraan T. C., Wurdinger T., Bouma G., Kraal G. (2012) Cutting edge: a variant of the IL-23R gene associated with inflammatory bowel disease induces loss of microRNA regulation and enhanced protein production. J. Immunol. 188, 1573–1577 [DOI] [PubMed] [Google Scholar]
- 11. Zhao J., Wei J., Mialki R. K., Mallampalli D. F., Chen B. B., Coon T., Zou C., Mallampalli R. K., Zhao Y. (2012) F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat. Immunol. 13, 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen B. B., Coon T. A., Glasser J. R., McVerry B. J., Zhao J., Zhao Y., Zou C., Ellis B., Sciurba F. C., Zhang Y., Mallampalli R. K. (2013) A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 14, 470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lejeune D., Dumoutier L., Constantinescu S., Kruijer W., Schuringa J. J., Renauld J. C. (2002) Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line: pathways that are shared with and distinct from IL-10. J. Biol. Chem. 277, 33676–33682 [DOI] [PubMed] [Google Scholar]
- 14. Jiang H., Lu Y., Yuan L., Liu J. (2011) Regulation of interleukin-10 receptor ubiquitination and stability by β-TrCP-containing ubiquitin E3 ligase. PLoS One 6, e27464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar K. G., Tang W., Ravindranath A. K., Clark W. A., Croze E., Fuchs S. Y. (2003) SCFHOS ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-α receptor. EMBO J. 22, 5480–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Radivojac P., Vacic V., Haynes C., Cocklin R. R., Mohan A., Heyen J. W., Goebl M. G., Iakoucheva L. M. (2010) Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78, 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. MacGurn J. A., Hsu P. C., Emr S. D. (2012) Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81, 231–259 [DOI] [PubMed] [Google Scholar]
- 18. Doble B. W., Woodgett J. R. (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sutherland C. (2011) What are the bona fide GSK3 substrates? Int. J. Alzheimers Dis. 2011, 505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zou C., Butler P. L., Coon T. A., Smith R. M., Hammen G., Zhao Y., Chen B. B., Mallampalli R. K. (2011) LPS impairs phospholipid synthesis by triggering β-transducin repeat-containing protein (β-TrCP)-mediated polyubiquitination and degradation of the surfactant enzyme acyl-CoA:lysophosphatidylcholine acyltransferase I (LPCAT1). J. Biol. Chem. 286, 2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zou C., Ellis B. M., Smith R. M., Chen B. B., Zhao Y., Mallampalli R. K. (2011) Acyl-CoA:lysophosphatidylcholine acyltransferase I (Lpcat1) catalyzes histone protein O-palmitoylation to regulate mRNA synthesis. J. Biol. Chem. 286, 28019–28025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Litovchick L., Chestukhin A., DeCaprio J. A. (2004) Glycogen synthase kinase 3 phosphorylates RBL2/p130 during quiescence. Mol. Cell. Biol. 24, 8970–8980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foltz D. R., Santiago M. C., Berechid B. E., Nye J. S. (2002) Glycogen synthase kinase-3β modulates notch signaling and stability. Curr. Biol. 12, 1006–1011 [DOI] [PubMed] [Google Scholar]
- 24. Surjit M., Lal S. K. (2007) Glycogen synthase kinase-3 phosphorylates and regulates the stability of p27kip1 protein. Cell Cycle 6, 580–588 [DOI] [PubMed] [Google Scholar]
- 25. Kwiek N. C., Thacker D. F., Haystead T. A. (2007) Dual kinase-mediated regulation of PITK by CaMKII and GSK3. Cell. Signal. 19, 593–599 [DOI] [PubMed] [Google Scholar]
- 26. Uruno T., Liu J., Zhang P., Fan Y. x., Egile C., Li R., Mueller S. C., Zhan X. (2001) Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 3, 259–266 [DOI] [PubMed] [Google Scholar]
- 27. Bougnères L., Girardin S. E., Weed S. A., Karginov A. V., Olivo-Marin J. C., Parsons J. T., Sansonetti P. J., Van Nhieu G. T. (2004) Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J. Cell Biol. 166, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao J., Wei J., Mialki R., Zou C., Mallampalli R. K., Zhao Y. (2012) Extracellular signal-regulated kinase (ERK) regulates cortactin ubiquitination and degradation in lung epithelial cells. J. Biol. Chem. 287, 19105–19114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eldar-Finkelman H., Seger R., Vandenheede J. R., Krebs E. G. (1995) Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J. Biol. Chem. 270, 987–990 [DOI] [PubMed] [Google Scholar]
- 30. Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen B. B., Mallampalli R. K. (2009) Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol. Cell. Biol. 29, 3062–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Filali M., Cheng N., Abbott D., Leontiev V., Engelhardt J. F. (2002) Wnt-3A/β-catenin signaling induces transcription from the LEF-1 promoter. J. Biol. Chem. 277, 33398–33410 [DOI] [PubMed] [Google Scholar]