

Background: An efficient cardiac stress response requires a series of coordinated molecular changes.
Results: Steroid receptor coactivator-2 (SRC-2) controls the expression and activity of key cardiac transcription factors.
Conclusion: Dual regulation of cardiac transcription poises SRC-2 as a novel coordinator of the stress-responsive molecular network.
Significance: Identification of factors responsible for coordination of the cardiac stress response is critical for improved treatment.
Keywords: Cardiac Metabolism, Gene Regulation, Transcription Factor, Transcription Target Gene, Transcriptional Coactivator
Abstract
We have previously demonstrated the potential role of steroid receptor coactivator-2 (SRC-2) as a co-regulator in the transcription of critical molecules modulating cardiac function and metabolism in normal and stressed hearts. The present study seeks to extend the previous information by demonstrating SRC-2 fulfills this role by serving as a critical coactivator for the transcription and activity of critical transcription factors known to control cardiac growth and metabolism as well as in their downstream signaling. This knowledge broadens our understanding of the mechanism by which SRC-2 acts in normal and stressed hearts and allows further investigation of the transcriptional modifications mediating different types and degrees of cardiac stress. Moreover, the genetic manipulation of SRC-2 in this study is specific for the heart and thereby eliminating potential indirect effects of SRC-2 deletion in other organs. We have shown that SRC-2 is critical to transcriptional control modulated by MEF2, GATA-4, and Tbx5, thereby enhancing gene expression associated with cardiac growth. Additionally, we describe SRC-2 as a novel regulator of PPARα expression, thus controlling critical steps in metabolic gene expression. We conclude that through regulation of cardiac transcription factor expression and activity, SRC-2 is a critical transcriptional regulator of genes important for cardiac growth, structure, and metabolism, three of the main pathways altered during the cardiac stress response.
Introduction
There are many transcriptional changes in response to cardiac stress; some changes contribute directly to the stress response, and others are compensatory changes. Collectively, there are few gene targets overlapping between studies that are “key” factors in these responses (1), but there is some overlap among pathways that are affected.
Several recent studies highlighted a role for coordinated control of several main cardiac pathways through a transcription factor complex including myocyte enhancer factor 2 (MEF2)2 and GATA-4 (2, 3). Transcriptional cross-talk occurs between MEF2, GATA-4, and other cardiac transcription factors, including NK2 homeobox 5 (Nkx2.5), serum response factor (SRF), and Tbx5. This coordinated control shows extensive target gene overlap among the factors, as well as control of several targets of similar signaling pathways (2–7). Many of these pathways are altered with stress, and genetic mouse models of these transcription factors confirm a role for the factors in controlling aspects of the stress response including structural, hypertrophic, and apoptotic pathways (8–15). Nevertheless, little is known about the mechanisms responsible for coordinate control of DNA-bound transcription factors.
Steroid receptor coactivator-2 (SRC-2) belongs to a family of transcriptional coactivators, the steroid receptor coactivators, well characterized for their ability to increase transcription at target genes driven by diverse transcription factors (16). Previously, we have shown that loss of SRC-2 results in several gene expression changes that mimic those of a stressed heart despite maintaining normal function under unstressed conditions. Major pathways affected include metabolic, sarcomeric, and hypertrophic functions. The onset of cardiac stress via transverse aortic constriction results in decreased function in hearts lacking SRC-2, and a normal hypertrophic response does not occur (17). These data suggest an important role for SRC-2 in regulating cardiac gene expression, but fail to delineate genes directly controlled by SRC-2 from compensatory changes due to stress or what factor(s) SRC-2 works with on those genes because it cannot directly bind DNA.
To characterize the molecular actions of SRC-2 in the heart, we used our previously published microarray data combined with newly generated chromatin immunoprecipitation-sequencing (ChIP-Seq) data to identify SRC-2 targets and correlate these with cardiac actions of SRC-2. We identified a novel mechanism by which SRC-2 acts as a major regulator of the cardiac transcription program through control of widespread cardiac transcription factor expression, as well as through coactivation of key cardiac transcription factors MEF2, GATA-4, and Tbx5. Furthermore, SRC-2 promotes the adult metabolic transcriptional profile through control of peroxisome proliferator-activated receptor (PPAR) α expression through a mechanism that at least partially involves control of GATA-4 and Tbx5 activity. These activities together poise SRC-2 to control adult cardiac gene expression including genes involved in cardiac growth and metabolism under unstressed conditions. In addition, it serves as a coordinator of transcription during the cardiac stress response.
EXPERIMENTAL PROCEDURES
Animals
All animal experiments were approved by the Institutional Animal Care Research Committee at Baylor College of Medicine (Protocol AN-544 and AN-124). The generation of the SRC-2 knock-out (KO) mice has been described previously (18–20). Cardiac-specific SRC-2 KO mice were generated through standard cross-breeding between mice with a floxed SRC-2 gene (SRC-2f/f) (21) and mice expressing Cre recombinase under the Myh6 promoter (Jackson Laboratories) resulting in cardiomyocyte-specific deletion of SRC-2. This breeding results in Myh6-Cre (−)/SRC-2f/f (WT) and Myh6-Cre (+)/SRC-2f/f (CKO) genotypes. Only male, age-matched littermates (10–16 weeks old) mice were used. Animals were maintained in a temperature-controlled (23 °C) facility with a 12-h light/dark cycle. Mice on normal chow were fed 2920X Teklad Global rodent ad libitum with free access to food and water.
Plasmids and Reagents
The pCR3.1-SRC-2 plasmid has been described previously (22). The pCGN-GATA-4, PXP2-α-MHC-Luc, and PXP2–3xMCK MEF2 (M2RE)-Luc were a generous gift from Dr. Mona Nemer (6). pCMX-MEF2A, C, and D were a generous gift from Dr. Hung-Ying Kao (23). G5-Luc-skeletal α-actin and cardiac α-actin luciferase reporters have been described previously (4). pEntr-Tbx5-flbio was purchased through Addgene, provided by Dr. William Pu (plasmid 32968) (2). pXP1-Fgf10-Luc was purchased through Addgene, provided by Dr. Benoit Bruneau (plasmid 24994) (24). pGL2T2 (2xTbox)-Luc was a generous gift from Dr. Peter Hurlin (25). hPPARα promoter (pα(H-H)-pGL3 was a generous gift from Dr. Bart Staels (26). Adenovirus containing GFP and GFP-Cre recombinase (Ad5-CMV-GFP and Ad5-CMV-Cre-GFP, respectively) were purchased from the Vector Development Core at Baylor College of Medicine. For adenoviral HA-SRC-2, mouse SRC-2 was cloned into the pShuttle vector and used for adenovirus production by the Viral Vector Production Core Laboratory at Baylor College of Medicine. pAdeno-CMV-GATA4 and pAdenoG-HA-CMV-mTbx5 were purchased from Applied Biological Materials, Inc. siRNAs against SRC-2, MEF2A, C, and D, and Tbx5 were purchased from Dharmacon. siRNA against GATA-4 was purchased from Ambion.
Cell Culture
H9c2 cells (ATCC) were maintained at 37 °C at 5% CO2 in Dulbecco's Eagle's modified medium (DMEM) supplemented with 10% charcoal-stripped fetal bovine serum (FBS), 1 mm sodium pyruvate, 50 units/ml penicillin G, and 50 μg/ml streptomycin sulfate.
Gene Expression Analyses
RNA was isolated from either cells or frozen heart tissue using the RNeasy RNA Isolation kit or Fibrous Tissue kit, respectively, according to manufacturer's instructions (Qiagen). cDNA analysis was performed on 500–1000 ng of RNA using random primers and the Superscript III enzyme according to the manufacturer's instructions (Invitrogen). qPCR analyses were performed using the Taqman system with gene-specific primers and the Universal Probe Library (Roche Applied Science) on a One-Step Plus qPCR machine (ABI). Primer sequences are available upon request.
siRNA-mediated Knockdown
H9c2 cells were cultured overnight in antibiotic medium and then trypsinized and collected in fresh antibiotic-free medium. Cells were washed twice with 1× PBS and resuspended at a density of 1 × 107 cells/ml in R-buffer for electroporation using the Neon system and according to the manufacturer's instructions (Invitrogen). The indicated siRNAs were added to cells in R-buffer, and electroporation was carried out at 3 pulses of 1650 V with a width of 10. Cells were immediately placed into culture medium and harvested 48 h later.
Adult Cardiomyocyte Isolation
Adult cardiomyocytes were isolated as described previously (27). Briefly, freshly isolated hearts were perfused with a collagenase solution through aortic cannulation followed by mincing and calcium reintroduction. Cardiomyocytes from several mice were pooled, plated, and finally cultured at 2% CO2 in medium containing ITS medium supplement and 2,3-butanedione monoxime. Where indicated, adenovirus was introduced after overnight incubation of isolated myocytes, and cells were harvested 36–48 h later.
Chromatin Immunoprecipitation
Fresh adult heart tissue was isolated and ChIP analyses were carried out according to manufacturer's instructions using the SimpleChip Enzymatic Chromatin IP Kit (Cell Signaling Technology). Three hearts were pooled for each chromatin preparation. DNA shearing and micrococcalnuclease digestion were optimized according to manufacturer's instructions. Each IP was conducted with 3–4 μg of antibody. Beads and antibody were preincubated in 0.5% bovine serum albumin in 1× PBS overnight before the addition of chromatin. Final products were eluted in 50 μl and then diluted 2–3-fold before qPCR analysis with SYBR Green and gene-specific primers. Primer sequences are shown in Table 1. Negative control primers target an untranslated region of the genome that shows no SRC-2 recruitment in the heart in ChIP-Seq analyses.
TABLE 1.
Primer sequences for ChIP assays
| Target | Forward primer | Reverse primer |
|---|---|---|
| Untr10 | tacacatgaggcccaggatca | tggctccttcagtcctttatg |
| Hand2 | cactcccaatcgcacctta | ggtggtggcgacaagagt |
| Nkx2–5 | ttagactcagcataacagaatcagg | gctcctcgttagcctgaaaa |
| MEF2a | ggtccttcaaagagggaagc | gaaggagatgacggctgct |
| SRF | cggagtcgaaagactcagga | aaagagggcagggatagattg |
| MEF2d | gggtcatacagtgcaacacg | aacttgtgaatttggtgtgagttt |
| GATA-4 | cgtggggatctcaggaaa | ccttcggtttggaaaagagaat |
| Cxcl12 | agggaactctttggtccttttta | tcagtgtttcctgcctttcc |
| Adam19 | ttgcatcctaaaccctaacca | ggctgtataggttggggac |
| Itga6 | ttagggtaagaaaaggggacaat | ggtagggaacactgagtccttct |
| Serca2 | ctctcgttgaccccgaag | ggagaacgctcacacaaagac |
| Tnni1 | gctccgggttttcctaagtt | acacccctcctcctttgact |
| Actc1 | gaccctcagacaacccttctc | gacccactgcagacatggt |
| Myh6 | ccataagactaaggaagagcattga | caggctcaacgccaactc |
| Gadd45g | cagggttctcggtgcttg | aaatattgcctcgcgttgac |
| PPARa | aaatgggcatcgaggagag | ctggacggcagtgtctga |
ChIP-Seq
Adult cardiomyocytes were isolated as described above. Immediately after isolation, cells were formaldehyde-cross-linked and stored at −80 ºC. Cells from four hearts were pooled, and ∼1 × 105 cells were used for chromatin preparation. Chromatin preparation and immunoprecipitation reactions were carried out as described previously with 3 μg of anti-SRC-2 antibodies (28). Input DNA and enriched DNA fragments were used to generate ChIP-Seq libraries for deep sequencing on the Illumina GAII platform. Library preparations reagents were from Illumina's Genomic DNA Sample Preparation kit (1000181) and prepared following a modified version of the manufacturer's recommendations. Briefly, DNA fragments were end-repaired using T4 DNA polymerase to fill in 5′ overhangs, Klenow polymerase to remove 3′ overhangs, and T4PNK to phosphorylate the 5′-OH. For Illumina adaptor ligation, a single adenine nucleotide overhang must be added to the polished DNA. A 1:30 dilution of the adaptor oligonucleotide mix was used in the ligation step with T4 ligase. The resulting constructs with adaptors were amplified via 18 cycles of PCR enriching the DNA fragments with adaptors flanking each end. The amplified product was quantified using a spectrophotometer at 260 nm (Nanodrop) and an aliquot analyzed by gel electrophoresis on a 6% TBE gel for size validation. The validated DNA library was submitted at a 10 nm concentration for sequencing. Samples were sequenced using the Illumina GAII platform at the University of Houston.
ChIP-Seq Data Processing
ChIP-Seq data were aligned to the mm9 reference genome using bowtie (29) with parameters -t -v 2 -a -m 1 -best -strata -S. Reads, which mapped to multiple locations in the genome, were filtered using Samtools (30) for subsequent analysis. ChIP-Seq fragment length estimation and peaks identification were done sequentially using HOMER (31). Using annotatePeaks.pl from HOMER (31) gene-specific information (nearest gene, distance from nearest transcription start site (TSS)) was assigned to each peak based on the nearest annotated TSS of the peak. We also classified peaks into promoter, integenic, intron, and exon. The peak regions were processed using findMotifsGenome.pl (31) for enriched motifs (with parameters -cpg -len 3,4,5,6,7,8 -N 50000 -S 25). To identify locations of specific transcription factors (TFs) in peaks, we obtained the position weight matrices of these TFs from JASPAR. These were used together with HOMER (31) to identify TF locations in peaks.
Immunoprecipitation
H9c2 cells were untreated or infected with the indicated adenovirus for 48 h before harvesting. Cells were lysed in radioimmuneprecipitation assay buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) plus protease inhibitors. The resulting lysates were incubated with the indicated antibodies and protein G Dynabeads (Invitrogen) overnight in NETN (20 mm Tri-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 10% glycerol, 1 mm DTT, 0.1% Nonidet P-40) with protease inhibitors at 4 ºC. Immunoprecipitates were washed five times with NETN and analyzed via SDS-PAGE. The products were visualized with anti-HRP conjugates and ECL detection (Pierce). Antibodies used were anti-SRC-2 (A300-345A; Bethyl Laboratories), anti-TIF2 (BD Biosciences), anti-MEF2 (B-4, Santa Cruz Biotechnology), anti-GATA4 (G-4, Santa Cruz Biotechnology), and anti-Tbx5 (Invitrogen).
Immunohistochemistry
Immunohistochemistry on deparaffinized heart sections was performed by standard techniques. Endogenous peroxidases were blocked by incubation in a hydrogen peroxide/methanol solution followed by antigen retrieval in Tris-EDTA, pH 9.0 (Diagnostic BioCare). Sections were then blocked with Background Sniper (BioCare Medical). Primary antibody incubation was performed overnight at 4 ºC with anti-TIF2 antibodies (BD Biosciences) at a concentration of 1:75. Detection was performed with the PolyVue Detection kit (Diagnostic Biosystems) and staining with Stable DAB Plus (Diagnostic Biosystems) for 5 min at room temperature. DAB staining was enhanced using Sparkle (BioCare Medical) for 1 min at room temperature, and counterstaining was performed using CAT hematoxylin (BioCare Medical) for 15–30 s. All rinses were made using Tris-Tween buffer, pH 7.4. All reagents and kits were used according to manufacturer's instructions. Imaging was performed on an Olympus BX41 microscope.
Mitochondrial Oxygen Consumption Rate
Viable mitochondria were isolated from WT or SRC-2 CKO mice hearts using the Mitochondrial Isolation kit from Sigma-Aldrich per the manufacturer's instructions. 50–100 mg of tissue was used per heart. Mitochondria were kept on ice and used immediately for analysis of oxygen consumption rate with the Seahorse XF24 Analyzer following the manufacturer's recommended procedure for isolated mitochondria. Briefly, mitochondria were diluted to 0.05 μg/μl in 1× MAS buffer (70 mm sucrose, 220 mm mannitol, 10 mm KH2PO4, 5 mm MgCl2, 2 mm HEPES, 1 mm EGTA, and 0.2% (w/v) fatty acid-free BSA, pH 7.2, at 37 ºC) containing 10 mm succinate and 2 μm rotenone and plated. The plate was centrifuged at 2000 × g for 20 min at 4 ºC to adhere mitochondria. Injections were 50 μl of 40 mm ADP, 55 μl of 25 μg/μl oligomycin, 60 μl of 40 μm FCCP, and 65 μl of 40 μm antimycin A. All measurements were collected for 3 min.
RESULTS
SRC-2 Loss Affects Cardiac Transcription Factor Target Gene Expression
Our previous work has shown that whole body loss of SRC-2 results in disruption of the cardiac stress response with altered signaling in several stress-responsive pathways. Further, we previously observed a surprising decrease in expression of several cardiac transcription factors in the absence of SRC-2 (17). However, characterizing cardiac-specific actions of SRC-2 in these initial studies was limited by the use of a whole body KO model, allowing for secondary effects from SRC-2 loss in other tissues or cardiac cell types. Therefore, we created a cardiomyocyte-specific SRC-2 KO (CKO) CKO, with SRC-2 loss under the control of the previously described α-myosin heavy chain promoter (Fig. 1, A and B). We observed the same decreases in widespread cardiac transcription factor expression when SRC-2 is lost in the cardiomyocyte (Fig. 1C).
FIGURE 1.

Loss of SRC-2 disrupts cardiac transcription factor target gene expression. A, immunohistochemistry of heart sections taken from adult WT or SRC-2 CKO mice with anti-SRC-2 antibodies. B, qPCR analysis of mRNA expression of SRC-2 in hearts and liver RNA from adult WT or SRC-2 CKO mice. Individual gene expression is normalized to an 18S rRNA internal control. C, mRNA expression analysis via qPCR for cardiac transcription factors in RNA isolated from WT and SRC-2 CKO hearts as in A. D, gene set enrichment analysis (GSEA) transcription factor motif enrichment of genes altered in the heart from loss of SRC-2 (17) with a False Discovery Rate ≤ 0.15. Bold lanes highlight well characterized cardiac transcription factors. E, genes used in D were analyzed for MEF2, GATA-4, and Tbx5 binding by ChIP-Seq analysis in HL-1 cells (2). Statistical analysis was performed with Student's t test where * = p ≤ 0.05, ** = p ≤ 0.01, and *** = p ≤ 0.001.
Additionally, to help identify which transcription factor or factors SRC-2 may be working with in the heart, we conducted enrichment analyses for common transcription factor binding motifs in the proximal promoter regions on the genes whose expression changed from loss of SRC-2 in our previous microarray analysis (17). In support of the previously characterized pleiotropic nature of SRC-2 in transcriptional regulation, no single transcription factor-binding motif was highly enriched; however, several motifs showed moderate enrichment (Fig. 1D). Interestingly, we noticed enrichment for binding sites of several well characterized cardiac transcription factors (in bold) that are known to synergize in target regulation (2, 3), including MEF2 and GATA. Recent work has highlighted the importance of these factors in the control of adult cardiac gene expression through the identification of genomic targets of these factors in HL-1 atrial cardiomyocytes. This work also introduced Tbx5 as a new key regulator in the adult heart (2). In support of a role for enrichment of these factors in SRC-2 gene targets, we found that 57.6% of genes with expression changes resulting from loss of SRC-2 are genes targeted by these transcription factors (Fig. 1E).
We hypothesized that SRC-2 could be affecting expression of target genes of this complex through direct regulation of expression of the transcription factors themselves. To test whether the decrease in transcription factor expression observed in the CKO hearts is due to direct effects of SRC-2 loss, we investigated whether transient loss of SRC-2 also resulted in a decreased expression of the transcription factors. Indeed, both siRNA-mediated SRC-2 knockdown in H9c2 cultured cardiomyocytes and Cre-mediated excision of SRC-2 from isolated adult cardiomyocytes resulted in decreased expression of MEF2, GATA-4, Tbx5, and several other cardiac transcription factors (Fig. 2, A and B). Further, protein expression mimics the decreased mRNA in CKO hearts compared with WT controls (Fig. 2C). ChIP experiments show that SRC-2 localizes to the promoter regions of several of these transcription factors, further supporting its ability to regulate expression of these transcription factors directly (Fig. 2D). To test whether the decreased transcription factor expression in the absence of SRC-2 is enough to effect target gene expression, we analyzed gene expression in CKO hearts for genes that are not known to be targeted directly by SRC-2, but are bound by MEF2, GATA-4, and/or Tbx5. As expected, we observed decreased cardiac TF expression in the absence of SRC-2 (Fig. 2E), despite any observed SRC-2 binding (Fig. 2F, compare binding of SRC-2 to a target gene to the negative control). Together, these results indicate that SRC-2 loss has a strong effect on the gene network controlled by MEF2, GATA-4, and Tbx5 and strongly suggest that this control is at least in part through direct regulation of their expression.
FIGURE 2.

SRC-2 controls cardiac expression of several transcription factors. A, qPCR analysis of cardiac transcription factors in H9c2 cells after siRNA-mediated transient knockdown of SRC-2 compared with control knockdown. Individual genes are normalized to an 18S rRNA or GAPDH internal control. B, qPCR analysis of cardiac transcription factors as in A. RNA was isolated from isolated primary adult cardiomyocytes derived from SRC-2f/f animals and cultured after isolation for 48 h with adenovirus expression GFP (control) or Cre recombinase (to excise SRC-2). C, immunoblot analysis of tissue lysates from WT and SRC-2 CKO hearts for the indicated transcription factors. Quantitative analysis is derived from densitometry measurements from at least two independent blots of n ≥ 4 animals. D, ChIP analysis in chromatin isolated adult WT heart tissue with anti-SRC-2 or IgG antibodies for the indicated target gene regions by qPCR. Individual target genes are presented relative to input controls. Untr10′ is a negative control region void of SRC-2 localization. E, qPCR analysis of the indicated gene targets as in A. RNA was isolated from WT or SRC-2 CKO hearts. F, ChIP analysis as described in D for the indicated target genes. Statistical analysis was performed with Student's t test where * = p ≤ 0.05, ** = p ≤ 0.01, and *** = p ≤ 0.001.
SRC-2 Coactivates Cardiac Transcription Factors to Regulate Several Gene Networks
It has been shown that MEF2, GATA-4, and Tbx5 can autoregulate their own and each other's expression (2, 3), raising the possibility that SRC-2 may coactivate one or several of these factors, thereby directly controlling their expression, as well as the expression of the other factors. Due to the possibility for both direct and indirect targets, we performed ChIP-Seq analysis for identification of genome-wide binding sites of SRC-2 in isolated adult primary cardiomyocytes. As anticipated, SRC-2 binding sites are strongly enriched within 20 kb of the TSS of all genes, with strongest enrichment even closer to the TSS (Fig. 3A). Using the same previously published ChIP-Seq datasets for MEF2, GATA-4, and Tbx5 in HL-1 cells (2) (used for comparison with SRC-2 microarrays), we found that the binding peaks for SRC-2 overlap at least 1 base pair with a binding site for MEF2, GATA-4, and/or Tbx5 in at least 50% of genes targeted by SRC-2 in the −7.5 kb to +2.5 kb region (Fig. 3B). This overlap is decreased to approximately 30% as the region analyzed is expanded to −25 kb to +25 kb, which is supported by enrichment of binding sites for MEF2, GATA-4, and Tbx5 being proximal to the TSS (Fig. 3B and (2). In further support, using primers generated to analyze genomic locations identified by He et al. to bind MEF2, GATA-4, and/or Tbx5, we found by ChIP that SRC-2 localized to many of the same genomic regions (Fig. 3C) (2). If SRC-2 is coactivating expression of these transcription factors at the gene promoter, it is anticipated that loss of SRC-2 would decrease expression of the target gene. Not surprisingly, analysis of gene expression in SRC-2 KO hearts confirms decreased expression of these target genes (Fig. 3D).
FIGURE 3.

Chromatin occupancy of SRC-2 largely overlaps that of MEF2, GATA-4, and Tbx5. A, analysis of chromatin localization sites for SRC-2 derived from ChIP-Seq analysis of SRC-2 in isolated adult cardiomyocytes as described under “Experimental Procedures” for the distance of each peak relative to the TSS. B, representation of overlap resulting from overlapping binding sites of at least 1 base pair of all binding peaks from the SRC-2 ChIP-Seq overlaid with binding peaks of MEF2, GATA-4, and Tbx5 in HL-1 cells (2). The peaks analyzed were isolated from the indicated regions under each Venn diagram relative to the TSS. C, ChIP analysis as described in Fig. 2C for the indicated target genes. D, qPCR analysis of mRNA expression for the indicated target genes as described in Fig. 1B. Statistical analysis was performed with Student's t test where * = p ≤ 0.05 and ** = p ≤ 0.01.
To further investigate the possibility of SRC-2 coactivating MEF2, GATA-4, and Tbx5, we performed co-immunoprecipitation experiments and found that SRC-2 can interact with each of these factors (Fig. 4A). Additionally, co-expression of SRC-2 with the transcription factor leads to increased transactivation of previously characterized target gene reporters for each of these factors, as indicated by analysis of luciferase reporters (Fig. 4, B–D). Collectively, these results show that SRC-2 is able to bind to and coactivate MEF2, GATA-4, and Tbx5 and strongly suggest that these activities occur at gene promoters targeted by these factors.
FIGURE 4.

SRC-2 interacts with and coactivates MEF2, GATA-4, and Tbx5. A, co-immunprecipitation (IP) analysis performed with the indicated antibodies or IgG controls in H9c2 cardiomyocyte whole cell lysates. Whole cell lysates for MEF2 and Tbx5 immunoprecipitations were isolated from H9c2 cells with endogenous protein. Whole cell lysates for GATA-4 IP were isolated from H9c2 cells infected with adenovirus for HA-SRC-2 or GATA-4 as indicated. Immunoblots were performed with the indicated antibodies. + indicates the removal of irrelevant lanes. All lanes shown are from a single gel. B–D, transactivation assays performed in HeLa cells on the ability of the indicated factors to activate luciferase activity driven by the promoters. SRC-2 or transcription factors were expressed from transiently transfected expression plasmids. RLU, relative luciferase units.
SRC-2 Controls Metabolic Gene Expression through Regulation of PPARα Expression
One major component of the cardiac stress response that is perturbed by loss of SRC-2 in the heart is the metabolic gene signature (17). Because loss of MEF2 and GATA-4 in the adult heart has not been shown to have a metabolic landscape similar to that of SRC-2 (8–10, 12, 14) and knock-out of Tbx5 in the adult heart has not been characterized, we hypothesized that SRC-2 was controlling these pathways through an alternate transcription factor. Loss of PPARα expression in the heart closely resembles loss of SRC-2, and we previously reported decreased PPARα expression in SRC-2 KO hearts (17). This decreased mRNA expression also is observed in mice with cardiac-specific loss of SRC-2 (Fig. 5A). Similar to that observed for MEF2, GATA-4, and Tbx5, PPARα expression was also decreased through transient loss of SRC-2 either by siRNA or Cre-mediated excision in myocytes and paralleled decreased protein expression in CKO hearts (Fig. 5A). Furthermore, the transcription factor motif analysis we performed in the SRC-2 microarray (Fig. 1A) identified a direct repeat 1 DNA response element (listed under hepatocyte nuclear factor, which is the DNA motif recognized by PPARs (32). To investigate whether SRC-2 directly controls PPARα expression, we investigated the ability of SRC-2 to localize to the PPARα promoter and whether transient loss of SRC-2 decreases PPARα expression. Indeed, ChIP experiments confirm the ability of SRC-2 to associate with the PPARα promoter in isolated adult primary cardiomyocytes (Fig. 5B). Furthermore, SRC-2 is able to coactivate luciferase expression driven by the PPARα promoter (Fig. 5C).
FIGURE 5.

SRC-2 controls PPARα expression. A, qPCR analysis of mRNA expression for the PPARα as described in Figs. 1B and 2, A and D, for analysis of RNA isolated from cardiac-specific SRC-2 KO hearts, siRNA mediated SRC-2 knockdown in H9c2 cells, and Cre-mediated SRC-2 excision in adult primary cardiomyocytes, respectively. Immunoblot analyses of protein expression for PPARα were as described in Fig. 2C. B, ChIP analysis as described in Fig. 2C for the PPARα promoter. C, transactivation assay as described in Fig. 4B for luciferase driven by the PPARα promoter. RLU, relative luciferase units. D–F, qPCR analysis of mRNA expression for the indicated target genes as described in Fig. 1B. F, oxygen consumption rates (OCR versus time) of mitochondria isolated from WT and SRC-2 CKO hearts. Oxygen consumption rate was performed on the Seahorse XF24 analyzer. Injections were at the times indicated as follows: A, ADP; B, oligomycin (ATP coupler); C, FCCP (electron transport chain accelerator); and D, antimycin A (complex III inhibitor). Statistical analysis was performed with Student's t test where * = p ≤ 0.05, ** = p ≤ 0.01, and *** = p ≤ 0.001.
Our previous study suggests that loss of SRC-2 results in decreased expression of several genes involved in fatty acid oxidation (17). Many of these genes are regulated by PPARα and are also decreased in CKO hearts (Fig. 5D), suggesting that SRC-2 controls their expression through regulation of PPARα expression. Another major cardiac metabolism regulator, peroxisome proliferator-activated receptor γ, coactivator-1 (PGC-1α) strongly regulates mitochondrial and oxidative phosphorylation pathway genes, often through the nuclear receptor estrogen-related receptor α (ERRα). We did not observe any major changes in ERRα or any ERRα/PGC-1α mitochondrial protein targets (Fig. 5E). Furthermore, the ERRα/PGC-1α signaling axis strongly regulates mitochondrial biogenesis. In support of the unaltered gene expression of ERRα/PGC-1α targets from loss of cardiomyocyte SRC-2, we observed no major changes in mitochondrial number as assayed by citrate synthase expression in CKO hearts (Fig. 5F). Finally, we analyzed mitochondrial respiratory capacity using the Seahorse XF24 Analyzer on mitochondria isolated from CKO hearts. In this assay, the isolated mitochondria begin coupled in state 2 by the addition of the substrate succinate and complex I inhibitor rotenone. From this state, we observed no major differences in basal respiration between the two groups, nor in state 3 (ADP added), state 4o (oligomycin added), or maximal uncoupler-stimlated respiration (FCCP added) (Fig. 5G), supporting the idea that mitochondrial capacity is not impaired from loss of SRC-2.
Considering our results indicating SRC-2 coactivates MEF2, GATA-4, and/or Tbx5, we analyzed the promoter fragment used in luciferase experiments as well as the ChIP-Seq data from HL-1 cells (2) and found that SRC-2 appears to control PPARα by coactivating GATA-4 and/or Tbx5 on the PPARα promoter. Transactivation assays support a role for GATA-4- and Tbx5-driven expression of this promoter and coactivation by SRC-2, whereas MEF2 did not appear to have a robust effect (Fig. 6A). Control of PPARα by GATA-4 and Tbx5 is supported through transient knockdown of these factors in H9c2 cells resulting in decreased PPARα expression (Fig. 6B). Knockdown of SRC-2 does not significantly decrease PPARα expression, further suggesting a conserved pathway for these factors in controlling PPARα expression (Fig. 6B).
FIGURE 6.

SRC-2 control of PPARα may be mediated through GATA-4 and Tbx5. A, transactivation assay as described in Fig. 4B for luciferase driven by the PPARα promoter. B, qPCR analysis of mRNA expression for PPARα or the indicated targets as described in Fig. 2A for siRNA-mediated knockdown in H9c2 cells. Statistical analysis was performed with Student's t test where * = p ≤ 0.05, ** = p ≤ 0.01, and *** = p ≤ 0.001. NS indicates p > 0.05.
DISCUSSION
We have described a dual role for SRC-2 in controlling cardiac gene expression through coordinated control of expression of key cardiac transcription factors as well as direct coactivation of MEF2, GATA-4, and Tbx5 (Fig. 7). These results describe the first characterized mechanism for SRC-2 activity in the heart and define several new transcription factors that SRC-2 can regulate in a context-dependent manner. First, we show that SRC-2 can directly control transcription of a number of cardiac transcription factors, including, but not limited to, MEF2, GATA-4, and Tbx5. Importantly, loss of SRC-2 only decreases, but does not abrogate, expression of the factors. Therefore, loss or down-regulation of SRC-2 will result in a widespread dampening of transcription promoted by these factors through a decrease in the availability of the factors to bind to promoters and drive transcription. Included in these target promoters are those of several other transcription factors, leading to a vast array of genes and pathways affected by loss of SRC-2, as we previously observed (17).
FIGURE 7.
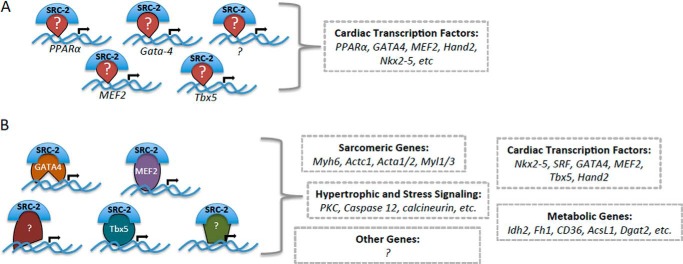
Model of SRC-2 action in cardiomyocytes. A, SRC-2 directly controls cardiac transcription factor expression. This may be through direct regulation of activity of the transcription factors through a feed-forward regulatory loop or through other unidentified transcription factors. B, SRC-2 coactivates cardiac transcription factors to control activation of their target genes. This is anticipated to extend to other unidentified transcription factors to encompass the whole subset of SRC-2 target genes.
Second, we show that SRC-2 can bind and coactivate transcription driven by MEF2, GATA-4, and Tbx5. This second layer of regulation further controls expression of target genes of these factors. Whereas the presence of the transcription factor at a specific site may be sufficient to induce a basal level of transcription, the absence of SRC-2 at those genes would result in an inability to increase transcription of the target when required. This lack of coactivation may be especially evident in response to signal-dependent transcription programs, such as those activated during cardiac stress. As a result, loss of SRC-2 would likely lead to an unorganized, ill timed, or decreased ability to respond to the stress, similar to the observation under transverse aortic constriction in SRC-2 KO animals (17). Further investigation into these specific signaling pathways will be required to identify specific roles for SRC-2 interplay with these factors at specific sets of target genes.
Our earlier work described widespread genomic remodeling in the absence of SRC-2 (17), which is supported by the mechanisms described in these studies for both direct and indirect control of gene expression by SRC-2. These pathways include those regulated by MEF2, GATA-4, and Tbx5 including sarcomeric and hypertrophic regulatory pathways (8, 10, 12, 14, 33). For example, Cxcl12, Adam19, and Itga6 (Fig. 2E) are all involved in chemokine/membrane receptor signaling, which in the heart is intimately linked with signal transduction and beating, which rely on calcium and ion transients involving genes such as Serca2 and Tnni3 (Fig. 3D). Understanding how SRC-2 controls these pathways in a context- and signal-dependent manner is expected to yield valuable information about coordination of cellular communication and maintenance of electrical conduction during stress. Furthermore, as shown previously (2, 3), cooperation among several of these transcription factors at certain targets may add another layer of complexity to SRC-2 regulation. This cooperation involves not only binding sites and cross-talk among multiple transcription factors at the same promoter (2, 3), but also recruitment of one transcription factor by the others for synergistic activity. For example, GATA-4 interacts with Hand2, MEF-2, Nkx2.5, and SRF, among other factors, to cooperatively regulate gene expression at many cardiac targets (for review, see Ref. 34). Interestingly, SRF has been shown capable of coactivating and synergizing with GATA-4 and belongs to the same (MADS)-box family of transcription factors as MEF-2 (35). Whereas our current analyses enriched for a binding site that more closely resembles that of MEF2 (Fig. 1D) (36, 37), we speculate that SRF also may be a key player in this transcription network whose activity also may be regulated by SRC-2. It is possible that these dynamic transcription factor complexes are further regulated through (or require interactions with) coactivator complexes that are SRC-2-dependent. Adding another layer of complexity, the transcription activity of MEF2 and GATA-4 is controlled through multiple posttranslational modifications, rendering them sensitive to several signaling pathways in the myocyte including calcium flux in the case of MEF2, and hypertrophic signaling for GATA-4 (for review, see Ref. 34). These modifications likely affect which factors the DNA-bound transcription factor interacts with, including the exchange of corepressors to coactivators as well as differential chromatin modifiers, but specifically which role these modifications play in regulating their interactions with SRC-2 remains to be determined. Furthermore, with the recent evidence that overexpression of GATA-4, MEF2, and Tbx5 can drive differentiation of fibroblasts to cardiomyocytes (38, 39), it is tempting to consider that introduction of SRC-2 or modulation of its activity could be beneficial in the efficacy of these factors in driving this differentiation program.
Our work also presents data in support of a novel control for the cardiac metabolic regulator PPARα by SRC-2, which suggests that the metabolic changes observed from SRC-2 loss are partly dependent on PPARα. PPARα is well characterized to control fatty acid oxidation in the heart (32), a pathway that is impaired by SRC-2 loss (17). SRC-2-driven control of PPARα activity appears to be partially mediated by SRC-2 control of GATA-4 and Tbx5. This regulation is likely signal-dependent and is not limited to these transcription factors. For example, PGC1-α/β are other known cardiac coactivators of the PPAR and ERR families of transcription factors in the heart. Interestingly, we did not observe any major changes to ERRα expression, target genes of ERRα/PGC-1α, mitochondrial number, or respiration capacity. This suggests that the SRC-2/PPARα axis of metabolic control functions independently of ERRα/PGC-1α control of mitochondrial integrity and function, but does not exclude PGC-1α control of PPARα at other target genes or direct control of PPARα by PGC-1α. Further, our data present the hypothesis that metabolic control during stress may be controlled through several interdependent mechanisms leading to many new questions as to how these pathways interact during stress conditions and contribute individually to the metabolic changes observed during stress. Additionally, whereas SRC-2 control of PPARα expression contributes to the decreased expression of fatty acid oxidation pathway genes, which is expected to contribute to the metabolic changes that we previously published, this phenotype is not solely limited to PPARα control. SRC-2 regulation of other metabolic targets is likely to contribute.
Our study was designed to assess the role of SRC-2 in cardiac muscle cells in an unstressed environment. Our previous paper defines a specific role for this transcription cofactor in coordinating the metabolic, growth and stress responses to cardiac overload but also described changes in unstressed SRC-2 KO mice. Whereas some of these changes may arise from metabolic consequences of SRC-2 deletion in other organs, the present report suggests that at least a subset of the metabolic alterations seen in the cardiac-specific SRC-2 deletion mice at rest are largely identical to those seen in the unstressed nonselective model. One exception is that the previously observed reduction of COX4 transcript was not observed. It is still possible that an unexpected abnormality in mitochondrial function, whether structural or the result of substrate metabolic responses, might surface under the proper stressors as these studies progress. These data provide the baseline metabolic state of the organ-specific SRC-2 model and describe one mechanism by which this state is regulated, through SRC-2 control of PPARα expression.
Taken together, this work provides novel mechanistic insight into SRC-2 activity in the cardiomyocyte. We show that SRC-2 is a key regulator upstream of numerous cardiac transcriptional pathways through dual control of the expression of several cardiac transcription factors as well as through coactivation of transcription activity. Whereas SRC-2 control of GATA-4, MEF2, and Tbx5 activity is likely to control cardiac growth, structural, and stress-responsive pathways, we show that metabolic gene expression controlled by SRC-2 occurs at least partially through direct regulation of PPARα expression. Future work into control of SRC-2 activity and binding is anticipated to elucidate how cardiac stress signaling pathways are intertwined and coordinately regulated during the stress response.
Acknowledgements
We thank Adam Dean, Pradip Saha, Atul Chopra, Billie Smith, Zahida Sayeeduddin, and Mohammed Sayeeduddin for technical expertise.
This work was supported, in whole or in part, by National Institutes of Health Grants 5P01-DK059820 from the NIDDK and 5R01-HD007857 from the NICHD, and the DUNN Foundation (to B. W. O.'M.); Grants 5R01-HL089902-02 and R01-HL113601 from the NHLBI and Center for the Advancement of Science in Space Request for Funding Proposals 2013-2 (to R. J. S.); R01-HL089792 from the NHLBI (to M. L. E.); R01-HL061483 from the NHLBI (to H. T.); and American Heart Association Postdoctoral Fellowship 12POST11830003 and NHLBI Grant 1K99-HL118159-01 (to E. L. R.). Histological analyses were supported by the Human Tissue Acquisition and Pathology Core and Baylor College of Medicine with funding from the National Institutes of Health Grant P30-CA125123 from the NCI. Seahorse analyses were supported by the Mouse Metabolic Core Core at Baylor College of Medicine under the Diabetes Research Center Grant P30 DK079638.
- MEF2
- myocyte enhancer factor 2
- ChIP-Seq
- chromatin immunoprecipitation-sequencing
- CKO
- cardiomyocyte-specific SRC-2 KO
- ERR
- estrogen-related receptor
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- Fgf10
- fibroblast growth factor 10
- Luc
- luciferase
- Myh6
- myosin heavy chain 6
- Nkx2.5
- NK2 homeobox 5
- PGC-1
- peroxisome proliferator-activated receptor γ, coactivator 1
- PPAR
- peroxisome proliferator-activated receptor
- qPCR
- quantitative PCR
- SRC
- steroid receptor coactivator
- SRF
- serum response factor
- TF
- transcription factor
- TSS
- transcription start site.
REFERENCES
- 1. Barth A. S., Kuner R., Buness A., Ruschhaupt M., Merk S., Zwermann L., Kääb S., Kreuzer E., Steinbeck G., Mansmann U., Poustka A., Nabauer M., Sültmann H. (2006) Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J. Am. Coll. Cardiol. 48, 1610–1617 [DOI] [PubMed] [Google Scholar]
- 2. He A., Kong S. W., Ma Q., Pu W. T. (2011) Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. U.S.A. 108, 5632–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schlesinger J., Schueler M., Grunert M., Fischer J. J., Zhang Q., Krueger T., Lange M., Tönjes M., Dunkel I., Sperling S. R. (2011) The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 7, e1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belaguli N. S., Sepulveda J. L., Nigam V., Charron F., Nemer M., Schwartz R. J. (2000) Cardiac tissue enriched factors serum response factor and GATA-4 are mutual coregulators. Mol. Cell. Biol. 20, 7550–7558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moore M. L., Wang G. L., Belaguli N. S., Schwartz R. J., McMillin J. B. (2001) GATA-4 and serum response factor regulate transcription of the muscle-specific carnitine palmitoyltransferase I β in rat heart. J. Biol. Chem. 276, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 6. Morin S., Charron F., Robitaille L., Nemer M. (2000) GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 19, 2046–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sepulveda J. L., Belaguli N., Nigam V., Chen C. Y., Nemer M., Schwartz R. J. (1998) GATA-4 and Nkx-2.5 coactivate Nkx-2 DNA binding targets: role for regulating early cardiac gene expression. Mol. Cell. Biol. 18, 3405–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bisping E., Ikeda S., Kong S. W., Tarnavski O., Bodyak N., McMullen J. R., Rajagopal S., Son J. K., Ma Q., Springer Z., Kang P. M., Izumo S., Pu W. T. (2006) Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc. Natl. Acad. Sci. U.S.A. 103, 14471–14476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. el Azzouzi H., van Oort R. J., van der Nagel R., Sluiter W., Bergmann M. W., De Windt L. J. (2010) MEF2 transcriptional activity maintains mitochondrial adaptation in cardiac pressure overload. Eur. J. Heart Fail. 12, 4–12 [DOI] [PubMed] [Google Scholar]
- 10. Kim Y., Phan D., van Rooij E., Wang D. Z., McAnally J., Qi X., Richardson J. A., Hill J. A., Bassel-Duby R., Olson E. N. (2008) The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J. Clin. Invest. 118, 124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Naya F. J., Black B. L., Wu H., Bassel-Duby R., Richardson J. A., Hill J. A., Olson E. N. (2002) Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 8, 1303–1309 [DOI] [PubMed] [Google Scholar]
- 12. Oka T., Maillet M., Watt A. J., Schwartz R. J., Aronow B. J., Duncan S. A., Molkentin J. D. (2006) Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ. Res. 98, 837–845 [DOI] [PubMed] [Google Scholar]
- 13. van Berlo J. H., Elrod J. W., Aronow B. J., Pu W. T., Molkentin J. D. (2011) Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 12331–12336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu J., Gong N. L., Bodi I., Aronow B. J., Backx P. H., Molkentin J. D. (2006) Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J. Biol. Chem. 281, 9152–9162 [DOI] [PubMed] [Google Scholar]
- 15. Zhang X., Azhar G., Chai J., Sheridan P., Nagano K., Brown T., Yang J., Khrapko K., Borras A. M., Lawitts J., Misra R. P., Wei J. Y. (2001) Cardiomyopathy in transgenic mice with cardiac-specific overexpression of serum response factor. Am. J. Physiol. Heart Circ. Physiol. 280, H1782–1792 [DOI] [PubMed] [Google Scholar]
- 16. Xu J., Wu R. C., O'Malley B. W. (2009) Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 9, 615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reineke E. L., York B., Stashi E., Chen X., Tsimelzon A., Xu J., Newgard C. B., Taffet G. E., Taegtmeyer H., Entman M. L., O'Malley B. W. (2012) SRC-2 coactivator deficiency decreases functional reserve in response to pressure overload of mouse heart. PLoS One 7, e53395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qi C., Zhu Y., Pan J., Yeldandi A. V., Rao M. S., Maeda N., Subbarao V., Pulikuri S., Hashimoto T., Reddy J. K. (1999) Mouse steroid receptor coactivator-1 is not essential for peroxisome proliferator-activated receptor α-regulated gene expression. Proc. Natl. Acad. Sci. U.S.A. 96, 1585–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gehin M., Mark M., Dennefeld C., Dierich A., Gronemeyer H., Chambon P. (2002) The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol. Cell. Biol. 22, 5923–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu J., Liao L., Ning G., Yoshida-Komiya H., Deng C., O'Malley B. W. (2000) The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. U.S.A. 97, 6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mukherjee A., Soyal S. M., Fernandez-Valdivia R., Gehin M., Chambon P., Demayo F. J., Lydon J. P., O'Malley B. W. (2006) Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol. Cell. Biol. 26, 6571–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lonard D. M., Nawaz Z., Smith C. L., O'Malley B. W. (2000) The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol. Cell 5, 939–948 [DOI] [PubMed] [Google Scholar]
- 23. Chakraborty S., Reineke E. L., Lam M., Li X., Liu Y., Gao C., Khurana S., Kao H. Y. (2006) α-Actinin 4 potentiates myocyte enhancer factor-2 transcription activity by antagonizing histone deacetylase 7. J. Biol. Chem. 281, 35070–35080 [DOI] [PubMed] [Google Scholar]
- 24. Agarwal P., Wylie J. N., Galceran J., Arkhitko O., Li C., Deng C., Grosschedl R., Bruneau B. G. (2003) Tbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryo. Development 130, 623–633 [DOI] [PubMed] [Google Scholar]
- 25. Hurlin P. J., Steingrìmsson E., Copeland N. G., Jenkins N. A., Eisenman R. N. (1999) Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 18, 7019–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pineda Torra I., Jamshidi Y., Flavell D. M., Fruchart J. C., Staels B. (2002) Characterization of the human PPARα promoter: identification of a functional nuclear receptor response element. Mol. Endocrinol. 16, 1013–1028 [DOI] [PubMed] [Google Scholar]
- 27. Baskin K. K., Taegtmeyer H. (2011) AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ. Res. 109, 1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee T. I., Johnstone S. E., Young R. A. (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1, 729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langmead B., Trapnell C., Pop M., Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Madrazo J. A., Kelly D. P. (2008) The PPAR trio: regulators of myocardial energy metabolism in health and disease. J. Mol. Cell. Cardiol. 44, 968–975 [DOI] [PubMed] [Google Scholar]
- 33. Akazawa H., Komuro I. (2003) Roles of cardiac transcription factors in cardiac hypertrophy. Circ. Res. 92, 1079–1088 [DOI] [PubMed] [Google Scholar]
- 34. Pikkarainen S., Tokola H., Kerkelä R., Ruskoaho H. (2004) GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 63, 196–207 [DOI] [PubMed] [Google Scholar]
- 35. Sepulveda J. L., Vlahopoulos S., Iyer D., Belaguli N., Schwartz R. J. (2002) Combinatorial expression of GATA4, Nkx2–5, and serum response factor directs early cardiac gene activity. J. Biol. Chem. 277, 25775–25782 [DOI] [PubMed] [Google Scholar]
- 36. Nurrish S. J., Treisman R. (1995) DNA binding specificity determinants in MADS-box transcription factors. Mol. Cell. Biol. 15, 4076–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu W., Huang X., Cheng J., Li Z., de Folter S., Huang Z., Jiang X., Pang H., Tao S. (2011) Conservation and evolution in and among SRF- and MEF2-type MADS domains and their binding sites. Mol. Biol. Evol. 28, 501–511 [DOI] [PubMed] [Google Scholar]
- 38. Inagawa K., Miyamoto K., Yamakawa H., Muraoka N., Sadahiro T., Umei T., Wada R., Katsumata Y., Kaneda R., Nakade K., Kurihara C., Obata Y., Miyake K., Fukuda K., Ieda M. (2012) Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 111, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 39. Qian L., Huang Y., Spencer C. I., Foley A., Vedantham V., Liu L., Conway S. J., Fu J. D., Srivastava D. (2012) In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598 [DOI] [PMC free article] [PubMed] [Google Scholar]