

Background: The tenascin-C-derived peptide TNIIIA2 is capable of activating β1-integrins.
Results: TNIIIA2 greatly enhanced cell survival and PDGF-dependent proliferation through potentiated and sustained activation of integrin α5β1, resulting in continuous proliferation even after reaching confluency.
Conclusion: TNIIIA2-induced integrin α5β1 activation causes deregulated cell growth.
Significance: These results offer a new insight into the physiological/pathological role of tenascin-C in tissues where it is highly expressed.
Keywords: Cell Adhesion, Cell Proliferation, Extracellular Matrix, Integrin, Tenascin, PDGF
Abstract
Tenascin-C is an adhesion modulatory matrix protein that is highly expressed in tumors; however, its biochemical activity involved in tumorigenesis is not fully understood. On the other hand, increasing evidence indicates the importance of integrin α5β1 in cancer development. We previously demonstrated that tenascin-C harbors a functional site that can be released as a proadhesive peptide such as TNIIIA2. Peptide TNIIIA2 is capable of inducing activation of β1-integrins including α5β1 via syndecan-4. In this study the proadhesive effect of TNIIIA2 was characterized by potentiated and sustained activation of integrin α5β1. Based on this effect, TNIIIA2 rendered nontransformed fibroblasts (NIH3T3) resistant to serum deprivation-elicited anoikis through activation of the Akt/Bcl-2 pathway. Moreover, TNIIIA2 hyperstimulated PDGF-dependent proliferation of NIH3T3 by activating integrin α5β1. Tenascin-C, a parental protein of TNIIIA2, also stimulated PDGF-dependent proliferation, which was blocked by a matrix metalloproteinase-2/9 inhibitor and an anti-TNIIIA2 function-blocking antibody, suggesting proteolytic exposure of the proadhesive effect of TNIIIA2. Mechanistic analyses revealed that TNIIIA2 induced a lateral association of PDGF receptor β with the molecular complex of activated integrin α5β1 and syndecan-4 in the membrane microdomains enriched with cholesterol/caveolin-1, resulting in prolonged activation of PDGF receptor β and the subsequent Ras/mitogen-activated protein kinase pathway in a PDGF-dependent manner. Of note, TNIIIA2 induced continuous proliferation in NIH3T3 in an integrin α5β1-dependent manner even after they formed a confluent monolayer. Thus, it was proposed that tenascin-C might be involved in deregulated cell growth through potentiated and sustained activation of integrin α5β1 after exposure of the proadhesive effect of TNIIIA2.
Introduction
Tenascin-C (TN-C)3 is one of the most intriguing extracellular matrix proteins and is characterized by its regulated expression. Expression of TN-C in normal adult tissues is generally low, whereas it increases transiently in pathological states including tumorigenesis (1–3). Taken together with the data of functional analyses using in vitro and in vivo experiments, it is considered that TN-C is a key determinant of the tumor stroma involved in tumor initiation and progression (4). In fact, most malignant tumors express TN-C, and its expression level correlates with a poor prognosis of disease free survival in patients with cancers such as glioma and lung and breast carcinomas (4). However, the reason why this correlation applies to some cancers but not others has not yet been clarified.
TN-C is also characterized by cell adhesion modulatory activity. TN-C acts as either an adhesive or an antiadhesive factor, depending on the cellular context (5–8). In particular, the antiadhesive effect of TN-C appears favorable for tumor cell proliferation and migration (9, 10). TN-C interferes with the adhesive interaction between syndecan-4 and the heparin-binding domain II of fibronectin, which blocks collaborative signaling induced by integrin α5β1 and syndecan-4 (11, 12). Inhibition of integrin α5β1-syndecan-4-mediated cell adhesion causes enhanced cell proliferation by several mechanisms that include disruption of the actin cytoskeleton through reduced tropomyosin1 expression, depression of Wnt signaling, and activation of MAP kinase signaling (13–15).
The hypothesis that the antiadhesive effect of TN-C is responsible for enhanced tumor cell proliferation is supported by several previous reports which show that integrin α5β1 serves as a tumor suppressor gene product (4, 16–19). These previous reports suggest that α5β1-mediated adhesion is a disadvantage to the proliferation of tumor cells, which clearly supports the potential advantage of the antiadhesive effect of TN-C on tumor cell proliferation. However, a number of recent studies have come to a conflicting conclusion, namely, that integrin α5β1 is associated with the expression of a malignant phenotype (20–23). More recently, integrin α5β1 emerged as a potential anticancer target because it is overexpressed in both tumor neovessels and tumor cells (24–28). Besides the antiadhesive effect, any additional biochemical activity must be defined to obtain a precise understanding of the role of TN-C in tumorigenesis.
We previously found that TN-C harbors a functional site within the fibronectin type III-like repeat A2 (29). Proteolytic cleavage with matrix-degrading proteinase, including matrix metalloproteinase-2 (MMP-2), can release a functional peptide(s) with proadhesive activity. A 22-mer peptide bearing the proadhesive site, TNIIIA2, has a potent ability to induce β1-integrin activation. Syndecan-4 serves as a membrane receptor for TNIIIA2-induced β1-integrin activation, and engagement with TNIIIA2 induces a physical association with β1-integrins, causing conformational changes in β1-integrin that result in functional activation (29). Araki et al. (30) later reported a similar mechanism of β1-integrin activation induced by the laminin peptide through syndecan-4. The mode of β1-integrin activation via syndecan-4 is entirely distinct from that through “inside-out” signaling (31). Therefore, as observed previously in apoptosis (32) and differentiation (33) of nonadherent hematopoietic tumor cells, β1-integrin activation induced by TNIIIA2 may elicit some specific effects in the survival and proliferation of nontransformed adherent cell types.
In this study we demonstrated that potentiated and sustained activation of integrin α5β1 induced by peptide TNIIIA2 causes anoikis resistance and hyperstimulation of PDGF-dependent proliferation in nontransformed fibroblasts, resulting in an attenuation of contact inhibition in cell proliferation. Our results offer a new insight into the physiological and pathological roles of TNC in tissues where it is highly expressed.
EXPERIMENTAL PROCEDURES
Materials
Peptide TNIIIA2 and its inactive control peptide, TNIIIA2mut, have been described previously (32). Antagonistic peptides for integrins, GRGDSP, and its control (GRGESP) for α5β1 (IWAKI, Tokyo), CS-1 for α4β1 (Operon Biotechnology, Tokyo), and cyclo-RGDfC (AnaSpec) for αvβ3 were purchased as indicated. AG1295, a selective inhibitor of PDGF receptor-tyrosine kinase, and AG1478, EGF receptor kinase-specific inhibitor, were obtained from Calbiochem. Methyl β-cyclodextrin (Mβ-CD) and phorbol myristate acetate were obtained from Sigma. PDGF-BB and BS3, a cross-linker, were purchased from WAKO Pure Chemicals, Amgen, and Pierce. The MMP-2/-9 inhibitor II, (2R)-((4-biphenylylsulfonyl)amino)-N-hydroxy-3-phenylpropionamide (BiPS) was obtained from Calbiochem.
Antibodies
A function-blocking antibody against the proadhesive site of peptide TNIIIA2 was prepared as follows: rabbits were immunized with a synthetic peptide (CATHYTITIRGVT) containing the active sequence coupled with thyroglobulin. The IgG fraction of rabbit serum was applied to Sepharose beads coupled with the synthetic peptide immunogen. Eluted IgG was used as the anti-TNIIIA2 antibody. Function-blocking monoclonal antibodies (mAbs) against mouse integrin subunit β1 (Ha2/5) (BD Biosciences), β3/CD61 (2C9.G2) (BD Biosciences), α4 (P1H4) (Chemicon), α5 (P1D6) (Chemicon), and αv (RMV-7) (Millipore) were obtained as indicated. mAbs against mouse integrin α4 (RI-2), α5 (5H10–27), and β3 (2C9.G2) were obtained from BD Pharmingen. Anti-phospho-PDGFR-β (Tyr-716), anti-phospho-PDGFR-β (Tyr-857), and anti-caveolin 1 (N-20) polyclonal antibodies were from Santa Cruz Biotechnology. Anti-PDGFR-β was from Cell Signaling Technology. mAbs against ERK1/2, phospho-ERK1/2, syndecan-4 (5G9), Bcl-2, Akt, and phospho-Akt (Ser-473) and mAbs against human (TS2/16) and mouse (9EG7) β1-integrin, which are able to activate β1-integrins, are described previously (29, 32).
Cell Culture
Nontransformed mouse fibroblasts NIH3T3 (provided by the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University) and human leukemia cell line K562 (obtained from RIKEN BioResource Center) were cultured as described previously (29, 33, 34).
Cell Adhesion Assay
Adhesion to fibronectin using K562 cells was performed as described previously (33).
Cell Survival and Proliferation
Cells suspended with serum-free medium were seeded on a 96-well plate coated with fibronectin (0.25–4 μg/ml) for NIH3T3 cells and allowed to adhere for 6 h. Cells were then stimulated with or without growth factors (PDGF) in the presence or absence of TNIIIA2 as indicated for 16 h. The number of viable cells was evaluated by using the Cell Counting kit (WST assay) as described previously (32).
Ras Activity Assay
Cells suspended with serum-free medium were seeded on a 6-well plate coated with fibronectin and cultured for 6 h as above. Cells were stimulated with PDGF (10 ng/ml) in the presence or absence of peptide TNIIIA2 (25 μg/ml) for the indicated times. Cell lysates were prepared, and their Ras activities were determined using the Ras Activation Assay kit (Upstate Biotechnology Inc.) according to the manufacturer's protocol.
PDGF Receptor Kinase Assay
Cells treated with PDGF in the presence or absence of peptide TNIIIA2 as described above were dissolved in PBS containing 0.5% Triton X-100 and immunoprecipitated with anti-PDGFR antibody. Tyrosine kinase activity was determined by the tyrosine kinase assay kit (Takara) according to the manufacturer's protocol.
Flow Cytometry
The activation status of β1-integrins on the cells was evaluated by using anti-active form of β1-integrin (9EG7) by flow cytometric analysis, as described previously (29).
Fractionation of the Membrane Domains Enriched with Cholesterol/Caveolin-1 by Sucrose Density Gradient Centrifugation
Cells were allowed to adhere to the fibronectin substrate for 6 h and then stimulated with or without PDGF (10 ng/ml) in the presence or absence of peptide TNIIIA2 (25 μg/ml). After incubation for 60 min, cells were harvested and sonicated for 5 min on ice in 500 mm sodium carbonate buffer (pH 11). Fractionation of the caveolin-rich membrane microdomains by sucrose density gradient centrifugation was performed as described previously (35) with some modifications. In brief, cell homogenates prepared as described above were mixed with an equal volume of 90% sucrose in 25 mm MES buffer (pH 6.5) containing 0.15 m NaCl and placed at the bottom of a centrifuge tube. A 5–35% sucrose gradient was layered above and centrifuged at 39,000 rpm (SW55Ti, Beckman) for 16 h at 4 °C. The gradient samples were divided into four fractions from the bottom of the tubes. Fractionated samples were dialyzed and concentrated by using an ultrafiltration centrifugal device (PES 10K, Thermo Scientific), and Western blotting was performed to detect caveolin-1, PDGF receptor β, and α5-integrin.
Focus Formation
Focus formation was observed following standard protocols (36) with some modifications: NIH3T3 cells were plated with or without PDGF (10 ng/ml) in the presence or absence of peptide TNIIIA2 (25 μg/ml) in either 12-well plates at a density of 4 × 104 cells/well or 96-well plates at a density of 6 × 103 cells/well. Cells were kept in DMEM with 5% calf serum with or without TNIIIA2. The culture medium was changed every 3 days. After the indicated periods, cells in the 12-well plates were stained by Giemsa to observe focus formation. Cells in 96-well plates were examined to evaluate the number of viable cells as described above.
RESULTS
Peptide TNIIIA2 Renders NIH3T3 Cells Anoikis-resistant and Hyperstimulates Their PDGF-dependent Proliferation
The effects of β1-integrin activation by peptide TNIIIA2 on cell survival and proliferation were investigated. The nontransformed fibroblasts, NIH3T3 cells, mainly express integrin α5β1 and αvβ3, with a low level of α4β1, as fibronectin receptors (data not shown). When NIH3T3 cells adhering to the fibronectin substrate were cultured under serum-deprived conditions, a decrease in the number of viable cells was evident (Fig. 1A). This decrease, which was sensitive to a general caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone, was prevented by increasing the concentration of fibronectin used for plate-coating (Fig. 1A). Activation of β1-integrin by 9EG7 prevented a decrease in cell viability (Fig. 1A), suggesting the involvement of detachment-induced apoptosis, so-called anoikis. Peptide TNIIIA2 rescued NIH3T3 cells from serum starvation-elicited apoptosis to a greater extent than that by 9EG7 (Fig. 1A). Consistent with previous studies (34), the anoikis resistance of cells adhered to the fibronectin substrate in response to TNIIIA2-induced β1-integrin activation was accompanied by Akt phosphorylation and Bcl-2 expression regardless of the presence or absence of inhibitors for receptor-type tyrosine kinase (RTK) of PDGF (AG1295) and EGF (AG1478) (Fig. 1, B and C). Experiments using function-blocking antibodies directed to integrin subunits (αv, α4, α5, β1, and β3) suggested that TNIIIA2-induced anoikis resistance was due to the activation of integrin α5β1 but not αvβ3 and α4β1 (Fig. 1D).
FIGURE 1.

Protection of NIH3T3 cells from serum deprivation-dependent apoptosis by TNIIIA2. A, NIH3T3 cells were seeded on a plate coated with the indicated concentration (μg/ml) of fibronectin and allowed to adhere. After completion of adhesion to the fibronectin substrate, cells were cultured with serum-free medium in the presence or absence of anti-apoptotic reagent, benzyloxycarbonyl-VAD (z-VAD; 50 μm), TNIIIA2 (25 or 50 μg/ml), normal IgG, or β1-integrin-activating mAb 9EG7 (20 μg/ml) for 1 day, and WST assays were conducted to detect viable cells. Each point represents the mean ± S.D. of triplicate determinations. One of three individual experiments is shown. *, p < 0.005 versus untreated cells on fibronectin (0.25 μg/ml) on day 0. **, p < 0.01 versus untreated cells on fibronectin (0.25 μg/ml) on day 1. B and C, NIH3T3 cells were cultured with serum-free medium in the presence or absence of TNIIIA2 (25 μg/ml) and in the presence or absence of tyrosine kinase inhibitor for PDGF receptor (AG1295, represented by 1295) or EGF-receptor (AG1478, represented by 1478). After the indicated period, cell lysates were subjected to immunoblot analysis to detect Akt phosphorylation (B) and Bcl-2 expression (C). D, cells adhering to the fibronectin were cultured with serum-free medium with or without TNIIIA2 in the presence or absence of normal IgG or anti-integrin function-blocking mAb (15 μg IgG/ml) directed to β1, β3, α4, α5, αv subunit, or a mixture of anti-αv and β3 (represented by αv+β3). Each point represents the mean ± S.D. of triplicate determinations. One of three individual experiments is shown. *, p < 0.005 versus untreated on day 0. **, p < 0.005 versus untreated on day 1. ***, p < 0.005 versus treated with normal IgG.
Proliferation of NIH3T3 cells was stimulated by PDGF in a dose-dependent manner but reached a plateau at around 10 ng/ml (Fig. 2A). TNIIIA2, which was previously demonstrated to activate β1-integrins of NIH3T3 cells (29), further stimulated cell proliferation with a submaximal dose of PDGF (Fig. 2A). TNIIIA2 alone without PDGF did not affect cell proliferation (Fig. 2B). TNIIIA2mut, a control peptide of TNIIIA2 inactive in β1-integrin activation (32), failed to enhance PDGF-dependent cell proliferation (Fig. 2B). The β1-integrin-activating mAb, 9EG7, also had the ability to enhance PDGF-dependent proliferation, whereas normal IgG and anti-β1-integrin mAb (2B1) with no function did not (Fig. 2B). This hyperstimulation of PDGF-dependent proliferation by TNIIIA2 was specifically abrogated by function-blocking mAbs directed to integrin subunits β1 and α5 but not to α4, αv, and β3 (Fig. 2C). Additionally, among the integrin antagonistic peptides, GRGDSP for α5β1, CS-1 for α4β1, and cyclo-RGDfC for αvβ3 (37), only GRGDSP was effective in abrogating TNIIIA2-induced hyperstimulation of cell proliferation (Fig. 2C). As shown in Fig. 2D, TN-C also stimulated PDGF-dependent cell proliferation, although only about one-half of that by TNIIIA2. Stimulation of proliferation by TN-C was abolished by BiPS, a specific inhibitor for MMP-2/9, and was restored by the addition of TNIIIA2 even in the presence of BiPS (Fig. 2D). Enhanced proliferation by TNC was also neutralized by anti-TNIIIA2 function-blocking antibody (represented as α-TNIII in Fig. 2D). These results suggested that TNC stimulated cell proliferation through proteolytic exposure of the TNIIIA2-related cryptic proadhesive site of the TN-C molecule. We previously detected the secretion of an active form of MMP-2 from NIH3T3 cells (38). Moreover, the TN-C-induced stimulation of cell proliferation was inhibited by the function-blocking mAb directed to the integrin α5 subunit but not by mAbs directed to the α4 and αv subunits (Fig. 2D) and thus closely resembled the stimulatory effect of TNIIIA2 on proliferation.
FIGURE 2.

Hyperstimulation of NIH3T3 cell proliferation through potentiated and sustained activation of β1-integrin by TNIIIA2. A, cells adhering to fibronectin were stimulated with the indicated concentrations of PDGF (left panel, circles) or with PDGF (10 ng/ml) in the presence of the indicated concentrations of TNIIIA2 (right panel, triangles) for 1 day. Cell proliferation and survival were evaluated by WST assays in which the viable cell number was determined using absorbance at 450 nm. The viable cell number on day 0 is shown by squares with dotted lines. Each point represents the mean ± S.D. of triplicate determinations. One of three individual experiments is shown. *, p < 0.001 versus untreated. B, cells adhering to the fibronectin were treated with or without PDGF (10 ng/ml) in the presence or absence of TNIIIA2 (50 μg/ml), TNIIIA2mut (mut), normal IgG (20 μg/ml), β1-integrin-activating mAb (9EG7) (20 μg/ml), or anti-β1-integrin with no function (2B1) for 1 day. WST assays were then performed. Each point represents the mean ± S.D. of triplicate determinations. *, p < 0.01 versus untreated; **, p < 0.01 versus treatment with PDGF alone; ***, p < 0.01 versus treated with normal IgG. C, effects of integrin antagonists on the hyperstimulation of PDGF-dependent cell proliferation by TNIIIA2 were examined. Cells adhering to the fibronectin were stimulated with PDGF (10 ng/ml) and TNIIIA2 (50 μg/ml) in the presence or absence of control IgG, anti-integrin function-blocking mAb (20 μg IgG/ml) directed to β1, β3, α4, α5, or αv subunit, or integrin antagonistic peptide, RGD (GRGDSP, 200 μg/ml) for α5β1, CS-1 (200 μg/ml) for α4β1, or RGDfC (cyclin RDGfC, 50 μg/ml) for αvβ3 for 1 day. Each point represents the mean ± S.D. of triplicate determinations. *, p < 0.01 versus without PDGF; **, p < 0.01 versus treatment with PDGF alone; ***, p < 0.01 versus treated with normal IgG. D, effect of TNC on NIH3T3 cell proliferation was examined as above (see A and B). Cells adhering to the fibronectin were stimulated with PDGF (10 ng/ml) and TNC (250 μg/ml) or TNIIIA2 (25 μg/ml) in the presence or absence of BiPS (200 nm), control IgG (20 μg/ml), anti-TNIIIA2 function-blocking antibody (αTNIIIA2) (20 μg/ml), or anti-integrin function-blocking mAb (20 μg IgG/ml) directed to β1, β3, α4, α5, or αv subunit. Each point represents the mean ± S.D. of triplicate determinations. *, p < 0.01 versus without PDGF; **, p < 0.01 versus treatment with PDGF alone; ***, p < 0.01 versus treated with normal IgG. #, p < 0.01 versus treatment with TNC plus BiPS.
Therefore, TNIIIA2 not only rendered NIH3T3 cells resistant to anoikis but also induced hyperstimulation of PDGF-dependent proliferation through activation of integrin α5β1. TN-C might acquire the ability to induce hyperstimulation of PDGF-dependent cell proliferation after proteolytic cleavage by matrix-degrading proteinases such as MMP-2/9.
Characterization of Proadhesive Nature of TNIIIA2
The question of how TNIIIA2 could exhibit such a potent stimulatory effect on cell survival/proliferation was addressed by characterizing the proadhesive nature of TNIIIA2 in comparison to other integrin-activating factors. Cell adhesion to fibronectin after β1-integrin activation was evaluated by using the human leukemia cell line K562, which exclusively expresses α5β1 in its inactive form as β1-integrin. Stem cell factor and phorbol myristate acetate, which activate β1-integrins through an inside-out pathway (33), promoted K562 cell adhesion, reaching a maximum within a 60-min incubation period and returning to basal level after 4 h (Fig. 3A). These factors induced fibronectin adhesion in only 20–40% of K562 cells. In contrast, TNIIIA2 and TS2/16, an anti-human β1-integrin mAb (25), both of which activate β1-integrins through conformational changes on the outer cell surface, induced potentiated and sustained adhesion to the fibronectin (Fig. 3A). Cell adhesion stimulated by TNIIIA2 remained high for at least 10 h under the conditions used (Fig. 3A). K562 cell adhesion to fibronectin induced by β1-integrin activation (29, 33) was abolished by cholesterol depletion with Mβ-CD and restored by supplementation with cholesterol (Fig. 3, B and C). Thus, the proadhesive effect of peptide TNIIIA2 was characterized by its potentiated and prolonged activation of β1-integrins in a manner that depended on the membrane microdomains enriched with cholesterol.
FIGURE 3.

Characterization of proadhesive nature of TNIIIA2. A, induced adhesion of K562 cells to the fibronectin by TNIIIA2. K562 cells were seeded on a 96-well plate coated with the fibronectin (10 μg/ml) in the presence of TNIIIA2 (25 μg/ml; circles), TS2/16 (20 μg/ml; squares), phorbol myristate acetate (50 ng/ml; triangles), or stem cell factor (50 ng/ml; inversed triangles). After culturing for the indicated time, cell adhesion to the fibronectin substrate was quantified as described previously (32, 33). Each point represents the mean ± S.D. of triplicate determinations. 1 of 3 individual experiments is shown. *, p < 0.005 versus untreated (0 h); **, p < 0.05 versus untreated (0 h). B, K562 cells were incubated with or without TNIIIA2 (25 μg/ml) in the presence or absence of Mβ-CD (10 mm) or Mβ-CD plus cholesterol (Chol; 16 mg/ml) inclusion complex for 30 min. Activation of β1-integrin was determined by flow cytometry using mAb 9EG7 recognizing the active β1 conformation specific epitope. C, to evaluate the effects on cell adhesion to the fibronectin substrate, K562 cells treated as above were seeded to 96-well plates coated with the fibronectin and incubated for 1 h. Cells adhering to fibronectin were fixed with formalin, and the numbers of cells were counted (C) as in Tanaka et al. (33). Data are representative of two individual experiments. *, p < 0.001 versus without TNIIIA2. **, p < 0.005 versus treated with TNIIIA2 alone. ***, p < 0.01 versus treated with TNIIIA2 and Mβ-CD.
Molecular Mechanism Underlying Hyperstimulation of PDGF-dependent Cell Proliferation by TNIIIA2
The molecular mechanism underlying the hyperproliferation in response to TNIIIA2-induced integrin α5β1 activation was investigated. Western blot analysis suggested that hyperstimulation of PDGF-dependent proliferation by TNIIIA2 might be caused by potentiated activation of PDGF receptor β. That is, combined stimulation with PDGF and TNIIIA2 markedly enhanced the phosphorylation of PDGF receptor β (Fig. 4A), which in turn induced the activation of signaling molecules of the Ras/MAP kinase pathway (Fig. 4, B–D). The increased phosphorylation and activation levels of PDGF receptor β (Tyr-716 and Tyr-857), Ras, MEK, and ERK1/2 were sustained for at least 2 h in the presence of TNIIIA2 (Fig. 4, B-D). Notably, like TNIIIA2, β1-integrin-activating mAb 9EG7 also enhanced the PDGF-dependent phosphorylation of PDGF receptor β (Fig. 4E), although this was less remarkable than that by TNIIIA2. Neither TNIIIA2 nor 9EG7 alone was able to stimulate the phosphorylation/activation of PDGF receptor β (Fig. 4E, marked with #).
FIGURE 4.

Intracellular signaling responsible for hyperstimulation of PDGF-dependent cell proliferation by TNIIIA2. A–D, NIH3T3 cells adhering to the fibronectin were incubated with PDGF (10 ng/ml) in the presence or absence of TNIIIA2 (25 μg/ml) for the indicated periods of time. In E, β1-integrin activating mAb 9EG7 was used instead of TNIIIA2. A and C–E were the results of immunoblot analysis using specific antibodies as indicated in each panel. # represents no stimulation by PDGF. B shows the results of Ras activation assays as described under “Experimental Procedures.” N/C, negative control; P/C, positive control. Data shown are representative of two or three individual experiments.
The potentiated phosphorylation of PDGF receptor β induced by TNIIIA2 and the resulting hyperproliferation of NIH3T3 cells were abrogated by A1295, a selective inhibitor of PDGF receptor-tyrosine kinase, but not by AG1478, an EGF receptor kinase-specific inhibitor (Fig. 5, A and B). TNIIIA2 enhanced the tyrosine kinase activity of PDGF receptor β in a PDGF-dependent manner (Fig. 5C). Supporting these results, TNIIIA2 was shown to promote the dimerization of PDGF receptor β on the cell surface in a PDGF-dependent manner (Fig. 5D). Importantly, all of these effects of TNIIIA2 were dependent on PDGF. As shown in Fig. 5, A--D (marked with #), TNIIIA2 alone showed little or no effect on the phosphorylation of PDGF receptor, cell proliferation, PDGF receptor-tyrosine kinase activity, or dimerization of the PDGF receptor monomer.
FIGURE 5.

TNIIIA2 hyperstimulates cell proliferation through enhanced activation of PDGF receptor β tyrosine kinase. A, cells adhering to the fibronectin were preincubated with or without AG1295, a selective inhibitor of PDGF receptor-tyrosine kinase, or AG1478, an EGF-receptor kinase-specific inhibitor for 1 h and then incubated in the presence or absence of PDGF (10 ng/ml) and/or TNIIIA2 (25 μg/ml) for 30 min. Cell lysates with an equal amount of proteins were subjected to Western blot analysis using antibodies recognizing phosphorylated Tyr-857 of PDGFR-β. Data are representative of two individual experiments. B, effect of PDGF receptor kinase inhibitor on the hyperstimulation of PDGF-dependent cell proliferation by TNIIIA2. Cells adhering to the fibronectin were preincubated with or without kinase inhibitor and cultured in the presence or absence of PDGF (10 ng/ml) and/or TNIIIA2 (25 μg/ml). The number of viable cells was evaluated by the WST assay. Each point represents the mean ± S.D. of triplicate determinations. *, p < 0.01 versus untreated (Control); **, p < 0.005 versus without kinase inhibitor. C, effect on tyrosine kinase activity. Cells adhering to the fibronectin were treated with or without PDGF (10 ng/ml) in the presence or absence of TNIIIA2 (25 μg/ml). Immunoprecipitates of these lysates by anti-PDGFR-β antibody were examined for tyrosine kinase activity as described under “Experimental Procedures.” Each point represents the mean ± S.D. of triplicate determinations. *, p < 0.01 versus untreated (Control); **, p < 0.01 versus with PDGF alone. D, effect of TNIIIA2 on the dimerization of PDGF receptor was examined. Cells adhering to the fibronectin were incubated with PDGF (10 ng/ml) in the presence or absence of TNIIIA2 (25 μg/ml). After washing once with HEPES-buffered saline (pH 8.0), cells were incubated with BS3 (0.1 mm) to covalently bridge the dimerization of the PDGF receptor monomers. After the addition of glycine (1 mm) to stop the reaction, cell lysates prepared with radioimmune precipitation assay buffer were subjected to immunoprecipitation with anti-PDGFR-β antibody, and Western blot analysis was performed using anti-PDGFR-β antibody. Data are representative of two individual experiments. In panels A–D, # represents no stimulation by PDGF.
In the same way as the effect on β1-integrin activation (see Fig. 3B), the membrane microdomains enriched with cholesterol were implicated in the potentiated activation of PDGF receptor β by TNIIIA2. First, hyperstimulation of PDGF receptor β phosphorylation/activation by PDGF together with TNIIIA2 was abrogated by cholesterol elimination with Mβ-CD and restored by re-supplementation with cholesterol (Fig. 6A). Second, an immunoprecipitation study showed that stimulation with PDGF in the presence of TNIIIA2 induced a physical association between integrin α5β1 and the phosphorylated/activated form of PDGF receptor β (Fig. 6B). Immunoprecipitation with anti-syndecan-4 antibody further showed that this physical association between integrin α5β1 and PDGF receptor β was mediated by syndecan-4, a membrane receptor of TNIIIA2 (Fig. 6C). In this immunoprecipitation study, the involvement of αv integrins was again ruled out. Third, sucrose density gradient centrifugation analysis of the membrane distribution of PDGF receptor β and α5-integrin might support the implied role of the cholesterol/caveolin-rich microdomain in TNIIIA2-induced hyperproliferation. Most PDGF receptors and α5-integrin remained at the bottom of the gradient, and little was found in the caveolin-rich membrane fraction in untreated cells (Fig. 7A), but stimulation by PDGF induced translocation of PDGF receptor β to the caveolin-rich fraction (Fig. 7B). Translocation of PDGF receptor β to the caveolin-rich fraction became more evident when cells were stimulated with PDGF in combination with TNIIIA2 (Fig. 7D).
FIGURE 6.
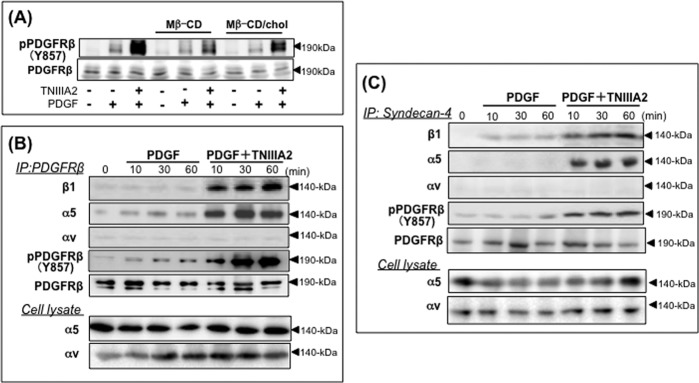
TNIIIA2 induces a physical association of integrin α5β1 with PDGF receptor β via syndecan-4. A, effect of cholesterol (Chol) elimination on TNIIIA2-induced enhanced activation of PDGF receptor β. Cells adhering to the fibronectin were incubated with PDGF (10 ng/ml) and/or TNIIIA2 (25 μg/ml) in the presence or absence of Mβ-CD (10 mm) or Mβ-CD-cholesterol (16 mg/ml) inclusion complex for 30 min and dissolved with modified radioimmune precipitation assay buffer (0.01 m Tris-HCl buffer, pH 8.0, 5 mm EDTA, 0.15 m NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and protease and phosphatase inhibitors). Cell lysates with an equal amount of proteins were subjected to Western blot analysis using antibodies recognizing phosphorylated Tyr-857 of PDGFR-β. B and C, an immunoprecipitation (IP) study was performed as described elsewhere (39), with some modifications. Cells treated with PDGF in the presence or absence of TNIIIA2 as indicated were dissolved by sonication in modified radioimmune precipitation assay buffer (39) and clarified by centrifugation (10,000 rpm, 5 min). Clear supernatants were incubated (4 °C, 8 h) with anti-PDGFR-β antibody (B) or anti-syndecan-4 (5G9 mAb) (C) followed by incubation (4 °C, 3 h) with protein G-Sepharose. After washing three times with radioimmune precipitation assay buffer, immune complexes adsorbed onto protein G-Sepharose were extracted with Laemmli buffer, and Western blot analysis was performed. Expression of integrin α5 and αv subunits was also confirmed by Western blotting of cell lysates (panels of Cell lysate in B and C). The antibodies used for Western blotting are shown on the left side of each panel. Data are representative of two individual experiments.
FIGURE 7.
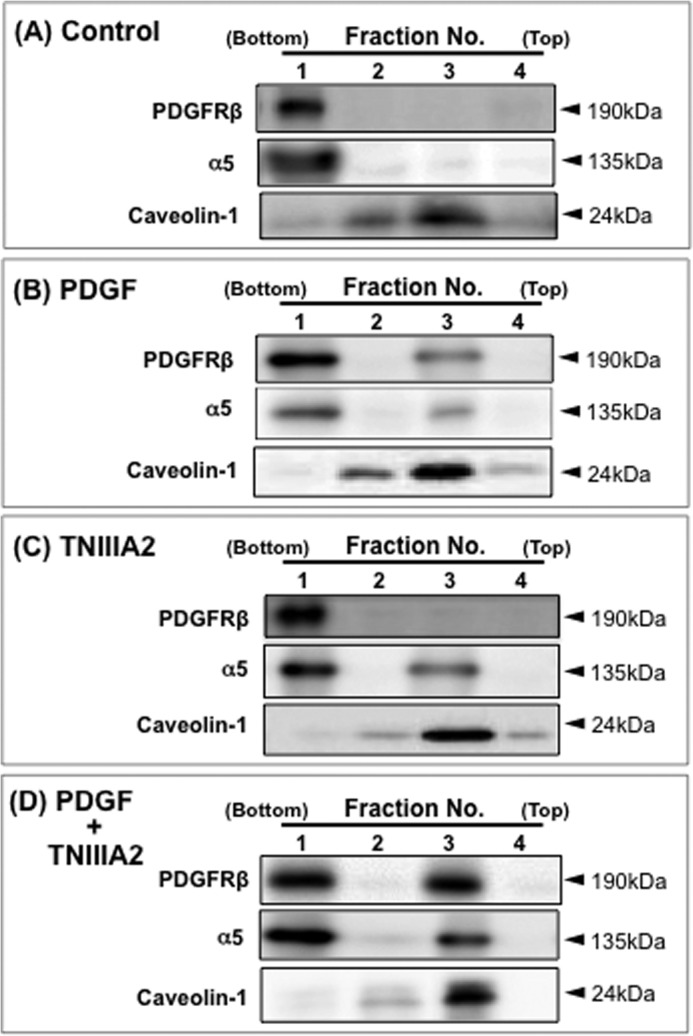
TNIIIA2 induces translocation of PDGF receptor β and β1-integrin into the caveolin-rich membrane microdomains. Cells were allowed to adhere to the fibronectin, stimulated with or without PDGF (10 ng/ml), TNIIIA2 (25 μg/ml) or a combination for 60 min, and then homogenized as described under “Experimental Procedures.” Cell homogenates were analyzed using sucrose floatation. After centrifugation, the gradient sample was divided into four fractions from the bottom of the tube, and Western blotting was performed on each fraction using anti-PDGFR-β, anti-α5 integrin, and anti-caveolin-1 antibodies. Data are representative of two individual experiments.
Taken together, TNIIIA2 facilitated the formation of a large molecular complex including syndecan-4 and activated forms of integrin α5β1 and PDGF receptor β in the membrane microdomains enriched with cholesterol and caveolin-1, which caused an enhanced activation of PDGF receptor β and its downstream MAP kinase signaling pathway, resulting in hyperstimulation of cell proliferation.
Physiological Relevance of TNIIIA2-induced Hyperstimulation of Cell Proliferation
Finally, we explored the physiological relevance of hyperstimulation of PDGF-dependent cell proliferation as a consequence of TNIIIA2-induced integrin α5β1 activation.
NIH3T3 cells actively replicated in the medium either with supplemental PDGF or without. However, cell growth was strongly suppressed from day 7 (Fig. 8A) when cells formed a confluent monolayer in a culture plate (Fig. 8B). The addition of PDGF had no remarkable effects on proliferative behavior (Fig. 8, A and B). However, when NIH3T3 cells were cultured with PDGF in combination with TNIIIA2, they continued to proliferate even after forming a confluent monolayer (Fig. 8A) and consequently formed fibroblast foci consisting of multilayered, randomly oriented cells (Fig. 8B, panel d). Focus formation was prevented by antagonizing integrin α5β1 with function-blocking mAb directed to integrin α5β1 (Fig. 8, A and 8B, panels e and f). Thus, NIH3T3 cells might acquire a transformed-like phenotype during culture in the presence of TNIIIA2, which is able to induce potentiated and prolonged activation of integrin α5β1.
FIGURE 8.

Deregulated cell proliferation induced by TNIIIA2 in integrin α5β1- and PDGF-dependent manner. Cell suspensions with or without TNIIIA2 (50 μg/ml) were seeded on 96- or 24-well plates coated with fibronectin and cultured with the medium containing calf serum (5%) under the following conditions: a, none (Control, open circles); b, +PDGF (10 ng/ml) (closed circles); c, +TNIIIA2 (50 μg/ml) (open triangles); d, +PDGF, TNIIIA2 and normal IgG (10 μg/ml) (closed triangles); e, +PDGF, TNIIIA2, and anti-α5 function-blocking mAb (10 μg/ml) (inverted triangles); f, +PDGF, TNIIIA2, and anti-αv function-blocking mAb (10 μg/ml) (rectangles). After culture for the indicated periods, cells in 96-well plates were examined by the WST assay (A), and cells in 24-well plates were stained with Giemsa (B) as described under “Experimental Procedures.” In A, each point represents the mean ± S.D. of triplicate determinations. 1 of 3 individual experiments is shown. *, p < 0.05 (closed triangle and rectangle versus open and inverted triangles).
DISCUSSION
Integrins are transmembrane adhesion receptors that control a variety of cellular processes through their activity to accept and transduce adhesion signals across the plasma membrane. The ability of integrin as an adhesion receptor is dependent on its conformational changes that specify the activation state (40). Therefore, the ability to alter the integrin activation state offers a potential explanation for the diverse cellular responses to integrin-mediated adhesion. The present study first demonstrated that the proadhesive effect of TNIIIA2 was characterized by potentiated and sustained activation of integrin α5β1. A lateral association with syndecan-4 seems to enable sustained stabilization of the active conformation of β1-integrins, including α5β1 (29). This unique effect of peptide TNIIIA2 was responsible for specific cellular responses observed in the present study.
A number of studies have shown that integrin signaling co-ordinates with RTK signaling in the regulation of cell survival and proliferation. TN-C is reportedly capable of participating in the integrin-dependent promotion of RTK signaling (41–45). For example, EGF-dependent survival and proliferation of smooth muscle cells are enhanced by TN-C through binding with its cognate receptor, integrin αvβ3 (41, 43). TN-C also enhances the cross-talk signaling of the integrin αvβ3/PDGF receptor β complex to promote proliferation and migration of smooth muscle cells (45). In all examples integrin αvβ3 serves as a mediator for the cross-talk signaling with the RTKs, although the active site in the TN-C molecule responsible for this effect has not yet been identified.
In contrast, hyperstimulation of PDGF-dependent cell proliferation induced by TNIIIA2 in our study was shown to be mediated by integrin α5β1 but not αvβ3. TNIIIA2-induced anoikis resistance was also mediated by integrin α5β1. Moreover, as a result of these effects, TNIIIA2 induced deregulated cell proliferation, in which NIH3T3 cells continued to proliferate even after forming a confluent monolayer in an integrin α5β1- and PDGF-dependent manner. Many studies indicate that integrin α5β1-mediated adhesion is an advantage to tumor cells for survival, proliferation, migration, and even chemoresistance (20–23, 46–49). Because integrin α5β1 expression positively correlates with tumor progression, it may be a promising therapeutic target for brain, breast, lung, and ovarian tumors (22–26). Activation of integrin α5β1 by TNIIIA2 may cause unfavorable effects in the host.
TN-C, a parental protein molecule of peptide TNIIIA2, also stimulated PDGF-dependent cell proliferation. A blocking study using the MMP-2/9 inhibitor BiPS and function-blocking Abs directed to TNIIIA2 and integrin subunits indicated that stimulation of cell proliferation by TN-C was mainly attributed to the activation of integrin α5β1 through proteolytic exposure of the TNIIIA2 effect. However, considering the difference between the abilities of TN-C and peptide TNIIIA2 to stimulate cell proliferation on a molar basis, the possibility that TN-C was functioning by another mechanism independent of the TNIIIA2 effect cannot be ruled out.
Increasing evidence indicates that the signaling association between RTKs and integrins induces enhanced activation of intracellular signaling (50, 51). Because this synergistic amplification of RTK-mediated signaling events by integrin could maximize cellular responses, the effect of integrins might be especially important in the active proliferation, migration, and invasion of tumor cells (50, 51). Considering the high expression of TN-C in tumor cells and tumor stroma, enhanced RTK signaling by TN-C needs to be investigated in relation to the role it plays in tumor initiation and progression. The present study showed an attenuation of contact inhibition in cell proliferation by TNIIIA2 in both a PDGF- and integrin α5β1-dependent manner. Of note, previous studies demonstrated that, on forced expression of PDGF, NIH3T3 cells undergo a loss of contact-inhibited growth and acquire the ability to grow in soft agar and form tumors in nude mice as a result of autocrine signaling (52, 53). Because coordinated input from RTKs and integrins is necessary for cell cycle progression and proliferation and because both types of receptors activate common members of the Ras/MAP kinase signaling pathway, it is not surprising that sustained activation of integrin α5β1 by TNIIIA2, in the presence of added PDGF, leads to the same result as that induced by stable expression of PDGF. An investigation is now in progress to define the effects of TNIIIA2 on PDGF-dependent malignancies expressing integrin α5β1, including glioblastoma.
Footnotes
- TN-C
- tenascin-C
- MMP-2
- matrix metalloproteinase-2
- Mβ-CD
- methyl β-cyclodextrin
- BiPS
- (2R)-((4-biphenylylsulfonyl)amino)-N-hydroxy-3-phenylpropionamide
- PDGFR
- PDGF receptor
- RTK
- receptor-type tyrosine kinase
- MAP
- mitogen-activated protein
- RGD
- Arg-Gly-Asp.
REFERENCES
- 1. Chiquet-Ehrismann R., Mackie E. J., Pearson C. A., Sakakura T. (1986) Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell 47, 131–139 [DOI] [PubMed] [Google Scholar]
- 2. Lightner V. A., Slemp C. A., Erickson H. P. (1990) Localization and quantitation of hexabrachion (tenascin) in skin, embryonic brain, tumors, and plasma. Ann. N.Y. Acad. Sci. 580, 260–275 [DOI] [PubMed] [Google Scholar]
- 3. Chiquet-Ehrismann R., Chiquet M. (2003) Tenascins: regulation and putative functions during pathological stress. J. Pathol. 200, 488–499 [DOI] [PubMed] [Google Scholar]
- 4. Orend G., Chiquet-Ehrismann R. (2006) Tenascin-C induced signaling in cancer. Cancer Lett. 244, 143–163 [DOI] [PubMed] [Google Scholar]
- 5. Chiquet-Ehrismann R., Kalla P., Pearson C. A., Beck K., Chiquet M. (1988) Tenascin interferes with fibronectin action. Cell 53, 383–390 [DOI] [PubMed] [Google Scholar]
- 6. Joshi P., Chung C,-Y., Aukhil I., Erickson H. P. (1993) Endothelial cells adhere to the RGD domain and the fibrinogen-like terminal knob of tenascin. J. Cell Sci. 106, 389–400 [DOI] [PubMed] [Google Scholar]
- 7. Murphy-Ullrich J. E. (2001) The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J. Clin. Invest. 107, 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsia H. C., Schwarzbauer J. E. (2005) Meet the tenascins: multifunctional and mysterious. J. Biol. Chem. 280, 26641–26644 [DOI] [PubMed] [Google Scholar]
- 9. Orend G. (2005) Potential oncogenic action of tenascin-C in tumorigenesis. Int. J. Biochem. Cell Biol. 37, 1066–1083 [DOI] [PubMed] [Google Scholar]
- 10. Huang W., Chiquet-Ehrismann R., Moyano J. V., Garcia-Pardo A., Orend G. (2001) Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 61, 8586–8594 [PubMed] [Google Scholar]
- 11. Midwood K. S., Valenick L. V., Hsia H. C., Schwarzbauer J. E. (2004) Coregiulation of fibronectin signaling and matrix contraction by tenascin-C and syndecan-4. Mol. Biol. Cell 15, 5670–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Woods A., McCarthy J. B., Furcht L. T., Couchman J. R. (1993) A synthetic peptide from the COOH-terminal heparin-binding domain of fibronectin promotes focal adhesion formation. Mol. Biol. Cell 4, 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lange K., Kammerer M., Hegi M. E., Grotegut S., Dittmann A., Huang W., Fluri E., Yip G. W., Götte M., Ruiz C., Orend G. (2007) Endothelin receptor type B counteracts tenascin-C-induced endothelin receptor type A-dependent focal adhesion and actin stress fiber disorganization. Cancer Res. 67, 6163–6173 [DOI] [PubMed] [Google Scholar]
- 14. Lange K., Kammerer M., Saupe F., Hegi M. E., Grotegut S., Fluri E., Orend G. (2008) Combined lysophosphatidic acid/platelet-derived growth factor signaling triggers glioma cell migration in a tenascin-C microenvironment. Cancer Res. 68, 6942–6952 [DOI] [PubMed] [Google Scholar]
- 15. Wenk M. B., Midwood K. S., Schwarzbauer J. E. (2000) Tenascin-C suppresses Rho activation. J. Cell Biol. 150, 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dedhar S., Argraves W. S., Suzuki S., Ruoslahti E., Pierschbacher M. D., (1987) Human osteosarcoma cells resistant to detachment by an Arg-Gly-Asp-containing peptide overproduce the fibronectin receptor. J. Cell Biol. 105, 1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giancotti F. G., Ruoslahti E. (1990) Elevated levels of the α5β1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell 60, 849–859 [DOI] [PubMed] [Google Scholar]
- 18. Plantefaber L. C., Hynes R. O. (1989) Changes in integrin receptors on oncogenically transformed cells. Cell 56, 281–290 [DOI] [PubMed] [Google Scholar]
- 19. Tennenbaum T., Yuspa S. H., Grover A., Castronovo V., Sobel M. E., Yamada Y., De Luca L. M. (1992) Extracellular matrix receptors and mouse skin carcinogenesis: altered expression linked to appearance of early markers of tumor progression. Cancer Res. 52, 2966–2976 [PubMed] [Google Scholar]
- 20. Roman J., Ritzenthaler J. D., Roser-Page S., Sun X., Han S. (2010) α5β1-Integrin expression is essential for tumor progression in experimental lung cancer. Am. J. Respir. Cell Mol. Biol. 43, 684–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caswell P. T., Spence H. J., Parsons M., White D. P., Clark K., Cheng K. W., Mills G. B., Humphries M. J., Messent A. J., Anderson K. I., McCaffrey M. W., Ozanne B. W., Norman J. C. (2007) Rab25 associates with α5β1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell. 13, 496–510 [DOI] [PubMed] [Google Scholar]
- 22. Kita D., Takino T., Nakada M., Takahashi T., Yamashita J., Sato H. (2001) Expression of dominant-negative form of Ets-1 suppresses fibronectin-stimulated cell adhesion and migration through down-regulation of integrin α5 expression in U251 glioma cell line. Cancer Res. 61, 7985–7991 [PubMed] [Google Scholar]
- 23. Nam J. M., Onodera Y., Bissell M. J., Park C. C. (2010) Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin α5β1 and fibronectin. Cancer Res. 70, 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adachi M., Taki T., Higashiyama M., Kohno N., Inufusa H., Miyake M. (2000) Significance of integrin α5 gene expression as a prognostic factor in node-negative non-small cell lung cancer. Clin. Cancer Res. 6, 96–101 [PubMed] [Google Scholar]
- 25. Lessey B. A., Albelda S., Buck C. A., Castelbaum A. J., Yeh I., Kohler M., Berchuck A. (1995) Distribution of integrin cell adhesion molecules in endometrial cancer. Am. J. Pathol. 146, 717–726 [PMC free article] [PubMed] [Google Scholar]
- 26. Maglott A., Bartik P., Cosgun S., Klotz P., Rondé P., Fuhrmann G., Takeda K., Martin S., Dontenwill M. (2006) The small α5β1 integrin antagonist, SJ749, reduces proliferation and clonogenicity of human astrocytoma cells. Cancer Res. 66, 6002–6007 [DOI] [PubMed] [Google Scholar]
- 27. Sawada K., Mitra A. K., Radjabi A. R., Bhaskar V., Kistner E. O., Tretiakova M., Jagadeeswaran S., Montag A., Becker A., Kenny H. A., Peter M. E., Ramakrishnan V., Yamada S. D., Lengyel E. (2008) Loss of E-cadherin promotes ovarian cancer metastasis via α 5-integrin, which is a therapeutic target. Cancer Res. 68, 2329–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin S., Cosset E. C., Terrand J., Maglott A., Takeda K., Dontenwill M. (2009) Caveolin-1 regulates glioblastoma aggressiveness through the control of α5β1 integrin expression and modulates glioblastoma responsiveness to SJ749, an α5β1 integrin antagonist. Biochim. Biophys. Acta 1793, 354–367 [DOI] [PubMed] [Google Scholar]
- 29. Saito Y., Imazeki H., Miura S., Yoshimura T., Okutsu H., Harada Y., Ohwaki T., Nagao O., Kamiya S., Hayashi R., Kodama H., Handa H., Yoshida T., Fukai F. (2007) A peptide derived from tenascin-C induces β1 integrin activation through syndecan-4. J. Biol. Chem. 282, 34929–34937 [DOI] [PubMed] [Google Scholar]
- 30. Araki E., Momota Y., Togo T., Tanioka M., Hozumi K., Nomizu M., Miyachi Y., Utani A. (2009) Clustering of syndecan-4 and integrin β1 by laminin α3 chain-derived peptide promotes keratinocyte migration. Mol. Biol. Cell 20, 3012–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faull R. J., Ginsberg M. H. (1996) Inside-out signaling through integrins. J. Am. Soc. Nephrol. 7, 1091–1097 [DOI] [PubMed] [Google Scholar]
- 32. Saito Y., Owaki T., Matsunaga T., Saze M., Miura S., Maeda M., Eguchi M., Tanaka R., Taira J., Kodama H., Goto S., Niitsu Y., Terada H., Fukai F. (2010) Apoptotic death of hematopoietic tumor cells through potentiated and sustained adhesion to fibronectin via VLA-4. J. Biol. Chem. 285, 7006–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanaka R., Owaki T., Kamiya S., Matsunaga T., Shimoda K., Kodama H., Hayashi R., Abe T., Harada Y. P., Shimonaka M., Yajima H., Terada H., Fukai F. (2009) VLA-5-mediated adhesion to fibronectin accelerates hemin-stimulated erythroid differentiation of K562 cells through induction of VLA-4 expression. J. Biol. Chem. 284, 19817–19825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matter M. L., Ruoslahti E. (2001) A signaling pathway from the α5β1 and αvβ3 integrins that elevates bcl-2 transcription. J. Biol. Chem. 276, 27757–27763 [DOI] [PubMed] [Google Scholar]
- 35. Riddell D. R., Sun X. M., Stannard A. K., Soutar A. K., Owen J. S. (2001) Localization of apolipoprotein E receptor 2 to caveolae in the plasma membrane. J. Lipid Res. 42, 998–1002 [PubMed] [Google Scholar]
- 36. Clark G. J., Cox A. D., Graham S. M., Der C. J. (1995) Biological assays for Ras transformation. Methods Enzymol. 255, 395–412 [DOI] [PubMed] [Google Scholar]
- 37. Markó K., Ligeti M., Mezo G., Mihala N., Kutnyánszky E., Kiss E., Hudecz F., Madarász E. (2008) A novel synthetic peptide polymer with cyclic RGD motifs supports serum-free attachment of anchorage-dependent cells. Bioconjug. Chem. 19, 1757–1766 [DOI] [PubMed] [Google Scholar]
- 38. Itagaki K., Naito T., Iwakiri R., Haga M., Miura S., Saito Y., Owaki T., Kamiya S., Iyoda T., Yajima H., Iwashita S., Ejiri S., Fukai F. (2012) Eukaryotic translation elongation factor 1A induces anoikis by triggering cell detachment. J. Biol. Chem. 287, 16037–16046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravid D., Leser G. P., Lamb R. A. (2010) A role of caveolin-1 in assembly and budding of the Paramyxovirus parainfluenza virus 5. J. Virol. 84, 9749–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liotta L. A., Kohn E. (2004) Anoikis: cancer and the homeless cell. Nature 430, 973–974 [DOI] [PubMed] [Google Scholar]
- 41. Jones P. L., Crack J., Rabinovitch M. (1997) Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the αvβ3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J. Cell Biol. 139, 279–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Castellon R., Caballero S., Hamdi H. K., Atilano S. R., Aoki A. M., Tarnuzzer R. W., Kenney M. C., Grant M. B., Ljubimov A. V. (2002) Effects of tenascin-C on normal and diabetic retinal endothelial cells in culture. Invest Ophthalmol. Vis. Sci. 43, 2758–2766 [PubMed] [Google Scholar]
- 43. Jones P. L., Jones F. S., Zhou B., Rabinovitch M. (1999) Induction of vascular smooth muscle cell tenascin-C gene expression by denatured type I collagen is dependent upon a β3 integrin-mediated mitogen-activated protein kinase pathway and a 122-base pair promoter element. J. Cell Sci. 112, 435–445 [DOI] [PubMed] [Google Scholar]
- 44. Perlstein I., Connolly J. M., Cui X., Song C., Li Q., Jones P. L., Lu Z., DeFelice S., Klugherz B., Wilensky R., Levy R. J. (2003) DNA delivery from an intravascular stent with a denatured collagen-polylactic-polyglycolic acid-controlled release coating: mechanisms of enhanced transfection. Gene Ther. 10, 1420–1428 [DOI] [PubMed] [Google Scholar]
- 45. Ishigaki T., Imanaka-Yoshida K., Shimojo N., Matsushima S., Taki W., Yoshida T. (2011) Tenascin-C enhances crosstalk signaling of integrin αvβ3/PDGFR-β complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J. Cell. Physiol. 226, 2617–2624 [DOI] [PubMed] [Google Scholar]
- 46. Danen E. H., Ten Berge P. J., Van Muijen G. N., Van't Hof-Grootenboer A. E., Bröcker E. B., Ruiter D. J. (1994) Emergence of α5β1 fibronectin- and αvβ3 vitronectin-receptor expression in melanocytic tumour progression. Histopathology 24, 249–256 [DOI] [PubMed] [Google Scholar]
- 47. Natali P. G., Nicotra M. R., Di Filippo F., Bigotti A. (1995) Expression of fibronectin, fibronectin isoforms and integrin receptors in melanocytic lesions. Br. J. Cancer 71, 1243–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schiller J. H., Bittner G. (1995) Loss of the tumorigenic phenotype with in vitro, but not in vivo, passaging of a novel series of human bronchial epithelial cell lines: possible role of an α5/β1-integrin-fibronectin interaction. Cancer Res. 55, 6215–6221 [PubMed] [Google Scholar]
- 49. Kanda R., Kawahara A., Watari K., Murakami Y., Sonoda K., Maeda M., Fujita H., Kage M., Uramoto H., Costa C., Kuwano M., Ono M. (2013) Erlotinib resistance in lung cancer cells mediated by integrin β1/Src/Akt-driven bypass signaling. Cancer Res. 73, 6243–6253 [DOI] [PubMed] [Google Scholar]
- 50. Guo W., Giancotti F. G. (2004) Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816–826 [DOI] [PubMed] [Google Scholar]
- 51. Soung Y. H., Clifford J. L., Chung J. (2010) Crosstalk between integrin and receptor tyrosine kinase signaling in breast carcinoma progression. BMB Rep. 43, 311–318 [DOI] [PubMed] [Google Scholar]
- 52. Fleming T. P., Matsui T., Aaronson S. A. (1992) Platelet-derived growth factor (PDGF) receptor activation in cell transformation and human malignancy. Exp. Gerontol. 27, 523–532 [DOI] [PubMed] [Google Scholar]
- 53. Lokker N. A., Sullivan C. M., Hollenbach S. J., Israel M. A., Giese N. A. (2002) Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 62, 3729–3735 [PubMed] [Google Scholar]