

Background: Histone demethylase JARID1B is a potential oncoprotein, but its in vivo roles are not well understood.
Results: Mice lacking JARID1B showed delayed mammary gland development and decreased female fertility.
Conclusion: JARID1B promotes luminal lineage transcription programs in the mammary gland and fine-tunes systemic estrogen level.
Significance: JARID1B functions in cancer are likely linked to its roles in gene regulation and mammary gland development.
Keywords: Breast Cancer, Epigenetics, Estrogen, Histone Methylation, Mouse Genetics, FOXA1, GATA3, KDM5B, Luminal Lineage, Mammary Gland Development
Abstract
The JmjC domain-containing H3K4 histone demethylase jumonji AT-rich interactive domain 1B (JARID1B) (also known as KDM5B and PLU1) is overexpressed in breast cancer and is a potential target for breast cancer treatment. To investigate the in vivo function of JARID1B, we developed Jarid1b−/− mice and characterized their phenotypes in detail. Unlike previously reported Jarid1b−/− strains, the majority of these Jarid1b−/− mice were viable beyond embryonic and neonatal stages. This allowed us to further examine phenotypes associated with the loss of JARID1B in pubertal development and pregnancy. These Jarid1b−/− mice exhibited decreased body weight, premature mortality, decreased female fertility, and delayed mammary gland development. Related to these phenotypes, JARID1B loss decreased serum estrogen level and reduced mammary epithelial cell proliferation in early puberty. In mammary epithelial cells, JARID1B loss diminished the expression of key regulators for mammary morphogenesis and luminal lineage specification, including FOXA1 and estrogen receptor α. Mechanistically, JARID1B was required for GATA3 recruitment to the Foxa1 promoter to activate Foxa1 expression. These results indicate that JARID1B positively regulates mammary ductal development through both extrinsic and cell-autonomous mechanisms.
Introduction
Histone methylation and demethylation have profound effects on transcriptional regulation and development (1, 2). JARID1B,2 also known as PLU1 or KDM5B, is a histone demethylase that converts tri- and dimethylated lysine 4 in histone H3 (H3K4me3/2) to the monomethylated form (H3K4me1) (3–5). JARID1B belongs to the JARID1 protein family, which also includes JARID1A/RBP2/KDM5A, JARID1C/SMCX/KDM5C, and JARID1D/SMCY/KDM5D (6). All the JARID1 proteins possess H3K4 histone demethylase activity in vitro and in vivo (3–5, 7, 8). Because trimethylated H3K4 positively correlates with active transcription, H3K4 histone demethylases can act as transcriptional repressors to silence gene expression (6). However, these enzymes can also activate gene expression through several possible mechanisms. For example, JARID1C was reported to activate enhancers by removing tri- and dimethyl marks on H3K4 at enhancers (9).
JARID1B is highly expressed in human breast tumors as well as many breast cancer cell lines (10, 11). Consistent with these findings, JARID1B contributes to proliferation of MCF-7 and 4T1 breast cancer cells in vitro and in vivo (4, 12). In addition to its demethylase function, JARID1B can form a complex with HDAC4 (13) and LSD1/NuRD (14) to mediate transcriptional repression. Its known repressed target genes in breast cancer include BRCA1, CAV1, and CCL4 (4, 14). JARID1B is also overexpressed in cancers of the prostate, lung, and bladder (15, 16). More recently, JARID1B came into the spotlight for its association with a slow cycling cell population and drug resistance in melanoma (17, 18).
The roles of JARID1B in mouse development remain controversial. The first reported strain of Jarid1b−/− mice was embryonic lethal between E4.5 and E7.5 (12). In contrast, a strain of Jarid1b−/− mice generated using a different targeting construct showed neonatal lethality due to abnormal respiratory function as well as skeletal and neuronal development defects (19). These defects were linked to the accumulation of H3K4me3 and activation of neural master regulators like Pax6 and Otx2 (19). Another mouse strain that expresses JARID1B with ARID deletion (ΔARID) exhibited a largely normal phenotype with the exception of delayed mammary gland development (12).
We implemented another strategy to generate a different strain of Jarid1b−/− mice. These mice were mostly viable but smaller than their wild-type littermates. Female Jarid1b−/− mice also showed reduced fertility, lower uterine weight, and a delay in mammary development during puberty and early adulthood. These phenotypes were accompanied by a significant decrease in serum estrogen level. Furthermore, cells derived from Jarid1b−/− mammary glands ex vivo exhibited diminished expression of key regulators of mammary gland morphogenesis and luminal lineage specification along with increased expression of mammary stem cell signature. Mechanistically, we showed that JARID1B facilitated GATA3 recruitment to the promoter of genes involved in mammary development and activated their transcription. These findings revealed the critical roles of an epigenetic regulator in modulating the female reproductive system and maturation of the mammary epithelium during pubertal development.
EXPERIMENTAL PROCEDURES
Generation of Jarid1b−/− Mice
Jarid1b−/− mice were generated using embryonic stem (ES) cell RRO123 derived from 129/Ola mouse strain from the BayGenomics gene trap resource. Insertion of the gene trap cassette led to a truncated fusion mRNA and protein product. The ES cells were injected into the C57BL/6 blastocysts using standard techniques to generate the chimeric mice, which were mated with C57BL/6 mice to generate Jarid1b+/− mice. Jarid1b+/− animals were further backcrossed with wild-type C57BL/6 and FVB/N mice purchased from The Jackson Laboratory for more than 10 generations. All mice were maintained in the research animal facility of Yale University and Dana-Farber Cancer Institute in accordance with National Institutes of Health guidelines. All procedures involving animals were approved by the Institutional Animal Care and Use Committees of Yale University and Dana-Farber Cancer Institute.
Genotyping
Genomic DNA was obtained from tail biopsies digested with alkaline lysis buffer (0.7 mm EDTA, 17.5 mm NaOH) and neutralized with 40 mm Tris-HCl. The 611-bp PCR product for the wild-type allele was amplified with the primers mPLU1-17 (5′-AGATTGTGGCCTAATTACACTG-3′) and mPLU1-18R (5′-CATCAAGATTCTCTTGAACTGG-3′). The 350-bp gene trap allele was amplified by mPLU1-17 and Genetrap6 (5′-CAGGGTTTTCCCAGTCACGAC-3′). The trio of primers was used for the PCR amplification with the following conditions: 94 °C for 5 min, 40 cycles of 94 °C for 15 s, 55 °C for 15 s, and 72 °C for 30 s, followed by 72 °C for 5 min. PCR products were resolved on 2% agarose gels.
Isolation and Culture Condition of Mouse Embryonic Fibroblasts (MEFs) and Mammary Epithelial Cells (MECs)
MEFs were isolated from 12.5–14.5-day-postcoitum embryos using standard procedures and maintained in DMEM (CellGro) containing 10% FBS (Invitrogen). MECs were isolated using a protocol adapted from a previous publication (20). Briefly, the third, fourth, and fifth pair of mammary glands were isolated from Jarid1b+/+ and Jarid1b−/− female virgin mice between 6 and 11 weeks of age from the same litter. Finely minced glands were digested with collagenase IV (C5138, Sigma) in RPMI 1640 medium (Invitrogen) with 5% FBS, 10 mm Hepes, and 1% penicillin/streptomycin (Invitrogen). Organoids were pelleted through differential centrifugation, and single cell suspensions were obtained by dissociating the organoids in 0.25% trypsin, EDTA (Invitrogen). The single cells were further treated with 100 μg/ml DNase I (D4263, Sigma) and filtered through a 70-μm cell strainer prior to being snap frozen or cultured in MEC growth medium (DMEM:Ham's F-12 medium (1:1) (Invitrogen) with 10% FBS, 1% penicillin/streptomycin, 5 μg/ml insulin (Invitrogen), 50 μg/ml gentamicin (Sigma), 1 μg/ml hydrocortisone (Sigma), and 10 ng/ml mouse epidermal growth factor (EGF) (AF-100-15, Peprotech)). Immortalized MECs were generated using SV40 large T antigen as described previously (21) and cultured in MEC growth medium with 1 μg/ml puromycin.
RNA Purification and Reverse Transcription and Quantitative PCR (RT-qPCR)
Total RNA was extracted from cultured mouse cells with the RNeasy Plus Mini kit (74134, Qiagen). cDNA was obtained using the High Capacity cDNA Reverse Transcription kit (4368814, Applied Biosystems) according to the manufacturer's instruction. Quantitative PCR was performed with a CFX-96 (Bio-Rad) using RT2 SYBR Green (330500, SA Biosciences) or Fast SYBR Green Master Mix (43856414, Applied Biosystems) according to the manufacturers' instructions. All values were normalized to the level of Gapdh abundance. Data are the average of triplicate experiments ±S.E. All primers used are described in supplemental Table 1.
Western Blotting
Mouse cells were collected following digestion with 0.25% trypsin, EDTA and lysed on ice with high salt lysis buffer (50 mm Tris-HCl (pH 7.9), 0.1 mm EDTA (pH 8.0), 320 mm NaCl, 0.5% Nonidet P-40, 10% glycerol) with 1× protease inhibitor mixture (11873580001, Roche Applied Science). Total protein extract was resolved on a 6% polyacrylamide gel and blotted with antibodies specific for JARID1A (mAB3876, Cell Signaling Technology), JARID1B (A301-813A, Bethyl Laboratories for Fig. 1D; HPA027179, Sigma for Fig. 1E; and DAIN, kindly provided by Dr. Kristian Helin (22) for Fig. 8E), GATA3 (558686, BD Pharmingen), and vinculin (V9131, Sigma).
FIGURE 1.

JARID1B expression is disrupted in Jarid1b−/− mouse. A, schematic showing that the JARID1B gene trap cassette was inserted between exons 2 and 3 of the endogenous Jarid1b locus. The location of the insertion site in the protein is depicted below. SA, splice acceptor; pA, polyadenylation sequence; PHD, plant homeodomain; β-geo, β-galactosidase and neomycin phosphotransferase. B, PCR genotyping of Jarid1b+/+, Jarid1b+/−, and Jarid1b−/− mice. The PCR product for the wild-type allele is 611 bp and for the knock-out allele is 350 bp. C, RT-qPCR analysis of Jarid1b using primers corresponding to the indicated exons. Error bars represent S.E. D, Western blot analysis of Jarid1b+/+, Jarid1b+/−, and Jarid1b−/− MEFs using the indicated antibodies. A nonspecific band is observed at 150 kDa (*). E, Western blot analysis of Jarid1b+/+ and Jarid1b−/− MECs using the indicated antibodies.
FIGURE 8.

JARID1B activates Foxa1 gene expression by recruiting GATA3 to the Foxa1 promoter. A, schematic of the Foxa1 promoter and potential binding sites of JARID1B (green) and GATA3 (blue) identified by their known DNA binding motif. Position and coverage of ChIP primers used are shown in red. B and C, ChIP quantification of JARID1B (B) and GATA3 (C) enrichment in immortalized Jarid1b+/+ and Jarid1b−/− MECs. PCR primers were designed to identify genomic regions −1.5, −0.5, 0.3, and 9.8 kb relative to the Foxa1 TSS. D, RT-qPCR analysis of Jarid1b, Foxa1, and Gata3 in immortalized Jarid1b+/+ and Jarid1b−/− MECs. E, Western blot analysis of immortalized Jarid1b+/+ and Jarid1b−/− MECs using the indicated antibodies. F, G, and H, ChIP analysis of Pol II (F), H3K4me3 (G), and H3K4me1 (H) levels at the indicated genomic regions relative to the Foxa1 TSS. Error bars represent S.E.
Fertility Test
For females, virgin Jarid1b+/+ and Jarid1b−/− female littermates between 6 and 8 weeks, 9 and 12 weeks, and 3 and 8 months were placed in the same cages with wild-type males. After 14 days, females were separated into different cages and monitored for delivery dates and number of pups. For males, virgin Jarid1b+/+ and Jarid1b−/− male littermates between 6 and 8 weeks, 9 and 12 weeks, and 3 and 8 months were housed with wild-type females in separate cages for 14 days. Females were monitored after separation, and delivery dates were recorded.
Mammary Gland Whole Mounts
The fourth pair of mammary glands were isolated from female virgin Jarid1b+/+ and Jarid1b−/− mice, fixed in acidic ethanol (75% ethanol, 25% acetic acid), and stained overnight with carmine solution (0.2% carmine, 0.5% aluminum potassium sulfate). Dehydration with graded ethanol and defatting with acetone were subsequently performed and followed by overnight clearing in xylene. The processed glands were mounted with Cytoseal 60 (Thermo Scientific) and air-dried before images were taken.
Serum Estrogen Level Test
Immediately following euthanasia, 1 ml of blood was collected via cardiac puncture into MiniCollect Serum Separator Tubes (22-030-401, ThermoFisher). Samples were spun down for 1 min at 12,000 rpm in an Eppendorf 5424 centrifuge and then transferred to new tubes. Serum was subsequently processed with the Mouse/Rat Estradiol ELISA kit (ES180S-100, Calbiotech) according to the manufacturer's protocol. Estradiol levels were analyzed in 96-well format with a Biotek Synergy MX plate reader.
Histological Analysis
The fourth pair of mammary glands were dissected from 7.5-week-old female virgin Jarid1b+/+ and Jarid1b−/− mice and fixed overnight in 10% neutral buffered formalin. Samples were then submitted to Yale Pathology Tissue Services for embedding and hematoxylin and eosin (H&E), Ki67, and TUNEL staining using standard protocols.
Fluorescence-activated Cell Sorting (FACS)
Fresh MECs were isolated and processed into single cell suspensions, which were subsequently blocked in PBS containing 0.5% BSA and 2 mm EDTA and stained at 4 °C for 15 min with the following antibodies: CD31-biotin (558737, BD Pharmingen), CD45-biotin (553077, BD Pharmingen), CD29-allophycocyanin (102206, Biolegend), CD24-FITC (101814, Biolegend), and CD61-phycoerythrin (553347, BD Pharmingen), and streptavidin-Alexa Fluor 750 (Invitrogen). DAPI (Invitrogen) was used to eliminate dead cells from the analysis. Cells were analyzed using a BD LSRFortessa cell analyzer.
Gene Expression Profiling and Gene Set Enrichment Analysis (GSEA)
Total RNA samples were isolated from three pairs of 80% confluent Jarid1b+/+ and Jarid1b−/− early passage MECs using an RNeasy Plus Mini kit (74134, Qiagen). Gene expression profiling was performed using a MouseWG-6 v2.0 Beadchip kit (BD-201-0202, Illumina) by the Yale Center for Genome Analysis. Non-normalized gene expression profiling data exported from Illumina Genome Studio were processed using the Lumi R package. The differentially expressed gene list from pairwise comparison based on littermates was generated using the Limma R package. Using a 1.5-fold difference as the cutoff, there were 64 up-regulated and 54 down-regulated genes. The raw data were deposited in the NCBI Gene Expression Omnibus database under accession number GSE50479. Gene set enrichment analysis was done with GSEA v2.0 software. Gene sets were generated from published gene signatures as referenced. Statistical significance was assessed by comparing the enrichment score with enrichment results generated from 10,000 random permutations of the gene set.
Identification of Transcription Factor Binding Sites with Bioinformatics Tools
The Foxa1 promoter was analyzed for JARID1B binding sites using MatInspector software (Genomatix) (23) and for GATA3 binding sites using Transfac Match 1.0 software (BIOBASE) (24).
Chromatin Immunoprecipitation (ChIP)
Cells were grown to 90% confluence in 150-mm tissue culture dishes. All subsequent cross-linking and immunoprecipitation experiments were carried out as described previously (25, 26). Briefly, 37% formaldehyde was directly added to medium to 10% final concentration and swirled for 10 min followed by quenching with 0.125 m glycine. Cells were harvested on ice and stored in −80 °C overnight in ChIP Lysis Buffer 1 (50 mm Hepes-KOH (pH 7.5), 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% Nonidet P-40, 0.25% Triton X-100). On the following day, cell lysates were thawed and centrifuged for 10 min at 3,000 rpm in an Eppendorf 5810R centrifuge at 4 °C. Cell pellets were subsequently resuspended in ChIP Lysis Buffer 2 (200 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 10 mm Tris-HCl (pH 8.0)), spun down, and resuspended in Lysis Buffer 3 (1 mm EDTA, 0.5 mm EGTA, 10 mm Tris-HCl (pH 8.0), 100 mm NaCl, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine). Cell lysates were sonicated with a 15-s burst and 30 s between pulses for a total of 15 min on ice using a Bioruptor UCD-200 (B01010002, Diagenode) to obtain chromatin fragments from 0.5 to 1 kilobase (kb). A 1:10 volume of 10% Triton X-100 was added to each lysate, and 50 μl was set aside as input. Prewashed and antibody-bound Dynabeads magnetic beads (10003D, Invitrogen) were added to each ChIP lysate and incubated overnight at 4 °C. The antibodies used were JARID1B (ab50958-100, Abcam), GATA3 (sc-268, Santa Cruz Biotechnology), polymerase II (Pol II) (MMS-126R, Covance), H3K4me1 (ab8895-100, Abcam), and H3K4me3 (ab8580-100, Abcam). For each ChIP lysate, 2 μg of antibody was used. ChIP lysates were washed eight times with radioimmune precipitation assay buffer (50 mm Hepes-KOH (pH 7.5), 1 mm EDTA, 0.7% sodium deoxycholate, 1% Nonidet P-40, 0.5 m LiCl) and once with TE buffer with 50 mm NaCl prior to being eluted and reverse cross-linked in elution buffer (50 mm Tris-HCl (pH 8.0), 10 mm EDTA, 1% SDS) at 65 °C overnight. On the next day, eluted chromatin was treated with 0.2 μg/μl RNase A for 1 h at 37 °C and 0.2 μg/μl proteinase K for 2 h at 55 °C followed by DNA purification with the QIAquick PCR Purification kit (28104, Qiagen). Chromatin enrichment was analyzed by quantitative PCR with a CFX-96 (Bio-Rad). Data are the average of triplicate experiments ±S.E. All ChIP primers used are described in supplemental Table 1.
Statistical Analysis
All data were assessed with two-tailed Student's t test with the exception of the Mendelian genetics ratio, which was analyzed by the χ2 test, and the Kaplan-Meier survival curve, which was analyzed by log rank (Mantel-Cox) test. Results with p values <0.05 were considered significant.
RESULTS
Loss of Jarid1b Leads to Reduced Body Weight and Higher Mortality Rate
To determine the in vivo function of JARID1B, we generated a new Jarid1b−/− mouse strain using a gene trap ES cell line. In this ES cell line, a β-geo (β-galactosidase and neomycin phosphotransferase)-containing gene trap cassette was inserted between exons 2 and 3 of the Jarid1b gene, which led to premature termination before the ARID (Fig. 1A). Wild-type (Jarid1b+/+), heterozygous (Jarid1b+/−), and knock-out (Jarid1b−/−) animals can be readily distinguished by allele-specific PCR (Fig. 1B). In Jarid1b−/− MEFs, JARID1B mRNA could be detected with primers 5′ to the insertion site but not with primers encompassing or 3′ to the insertion site (Fig. 1C). Consistent with these findings, Western blot analysis using an antibody against the C terminus of JARID1B showed that the expression level of the full-length JARID1B protein was markedly reduced in Jarid1b+/− MEFs and undetectable in Jarid1b−/− MEFs (Fig. 1D). Similarly, Western blot analysis using a different antibody against the C terminus of JARID1B showed that full-length JARID1B protein was also undetectable in Jarid1b−/− MECs (Fig. 1E). Interestingly, the expression of JARID1A protein increased in Jarid1b−/− MECs. These results indicate that deletion of Jarid1b using this strategy results in the loss of full-length JARID1B mRNA and protein.
In the mixed genetic background, Jarid1b−/− mice were viable and appeared grossly normal. These animals were born largely at the expected Mendelian ratio (Table 1), demonstrating a dispensable role for JARID1B in embryonic development. Both male and female Jarid1b−/− mice weighed less than their Jarid1b+/+ littermates, and this difference persisted from preweaning age through early adulthood (Fig. 2, A and B). In contrast, Jarid1b+/− mice weighed similarly to their Jarid1b+/+ littermates (data not shown).
TABLE 1.
The number of Jarid1b+/+, Jarid1b+/−, and Jarid1b−/− newborn mice in the indicated genetic background
| Genetic background | +/+ |
+/− |
−/− |
Total | p value | |||
|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |||
| Mixed | 53 | 26.6 | 97 | 48.7 | 49 | 24.6 | 199 | 0.867 |
| FVB/N | 61 | 31.9 | 100 | 52.4 | 30 | 15.7 | 191 | 0.005 |
| C57BL/6 | 42 | 28.4 | 75 | 50.7 | 31 | 20.9 | 148 | 0.436 |
| Total | 156 | 29.0 | 272 | 50.6 | 110 | 20.4 | 538 | 0.019 |
FIGURE 2.

Jarid1b−/− mice exhibit reduced body weight and higher incidence of adult mortality. A and B, weight of male (A) and female (B) Jarid1b+/+ and Jarid1b−/− mice of the indicated age. For males, n = 24 for Jarid1b+/+ and n = 23 for Jarid1b−/−; for females, n = 28 for Jarid1b+/+ and n = 40 for Jarid1b−/−. D, days; M, months. Error bars represent S.E. C and D, Kaplan-Meier survival curve of male (C) and female (D) Jarid1b+/+ and Jarid1b−/− animals. For males, n = 44 for Jarid1b+/+ and n = 58 for Jarid1b−/−; for females, n = 43 for Jarid1b+/+ and n = 67 for Jarid1b−/−. *, p < 0.05; **, p < 0.01.
The early lethality phenotypes reported in other Jarid1b−/− strains (12, 19) suggest that genetic background could contribute to the differences in phenotypes. We then backcrossed these animals to both FVB/N and C57BL/6 backgrounds. Neonatal lethality was observed in FVB/N background but not in C57BL/6 background (Table 1). In both FVB/N and mixed genetic backgrounds, both male and female Jarid1b−/− animals displayed a higher mortality rate than their Jarid1b+/+ and Jarid1b+/− littermates due to unknown causes (Fig. 2, C and D, and data not shown). No obvious organ defects were observed by necropsy at the time of death (data not shown), and the affected animals appeared otherwise normal prior to the onset of death. More detailed analyses will be required to determine the subtle changes that led to the death of these animals.
Loss of Jarid1b Causes Reduced Female Fertility and Delayed Mammary Gland Development
In FVB/N background, Jarid1b−/− males were able to mate and father offspring at a slightly lower efficiency than Jarid1b+/+ males (Table 2). Interestingly, Jarid1b−/− females exhibited a significantly reduced level of fertility compared with their Jarid1b+/+ littermates (Table 2). Jarid1b−/− females showed lower a pregnancy rate when mated with wild-type males from the time they reach sexual maturity at 6–8 weeks through 9–12 weeks. Remarkably, Jarid1b−/− females older than 3 months were infertile for the limited number of mice we examined. These results suggest that JARID1B contributes to the development and proper function of the female reproductive system.
TABLE 2.
Jarid1b−/− females in FVB/N background show lower fertility rate
| Genotype | No. pregnant/no. mated |
% Pregnancy |
||||
|---|---|---|---|---|---|---|
| 6–8 weeks | 9–12 weeks | 3–8 months | 6–8 weeks | 9–12 weeks | 3–8 months | |
| Male | ||||||
| +/+ | 8/8 | 5/5 | 4/4 | 100 | 100 | 100 |
| −/− | 6/9 | 5/5 | 3/4 | 67 | 100 | 75 |
| Female | ||||||
| +/+ | 4/4 | 4/5 | 4/5 | 100 | 80 | 80 |
| −/− | 2/5 | 3/5 | 0/4 | 40 | 60 | 0 |
Whole mount preparation of the fourth mammary glands from young Jarid1b−/− female mice between 5 and 6 weeks of age revealed a dramatic reduction in the number of terminal end buds (TEBs), secondary branching, and outgrowth length compared with their Jarid1b+/+ and Jarid1b+/− littermates (Fig. 3, A–D, and data not shown). These differences became less pronounced at 7–9 weeks and eventually disappeared in females older than 3 months of age. At 3 months of age, pubertal ductal outgrowth is complete, and TEBs were replaced by extensive side branches and ductal ends in both Jarid1b+/+ and Jarid1b−/− females. Despite the histological difference observed in early puberty, virtually no difference was observed between Jarid1b+/+ and Jarid1b−/− female littermates in the weight of their mammary fat pads normalized to the animal body weight (Fig. 3E).
FIGURE 3.

JARID1B is involved in the development of the mouse mammary gland. A, whole mount preparation of mouse mammary gland from female Jarid1b+/+ and Jarid1b−/− littermates in FVB/N background of the indicated age. Scale bar, 5 mm. The 3 × 3-mm boxed images at increased magnification are also shown at the corners. B, C, and D, quantification of the number of TEBs or ductal ends (in mice older than 8 weeks) (B), side branches (C), and outgrowth length (D) for virgin mice of the indicated age. TEBs, ductal ends, and side branches were counted with ImageJ in comparable areas of 3 × 3-mm squares for at least five pairs of female littermates per age group. Outgrowth lengths were measured from the center of the lymph node to the farthest reaching point of the visible epithelium by ImageJ. For 5–6 weeks, n = 9 for each genotype; for 7–9 weeks, n = 7; for ≥3 months, n = 5. Error bars represent S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, weight of Jarid1b+/+ and Jarid1b−/− mammary glands normalized to body weights. MG, mammary gland. wks, weeks.
Although most Jarid1b−/− females were infertile, it is worth noting that for those that became pregnant and subsequently delivered, their mammary glands were otherwise functional for nursing. Their lobuloalveolar structure formation proceeded normally in preparation for lactation, and involution took place without apparent defects postweaning (data not shown). These results suggest that JARID1B specifically promotes the rapid growth of epithelial cells that give rise to the ductal structure in the mammary fat pad during early puberty but not in alveolar development during pregnancy.
Loss of Jarid1b Leads to Reduced Serum Estrogen Level and Decreased Proliferation of Mammary Epithelial Cells
To investigate the mechanisms behind reduced female fertility and delayed mammary gland development, we started by examining the estrogen level. Serum 17β-estradiol levels were remarkably lower in Jarid1b−/− female mice than those in their Jarid1b+/+ female littermates, especially prior to 6 weeks of age (Fig. 4A). We also examined the uterus because it is an organ directly regulated by estrogen and can serve as a surrogate readout for serum estrogen level. Consistent with our serum testing results, Jarid1b−/− females had significantly lower uterine weight at 5–6 weeks but not after 7 weeks of age (Fig. 4B), which correlates with the reduced ductal growth observed in Jarid1b−/− animals at similar time points.
FIGURE 4.
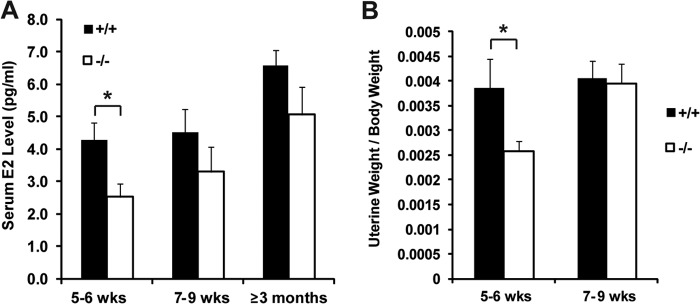
JARID1B is required for normal serum estrogen level. A, serum 17β-estradiol levels of Jarid1b+/+ and Jarid1b−/− females of the indicated age. E2, 17β-estradiol. For 5–6 weeks, n = 6 for each genotype; for 7–9 weeks, n = 5; for ≥3 months, n = 3. Error bars represent S.E. B, normalized uterine weights of Jarid1b+/+ and Jarid1b−/− animals of the indicated age. For 5–6 weeks, n = 8; for 7–9 weeks, n = 6. Error bars represent S.E. *, p < 0.05. wks, weeks.
To further investigate the in vivo mechanisms behind the delayed ductal morphogenesis of Jarid1b−/− mammary glands, we conducted histological analysis of mammary glands. H&E staining showed a normal distribution of ducts and end buds in the mammary glands of 5-week-old Jarid1b−/− mice (Fig. 5A). Preserved epithelial-basal organization indicates that Jarid1b−/− females have normal intraductal morphology despite delayed TEB formation and side branching. However, a lower percentage of cells stained positive for the proliferation marker Ki67 in Jarid1b−/− TEBs (Fig. 5, A and B). This suggests that the delay in ductal branching and outgrowth is caused by the lower percentage of proliferating epithelial cells in the Jarid1b−/− mammary glands compared with Jarid1b+/+ glands during early pubertal stages. In contrast, very few cells stained positive by TUNEL staining in either Jarid1b+/+ or Jarid1b−/− mammary gland, and no difference was observed in the percentage of apoptotic cells (Fig. 5, A and C). Therefore, altered proliferation but not apoptosis contributed to the phenotype observed in the Jarid1b−/− mammary gland.
FIGURE 5.

Normal histology and apoptosis in Jarid1b−/− mammary glands are accompanied by increased cell proliferation. A, representative H&E, Ki67, and TUNEL images of Jarid1b+/+ and Jarid1b−/− mammary glands. Scale bar, 50 μm. B and C, quantification of Ki67+ (B) and TUNEL+ (C) cells. At least three pairs of female littermates were used per group, and three independent TEBs or proliferative ends were selected from different fields of view per mammary gland for quantification. Positively stained cells were counted by ImageJ. Error bars represent S.E. *, p < 0.05. N.S., not significant.
JARID1B Is Critical for Expression of Key Genes Involved in Mammary Gland Development
To further identify genes regulated by JARID1B in mammary gland development, microarray analysis was performed using three pairs of Jarid1b+/+ and Jarid1b−/− primary MECs. Using a cutoff of 1.5-fold difference, 64 genes were up-regulated in Jarid1b−/− MECs, whereas another 54 genes were down-regulated (supplemental Table 2). We validated the expression changes of a number of differentially expressed genes using RT-qPCR (Fig. 6, A and B). Among the validated down-regulated genes in Jarid1b−/− MECs, several are known key regulators of pubertal mammary gland development, such as Foxa1, Wnt7b, Fgfr2, and Prlr. In particular, FOXA1 acts upstream of ERα in TEB formation and ductal invasion and plays an indispensable role in ERα-dependent transcription (27, 28). RT-qPCR experiments showed that expression of other previously identified mammary gland morphogenesis regulators, such as Elf5, Erbb2, Erbb3, Pgr, Stat5a, Tgfb1, and Esr1, were also significantly reduced upon JARID1B loss (Fig. 6C). Although lower systemic estrogen levels previously mentioned may have contributed to the delayed epithelial outgrowth in an endocrine fashion, persistent gene expression changes in mammary epithelial cells ex vivo reveal a cell-autonomous role of JARID1B. In fact, many well characterized genes in previously defined estrogen response signatures, such as Greb1, Xbp1, and Fos, all remained down-regulated in Jarid1b−/− MECs even after they were purified and cultured briefly in vitro (Fig. 6D).
FIGURE 6.

Key mammary development regulators are down-regulated in Jarid1b−/− mammary epithelial cells. A and B, validation of genes up-regulated (A) and down-regulated (B) in Jarid1b−/− MECs compared with Jarid1b+/+MECs. C, RT-qPCR analysis of known regulators of mammary gland morphogenesis. D, RT-qPCR analysis of selected estrogen-responsive genes. Error bars represent S.E.
JARID1B Loss Switches Transcription Program from Luminal Lineage to Stem Cell Lineage
To determine whether JARID1B controls mammary lineage-specific transcription programs, we conducted GSEA (29) using a set of gene signatures previously described for the mammary stem cell (MaSC)-enriched, luminal progenitor, mature luminal, and stromal subpopulations derived from mouse mammary glands (30). GSEA revealed strong positive correlations between JARID1B loss with genes down-regulated in the mature luminal cells and genes up-regulated in the MaSC-enriched population, whereas JARID1B loss correlated negatively with up-regulated genes in luminal cells as well as down-regulated genes in MaSC-enriched cells (Fig. 7A). This analysis suggests that JARID1B promotes gene expression changes toward the luminal cell lineage and represses stem cell-associated genes during mammary development. Several aforementioned key mammary development regulators, such as Foxa1 and Wnt7b, were also among the down-regulated luminal signatures.
FIGURE 7.

JARID1B loss leads to decreased luminal gene signature and enriched stem cell signature without major changes in cell population. A, GSEA of Jarid1b+/+ and Jarid1b−/− MECs using gene sets differentially expressed in mature luminal cells (top two panels) and mammary stem cell-enriched populations (bottom two panels). NES, normalized enrichment score; FDR, false discovery rate; DOWN, down-regulated; UP, up-regulated. B, flow cytometry analysis of representative Jarid1b+/+ and Jarid1b−/− MECs isolated from females at 7.5 weeks of age. The dot plot shows mature luminal (CD29loCD24+CD61−), MaSC-enriched (CD29hiCD24loCD61+), and luminal progenitor cells (CD29loCD24+CD61+). All cells shown are CD31−CD45−CD24+. C, quantification of each subpopulation in Jarid1b+/+/Jarid1b+/− and Jarid1b−/− MECs. For the combined Jarid1b+/+ and Jarid1b+/− group, n = 3 for Jarid1b+/+ and n = 2 for Jarid1b+/−; for Jarid1b−/−, n = 5. Error bars represent S.E.
To further determine the roles of JARID1B in lineage specification, we asked whether JARID1B loss is sufficient to switch the lineages of mammary epithelial cell subpopulations. To this end, we performed FACS analysis of MECs. Interestingly, compared with the Jarid1b+/+ and Jarid1b+/− MECs isolated from littermates, Jarid1b−/− MECs did not show significant changes of the mature luminal population, which is defined as CD29loCD24+CD61−. Similarly, the MaSC-enriched and luminal progenitor populations also did not change significantly upon JARID1B loss (Fig. 7, B and C). Therefore, the switch of transcription program from the luminal lineage to stem cell lineage in Jarid1b−/− MECs was insufficient to convert the cell identity of corresponding subpopulations; however, it may contribute to delayed mammary gland development.
JARID1B Is Critical for GATA3 Recruitment to Activate Foxa1 Expression
The significant down-regulation of the luminal transcription factor FOXA1 upon JARID1B loss raises the question of whether JARID1B can directly bind to the Foxa1 promoter to activate its expression. Based on a previously reported JARID1B binding motif (31), a predicted JARID1B binding site was identified at 0.4 kb upstream of the Foxa1 transcriptional start site (TSS) (Fig. 8A). Consistent with our bioinformatics prediction, ChIP analysis of immortalized MECs showed that JARID1B enrichment peaked near 0.5 kb upstream of Foxa1 TSS in wild-type MECs but not at an intergenic region about 9.8 kb downstream of the Foxa1 TSS (Fig. 8B).
Consistent with a previous study showing that the master luminal lineage regulator GATA3 binds the Foxa1 promoter to enhance Foxa1 expression in mammary glands (32), we identified several potential GATA3 binding sites near 1.6 kb and another at 0.77 kb upstream of Foxa1 TSS based on the known GATA3 DNA binding motif (Fig. 8A). ChIP experiments confirmed the enrichment of GATA3 at 1.5 and 0.5 kb upstream but not downstream of Foxa1 TSS in Jarid1b+/+ MECs (Fig. 8C). In contrast, GATA3 binding at the Foxa1 promoter was not observed in Jarid1b−/− MECs (Fig. 8C), suggesting that JARID1B is required for the recruitment of GATA3 to the Foxa1 promoter. These changes were not simply due to a decreased expression level of Gata3 in immortalized Jarid1b−/− MECs because its mRNA and protein levels were comparable between immortalized Jarid1b+/+ and Jarid1b−/− MECs (Fig. 8, D and E).
In correlation with dampened Foxa1 expression (Figs. 6B and 8C), the recruitment of RNA Pol II to the Foxa1 TSS decreased dramatically in Jarid1b−/− MECs (Fig. 8F). As with Pol II recruitment, H3K4me3 enrichment at the Foxa1 TSS was also reduced in Jarid1b−/− MECs (Fig. 8G). In contrast, H3K4me1 levels near the Foxa1 TSS were unaffected by JARID1B loss (Fig. 8H). Therefore, at the Foxa1 promoter, JARID1B unlikely functions as an H3K4 demethylase but is rather involved in recruiting GATA3 and Pol II and maintaining high H3K4me3 levels for active transcription.
JARID1B Promotes Mammary Development Gene Expression through Diverse Mechanisms
To investigate whether other mammary development genes down-regulated in Jarid1b−/− MECs were regulated by JARID1B via a similar mechanism, we examined the levels of JARID1B, GATA3, and H3K4me3 at the promoters of Prlr, Wnt7b, Fgfr2, Elf5, Erbb3, Tgfb1, and Stat5a. As with Foxa1, the levels of H3K4me3 decreased significantly at all these promoters (Fig. 9A). However, JARID1B was only enriched at the promoters of Prlr, Fgfr2, and Stat5a (Fig. 9B). The lack of JARID1B binding to Wnt7b, Elf5, Erbb3, and Tgfb1 suggests that JARID1B indirectly affects their gene expression. Among this panel of genes, only Stat5a exhibited GATA3 enrichment at its promoter (Fig. 9C). These results demonstrate that JARID1B can activate Foxa1 and Stat5a expression by binding directly to their promoters and facilitating recruitment of the master luminal regulator GATA3. Conversely, JARID1B can also directly regulate the expression of a subset of mammotropic factors through GATA3-independent mechanisms, as in the case of Prlr and Fgfr2, likely by recruiting other transcriptional regulators.
FIGURE 9.
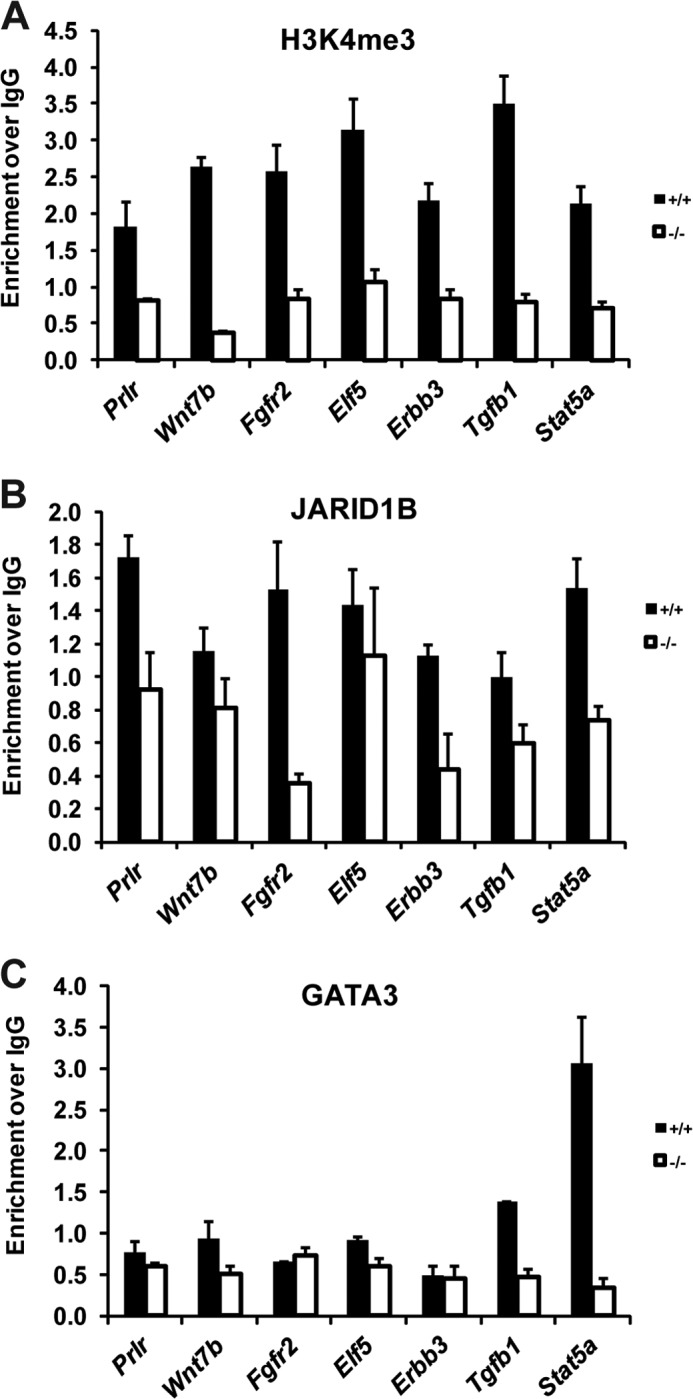
JARID1B directly regulates mammary development genes through both GATA3-dependent and -independent mechanisms. A, B, and C, ChIP analysis of H3K4me3 (A), JARID1B (B), and GATA3 (C) enrichment at the promoter regions of a panel of previously described mammary development genes. PCR primers were designed to identify genomic regions ranging from −2 to 0.3 kb relative to the TSS of each gene. Exact primer locations are noted in supplemental Table 1. Error bars represent S.E.
Collectively, our findings suggest that JARID1B facilitates pubertal mammary ductal outgrowth by promoting the systemic estrogen level and the expression of key mammary development regulators, such as FOXA1. By recruiting the master luminal regulator GATA3 to the Foxa1 promoter, JARID1B maintains proper levels of ERα-dependent transcription in the mammary gland and ensures timely lineage differentiation of epithelial cells (Fig. 10).
FIGURE 10.
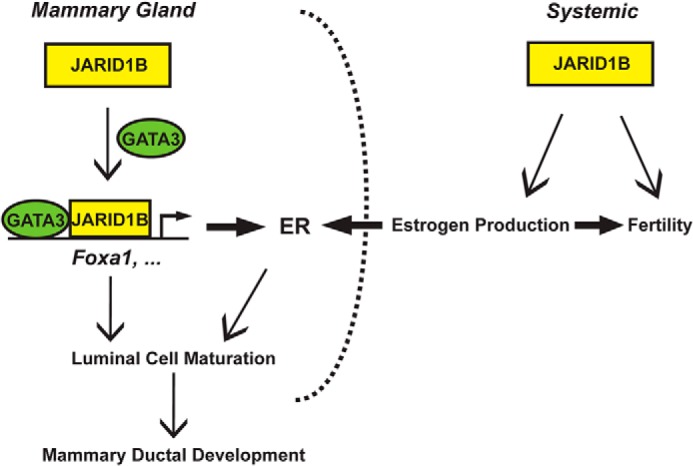
Proposed model for JARID1B regulation of mammary gland development and female fertility through both endocrine and cell-autonomous mechanisms. By facilitating GATA3 recruitment to its target promoters, JARID1B transcriptionally activates Foxa1 and other mammary development regulators to facilitate the differentiation of luminal epithelial cells toward ductal development.
DISCUSSION
Pubertal development of the mouse mammary gland reflects both endocrine actions of various sex and growth hormones in the body and cell-intrinsic properties of mammary epithelial cells (33, 34). Using a systemic knock-out mouse for histone demethylase JARID1B, we identified a role for this epigenetic regulator specific to the mammary gland and several components of the female reproductive system. In the absence of JARID1B, female animals exhibited delays in mammary gland development and a decreased fertility rate. We found reduced estrogen levels in circulating blood, a smaller population of proliferating cells, and an altered transcription program in the mammary epithelium of Jarid1b−/− females.
The mammary epithelium undergoes estrogen-independent preliminary growth at the embryonic and neonatal stages followed by a well characterized steroid hormone-dependent pubertal stage (35). As the primary receptor for estrogen, ERα plays a quintessential role in the developing mammary gland as demonstrated by ERα knock-out mouse models (36). FOXA1 is a transcription factor that functions upstream of ERα and is essential to a subset of ERα-dependent transcription by modulating ERα binding to many of its target genes. In fact, FOXA1 was shown to be indispensable in long range chromatin remodeling and ERα access of target genes, such as cyclin D1 (37). Furthermore, FOXA1 function in the mammary gland is critical for ductal invasion during puberty but not essential for alveologenesis in pregnancy and lactation (28), which is remarkably similar to the phenotype observed in our Jarid1b−/− animals. The persistent down-regulation of FOXA1 in the Jarid1b−/− mammary epithelium uncovered by our study also illustrates that a transcription factor with unique chromatin remodeling functions can be directly modulated by another epigenetic regulator.
The luminal master regulator GATA3 acts upstream of FOXA1 by directly regulating its expression in the mammary epithelium (32) and by modulating ERα binding (38). Conditional deletion of GATA3 in the mammary gland abolishes TEB formation during puberty, whereas acute loss of GATA3 leads to undifferentiated luminal cell expansion followed by apoptosis (32). In this study, we demonstrated that JARID1B is critical for the recruitment of GATA3 to the Foxa1 promoter and its transcriptional activation. As an H3K4me3/2 demethylase, JARID1B binding at the gene promoters can lead to decreased gene expression (19, 39), but its recruitment to the intragenic regions (40) or enhancers (41) was shown to activate gene expression. In mammary epithelial cells, JARID1B loss induced a decrease rather than an increase in H3K4me3 levels at many of its target promoters. Therefore, these target genes are likely regulated through histone demethylase-independent mechanisms by JARID1B. In fact, we showed that JARID1B acts as a transcriptional activator by recruiting GATA3 to its target genes, such as Foxa1 and Stat5a.
ERα, FOXA1, and GATA3 are all factors leading to luminal epithelial differentiation as well as markers for luminal subtypes of breast cancer (28, 32, 42, 43). These factors and many other luminal genes showed various levels of decrease in Jarid1b−/− MECs as demonstrated by GSEA, revealing the role of JARID1B in the maintenance of the luminal cell identity. Surprisingly, the depletion of the luminal signature and concurrent enrichment of the MaSC signature upon JARID1B loss are insufficient to perturb the corresponding epithelial cell populations characterized by FACS analysis of cell surface markers. Therefore, the overall delay in ductal invasion observed by histology cannot be simply attributed to the decrease of certain epithelial cell populations in the mammary epithelium. It is likely that mammary development delay was caused by changes of the proliferative property in subpopulations following a transcriptional switch. At the same time, this finding highlights the essence of luminal lineage genes, rather than stem cell-associated genes, in promoting the branching and outgrowth of pubertal mammary ducts.
In previously generated Jarid1b−/− mouse models, embryonic lethality and severe neonatal defects hindered the study of JARID1B functions in the mammary gland and other adult tissues (12, 19). The reduced number of viable Jarid1b+/− mice reported by Catchpole et al. (12) suggests that the embryonic lethality they observed in association with disruption of the Jarid1b locus might be due to the neomycin cassette carried by the ES clones that were used to generate the line. A different knock-out strain largely survives embryonic development but rather presents severe mortality in Jarid1b−/− newborns in the C57BL/6 background (19). In contrast, we were able to generate Jarid1b−/− mice that are largely viable, which allowed for the study of developmental phenotypes in adults. The less severe phenotypes of our Jarid1b−/− mice may be related to the difference in mouse genetic background and colony maintenance. It is worth noting that although we did not observe prevalent neonatal lethality in the C57BL/6 background as described by Albert et al. (19) we did record a lower than expected number of Jarid1b−/− pups born in the FVB/N background (Table 1) as well as a disproportionally high rate of preweaning mortality of Jarid1b−/− newborns (data not shown).
The delayed mammary gland development defect in our Jarid1b−/− mice is similar to the phenotype of a different mouse strain that carries an ARID deletion of JARID1B (12). Both studies highlight the importance of the DNA-binding ARID for the specific function of JARID1B in the mammary glands. Using purified primary MECs, we were able to further attribute the phenotype to the down-regulation of ERα, FOXA1, and many other regulators of mammary morphogenesis. Despite displaying reduced size and body weight in concordance with many previous reports on mouse knock-out models (44), our Jarid1b−/− mice had mammary fat pads comparable by mass with Jarid1b+/+ animals when normalized to body weights. The aforementioned delay in the mammary ductal growth, but not in the mammary fat pad as a whole, likely suggests that JARID1B functions in the epithelium rather than the stroma. In contrast, other signaling factors like EGF receptor have vital functions in the stroma while likely being redundant in the epithelium (45).
Our studies also showed that Jarid1b−/− females have a significantly reduced fertility rate and thus provided a new animal model of female infertility. Although many female infertility genes have been discovered, JARID1B is the first known histone demethylase that affects female fertility. The ovary is the primary site of estrogen production and is also under the constant feedback and regulation of estrogen along with the uterus. It was reported that JARID1B is expressed at moderate levels in both mouse ovary and uterus (46). The lower uterine weight observed in our Jarid1b−/− animals correlates well with decreased serum estrogen levels; however, it may just be one of many factors that contributed to their reduced fertility. More detailed studies of the ovary and uterus are needed to characterize the full spectrum of changes in the female reproductive tract when JARID1B is lost.
All members of the JARID1 protein family can exert profound influences on their target gene transcription. Interestingly, both of our Jarid1a and Jarid1b knock-out mouse strains are viable, suggesting that the loss of one family member may be functionally compensated by other family members. We indeed observed elevated JARID1A protein level in Jarid1b−/− MECs (Fig. 1E). Similarly, increased expression of Jarid1b and recruitment of JARID1B to JARID1A target genes were observed in cells with reduced JARID1A expression (21, 47). To further characterize the roles of H3K4 demethylation in mouse development, it is therefore necessary to investigate the phenotypes of the combined Jarid1a and Jarid1b knock-out mice.
JARID1B was initially identified as a potential downstream target of HER2/ERBB2, which is also required for mammary ductal morphogenesis (48). This connection suggests that JARID1B may act downstream of HER2 in both mammary gland development and breast cancer. Consistent with these findings, depletion of JARID1B by shRNA or inhibition of JARID1B with a small molecule inhibitor reduced the proliferation rate of HER2+ UACC-812 breast cancer cells (49). In addition, knockdown of JARID1B reduced the tumor formation potential of MCF-7 luminal A breast cancer cells (12). Together with the link of JARID1B and FOXA1 to luminal and HER2+ breast cancer, these findings suggest that JARID1B can be targeted to suppress these two subtypes of breast cancer.
Supplementary Material
Acknowledgments
We thank Dr. William Kaelin for generous support to generate the Jarid1b−/− strain and critical reading of the manuscript; Drs. David Stern and John Wysolmerski for advice on the project and critical reading of the manuscript; and members of the Kaelin, Polyak, Stern, Wysolmerski, and Yan laboratories for kind help and valuable discussions. We thank Dr. Carmen Booth, Dr. Malini Harigopal, Yale Research Histology (Department of Pathology), and Yale Mouse Research Pathology (Section of Comparative Medicine) for kind help on histological analysis. We thank Dr. Kristian Helin for providing the DAIN JARID1B antibody.
This work was supported by an Alexander and Margaret Stewart Trust fellowship (to Q. Y.), a Breast Cancer Alliance young investigator grant (to Q. Y.), a Melanoma Research Foundation career development award (to Q. Y.), United States Department of Defense Peer-reviewed Cancer Research Program Career Development Award W81XWH-13-1-0235 (to Q. Y.), and American Cancer Society Research Scholar Grant RSG-13-384-01-DMC (to Q. Y.). This work was also supported in part by National Institutes of Health Grants R01 CA076120 (to W. G. K.) and P01 CA080111 (to K. P.) from the NCI and F32 CA156991, an NCI fellowship (to S. J. H.).

This article contains supplemental Tables 1 and 2.
- JARID1
- jumonji AT-rich interactive domain 1
- H3K4me3
- trimethylated lysine 4 in histone H3
- H3K4me2
- dimethylated lysine 4 in histone H3
- H3K4me1
- monomethylated lysine 4 in histone H3
- ARID
- AT-rich interactive domain
- ΔARID
- ARID deletion
- ERα
- estrogen receptor α
- FOXA1
- forkhead box A1
- GSEA
- gene set enrichment analysis
- KDM5B
- lysine (K)-specific demethylase 5B
- MEC
- mammary epithelial cell
- MEF
- mouse embryonic fibroblast
- TEB
- terminal end bud
- TSS
- transcriptional start site
- qPCR
- quantitative PCR
- Pol II
- polymerase II
- MaSC
- mammary stem cell.
REFERENCES
- 1. Kooistra S. M., Helin K. (2012) Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 [DOI] [PubMed] [Google Scholar]
- 2. Blair L. P., Yan Q. (2012) Epigenetic mechanisms in commonly occurring cancers. DNA Cell Biol. 31, Suppl. 1, S49–S61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Christensen J., Agger K., Cloos P. A., Pasini D., Rose S., Sennels L., Rappsilber J., Hansen K. H., Salcini A. E., Helin K. (2007) RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128, 1063–1076 [DOI] [PubMed] [Google Scholar]
- 4. Yamane K., Tateishi K., Klose R. J., Fang J., Fabrizio L. A., Erdjument-Bromage H., Taylor-Papadimitriou J., Tempst P., Zhang Y. (2007) PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 25, 801–812 [DOI] [PubMed] [Google Scholar]
- 5. Iwase S., Lan F., Bayliss P., de la Torre-Ubieta L., Huarte M., Qi H. H., Whetstine J. R., Bonni A., Roberts T. M., Shi Y. (2007) The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128, 1077–1088 [DOI] [PubMed] [Google Scholar]
- 6. Blair L. P., Cao J., Zou M. R., Sayegh J., Yan Q. (2011) Epigenetic regulation by lysine demethylase 5 (KDM5) enzymes in cancer. Cancers 3, 1383–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klose R. J., Yan Q., Tothova Z., Yamane K., Erdjument-Bromage H., Tempst P., Gilliland D. G., Zhang Y., Kaelin W. G., Jr. (2007) The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128, 889–900 [DOI] [PubMed] [Google Scholar]
- 8. Tahiliani M., Mei P., Fang R., Leonor T., Rutenberg M., Shimizu F., Li J., Rao A., Shi Y. (2007) The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 447, 601–605 [DOI] [PubMed] [Google Scholar]
- 9. Outchkourov N. S., Muiño J. M., Kaufmann K., van Ijcken W. F., Groot Koerkamp M. J., van Leenen D., de Graaf P., Holstege F. C., Grosveld F. G., Timmers H. T. (2013) Balancing of histone H3K4 methylation states by the Kdm5c/SMCX histone demethylase modulates promoter and enhancer function. Cell Rep. 3, 1071–1079 [DOI] [PubMed] [Google Scholar]
- 10. Barrett A., Madsen B., Copier J., Lu P. J., Cooper L., Scibetta A. G., Burchell J., Taylor-Papadimitriou J. (2002) PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: a new cancer/testis antigen? Int. J. Cancer 101, 581–588 [DOI] [PubMed] [Google Scholar]
- 11. Lu P. J., Sundquist K., Baeckstrom D., Poulsom R., Hanby A., Meier-Ewert S., Jones T., Mitchell M., Pitha-Rowe P., Freemont P., Taylor-Papadimitriou J. (1999) A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J. Biol. Chem. 274, 15633–15645 [DOI] [PubMed] [Google Scholar]
- 12. Catchpole S., Spencer-Dene B., Hall D., Santangelo S., Rosewell I., Guenatri M., Beatson R., Scibetta A. G., Burchell J. M., Taylor-Papadimitriou J. (2011) PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int. J. Oncol. 38, 1267–1277 [DOI] [PubMed] [Google Scholar]
- 13. Barrett A., Santangelo S., Tan K., Catchpole S., Roberts K., Spencer-Dene B., Hall D., Scibetta A., Burchell J., Verdin E., Freemont P., Taylor-Papadimitriou J. (2007) Breast cancer associated transcriptional repressor PLU-1/JARID1B interacts directly with histone deacetylases. Int. J. Cancer 121, 265–275 [DOI] [PubMed] [Google Scholar]
- 14. Li Q., Shi L., Gui B., Yu W., Wang J., Zhang D., Han X., Yao Z., Shang Y. (2011) Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 71, 6899–6908 [DOI] [PubMed] [Google Scholar]
- 15. Xiang Y., Zhu Z., Han G., Ye X., Xu B., Peng Z., Ma Y., Yu Y., Lin H., Chen A. P., Chen C. D. (2007) JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 19226–19231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayami S., Yoshimatsu M., Veerakumarasivam A., Unoki M., Iwai Y., Tsunoda T., Field H. I., Kelly J. D., Neal D. E., Yamaue H., Ponder B. A., Nakamura Y., Hamamoto R. (2010) Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 9, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roesch A., Fukunaga-Kalabis M., Schmidt E. C., Zabierowski S. E., Brafford P. A., Vultur A., Basu D., Gimotty P., Vogt T., Herlyn M. (2010) A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roesch A., Vultur A., Bogeski I., Wang H., Zimmermann K. M., Speicher D., Körbel C., Laschke M. W., Gimotty P. A., Philipp S. E., Krause E., Pätzold S., Villanueva J., Krepler C., Fukunaga-Kalabis M., Hoth M., Bastian B. C., Vogt T., Herlyn M. (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 23, 811–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Albert M., Schmitz S. U., Kooistra S. M., Malatesta M., Morales Torres C., Rekling J. C., Johansen J. V., Abarrategui I., Helin K. (2013) The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet. 9, e1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Welm B. E., Dijkgraaf G. J., Bledau A. S., Welm A. L., Werb Z. (2008) Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2, 90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin W., Cao J., Liu J., Beshiri M. L., Fujiwara Y., Francis J., Cherniack A. D., Geisen C., Blair L. P., Zou M. R., Shen X., Kawamori D., Liu Z., Grisanzio C., Watanabe H., Minamishima Y. A., Zhang Q., Kulkarni R. N., Signoretti S., Rodig S. J., Bronson R. T., Orkin S. H., Tuck D. P., Benevolenskaya E. V., Meyerson M., Kaelin W. G., Jr., Yan Q. (2011) Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc. Natl. Acad. Sci. U.S.A. 108, 13379–13386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmitz S. U., Albert M., Malatesta M., Morey L., Johansen J. V., Bak M., Tommerup N., Abarrategui I., Helin K. (2011) Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 30, 4586–4600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cartharius K., Frech K., Grote K., Klocke B., Haltmeier M., Klingenhoff A., Frisch M., Bayerlein M., Werner T. (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21, 2933–2942 [DOI] [PubMed] [Google Scholar]
- 24. Matys V., Kel-Margoulis O. V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K., Voss N., Stegmaier P., Lewicki-Potapov B., Saxel H., Kel A. E., Wingender E. (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Oevelen C., Wang J., Asp P., Yan Q., Kaelin W. G., Jr., Kluger Y., Dynlacht B. D. (2008) A role for mammalian Sin3 in permanent gene silencing. Mol. Cell 32, 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao J., Liu Z., Cheung W. K., Zhao M., Chen S. Y., Chan S. W., Booth C. J., Nguyen D. X., Yan Q. (2014) Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 6, 868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lupien M., Eeckhoute J., Meyer C. A., Wang Q., Zhang Y., Li W., Carroll J. S., Liu X. S., Brown M. (2008) FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132, 958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernardo G. M., Lozada K. L., Miedler J. D., Harburg G., Hewitt S. C., Mosley J. D., Godwin A. K., Korach K. S., Visvader J. E., Kaestner K. H., Abdul-Karim F. W., Montano M. M., Keri R. A. (2010) FOXA1 is an essential determinant of ERα expression and mammary ductal morphogenesis. Development 137, 2045–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim E., Wu D., Pal B., Bouras T., Asselin-Labat M. L., Vaillant F., Yagita H., Lindeman G. J., Smyth G. K., Visvader J. E. (2010) Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 12, R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scibetta A. G., Santangelo S., Coleman J., Hall D., Chaplin T., Copier J., Catchpole S., Burchell J., Taylor-Papadimitriou J. (2007) Functional analysis of the transcription repressor PLU-1/JARID1B. Mol. Cell. Biol. 27, 7220–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kouros-Mehr H., Slorach E. M., Sternlicht M. D., Werb Z. (2006) GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127, 1041–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hennighausen L., Robinson G. W. (2001) Signaling pathways in mammary gland development. Dev. Cell 1, 467–475 [DOI] [PubMed] [Google Scholar]
- 34. Hynes N. E., Watson C. J. (2010) Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb. Perspect. Biol. 2, a003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sternlicht M. D., Kouros-Mehr H., Lu P., Werb Z. (2006) Hormonal and local control of mammary branching morphogenesis. Differentiation 74, 365–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bocchinfuso W. P., Lindzey J. K., Hewitt S. C., Clark J. A., Myers P. H., Cooper R., Korach K. S. (2000) Induction of mammary gland development in estrogen receptor-α knockout mice. Endocrinology 141, 2982–2994 [DOI] [PubMed] [Google Scholar]
- 37. Hurtado A., Holmes K. A., Ross-Innes C. S., Schmidt D., Carroll J. S. (2011) FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 43, 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Theodorou V., Stark R., Menon S., Carroll J. S. (2013) GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 23, 12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klein B. J., Piao L., Xi Y., Rincon-Arano H., Rothbart S. B., Peng D., Wen H., Larson C., Zhang X., Zheng X., Cortazar M. A., Peña P. V., Mangan A., Bentley D. L., Strahl B. D., Groudine M., Li W., Shi X., Kutateladze T. G. (2014) The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 6, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xie L., Pelz C., Wang W., Bashar A., Varlamova O., Shadle S., Impey S. (2011) KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 30, 1473–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kidder B. L., Hu G., Zhao K. (2014) KDM5B focuses H3K4 methylation near promoters and enhancers during embryonic stem cell self-renewal and differentiation. Genome Biol. 15, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Visvader J. E. (2009) Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 23, 2563–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamaguchi N., Ito E., Azuma S., Honma R., Yanagisawa Y., Nishikawa A., Kawamura M., Imai J., Tatsuta K., Inoue J., Semba K., Watanabe S. (2008) FoxA1 as a lineage-specific oncogene in luminal type breast cancer. Biochem. Biophys. Res. Commun. 365, 711–717 [DOI] [PubMed] [Google Scholar]
- 44. Reed D. R., Lawler M. P., Tordoff M. G. (2008) Reduced body weight is a common effect of gene knockout in mice. BMC Genet. 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wiesen J. F., Young P., Werb Z., Cunha G. R. (1999) Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development 126, 335–344 [DOI] [PubMed] [Google Scholar]
- 46. Madsen B., Spencer-Dene B., Poulsom R., Hall D., Lu P. J., Scott K., Shaw A. T., Burchell J. M., Freemont P., Taylor-Papadimitriou J. (2002) Characterisation and developmental expression of mouse Plu-1, a homologue of a human nuclear protein (PLU-1) which is specifically up-regulated in breast cancer. Gene Expr. Patterns 2, 275–282 [DOI] [PubMed] [Google Scholar]
- 47. Islam A. B., Richter W. F., Lopez-Bigas N., Benevolenskaya E. V. (2011) Selective targeting of histone methylation. Cell Cycle 10, 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jackson-Fisher A. J., Bellinger G., Ramabhadran R., Morris J. K., Lee K. F., Stern D. F. (2004) ErbB2 is required for ductal morphogenesis of the mammary gland. Proc. Natl. Acad. Sci. U.S.A. 101, 17138–17143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sayegh J., Cao J., Zou M. R., Morales A., Blair L. P., Norcia M., Hoyer D., Tackett A. J., Merkel J. S., Yan Q. (2013) Identification of small molecule inhibitors of jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem. 288, 9408–9417 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.