

Background: δ-Opioid receptor (δOR) F27C polymorphism affects receptor cell surface targeting.
Results: If lacking N-glycans, Cys27, but not Phe27, is extensively targeted to endoplasmic reticulum-associated degradation and can escape to the cell surface in nonnative conformation.
Conclusion: δOR relies on N-glycan-dependent calnexin-mediated quality control (QC) for correct folding, whereas N-glycan-independent QC is a backup system.
Significance: Alternative QC pathways monitor nascent GPCRs.
Keywords: ER Quality Control, ER-associated Degradation, Genetic Polymorphism, Glycosylation, Ubiquitination, G Protein-coupled Receptor (GPCR), Calnexin, Opioid Receptor, Pharmacological Chaperone
Abstract
Quality control (QC) in the endoplasmic reticulum (ER) scrutinizes newly synthesized proteins and directs them either to ER export or ER-associated degradation (ERAD). Here, we demonstrate that the human δ-opioid receptor (hδOR) is subjected to ERQC in both N-glycan-dependent and -independent manners. This was shown by investigating the biosynthesis and trafficking of wild-type and non-N-glycosylated F27C variants in metabolic pulse-chase assays coupled with flow cytometry and cell surface biotinylation. Both QC mechanisms distinguished the minute one-amino acid difference between the variants, targeting a large fraction of hδOR-Cys27 to ERAD. However, the N-glycan-independent QC was unable to compensate the N-glycan-dependent pathway, and some incompletely folded non-N-glycosylated hδOR-Cys27 reached the cell surface in conformation incompatible with ligand binding. The turnover of receptors associating with the molecular chaperone calnexin (CNX) was significantly slower for the hδOR-Cys27, pointing to an important role of CNX in the hδOR N-glycan-dependent QC. This was further supported by the fact that inhibiting the co-translational interaction of hδOR-Cys27 precursors with CNX led to their ERAD. Opioid receptor pharmacological chaperones released the CNX-bound receptors to ER export and, furthermore, were able to rescue the Cys27 variant from polyubiquitination and retrotranslocation to the cytosol whether carrying N-glycans or not. Taken together, the hδOR appears to rely primarily on the CNX-mediated N-glycan-dependent QC that has the capacity to assist in folding, whereas the N-glycan-independent mechanism constitutes an alternative, although less accurate, system for directing misfolded/incompletely folded receptors to ERAD, possibly in altered cellular conditions.
Introduction
Membrane-bound and secreted proteins that traverse the secretory pathway fold in the endoplasmic reticulum (ER).4 A majority of these proteins are co-translationally glycosylated with N-glycans that are added to asparagine residues in the Asn-Xaa-(Ser/Thr) (where Xaa ≠ Pro) sequons (1, 2). The N-glycans increase solubility and prevent aggregation of nascent glycoproteins and furthermore mediate interactions with lectin molecular chaperones and other factors in the ER. The interactions facilitate the folding of newly synthesized glycoproteins, prevent ER export of incorrectly folded or assembled proteins, and also assist in the targeting of terminally misfolded proteins for degradation (1–3). These mechanisms are collectively referred to as the glycoprotein ER quality control (ERQC). The corresponding mechanisms that monitor non-N-glycosylated proteins are far less known.
The importance of ER surveillance mechanisms is demonstrated by the various diseases associated with proteins that fail to pass the QC checkpoints (4). For example, mutant forms of a number of polytopic membrane proteins, such as G protein-coupled receptors (GPCRs) rhodopsin and V2 vasopressin and melanocortin-4 receptors, are characterized by ER retention, which is one of the underlying causes for retinitis pigmentosa, nephrogenic diabetes insipidus, and morbid obesity, respectively (5–7). The mutations, however, do not necessarily lead to functional inactivation, and some mutants can be rescued by membrane-permeable receptor-specific ligands, pharmacological chaperones that stabilize newly synthesized receptors and enhance their ER export (8–11). Similar examples are found among mutant soluble proteins, such as several lysosomal enzymes (12). Furthermore, even WT proteins may be targeted for degradation prematurely (13–16), indicating that the intricate ERQC may lead to the degradation of on-pathway folding intermediates. These findings underscore the need to understand the complex mechanisms and components of the ERQC in a more comprehensive manner.
The first lectin chaperone that a nascent glycoprotein encounters in the ER is calnexin (CNX), a membrane protein that is closely associated with the translocon and the oligosaccharyl transferase that adds the core N-glycan (N-acetylglucosamine2-mannose9-glucose3) to the nascent polypeptide chain (1, 2). CNX and its lumenal counterpart calreticulin (CRT) bind to oligosaccharides in their monoglucosylated form in a cycle of attempts to fold and refold the substrate protein, which is known as the CNX/CRT cycle (1, 2). CNX and CRT assist in folding by recruiting ER foldases ERp57 and cyclophilin B (17, 18). These enzymes, belonging to the protein disulfide isomerase and peptidyl prolyl cis/trans-isomerase families, respectively, catalyze rate-limiting steps in protein folding (19). While assisting in folding, CNX and CRT are also involved in the surveillance of the folding status of their substrates in an attempt to ensure that only those with correct conformation leave the ER. During the CNX/CRT cycle, the folding intermediates dissociate from CNX/CRT when the remaining glucose is removed from the N-glycan, enabling interaction with the folding sensor UDP-glucose:glycoprotein glucosyltransferase. If the substrate protein has not reached the native conformation, glucose is readded, and the cycle continues (1, 2). Substrates failing to reach the correct conformation in time are demannosylated and targeted for degradation in a series of events called as the ER-associated degradation (ERAD). The targeting involves a number of factors, like EDEMs (ER-degradation-enhancing mannosidase-like proteins), OS9, and XTP3-B/erlectin, the last two of which are lectins (2, 3, 20). Finally, the misfolded proteins are retrotranslocated to the cytosol, ubiquitinated, and degraded in proteasomes (20, 21).
The δ opioid receptor (δOR) is a member of the opioid receptor subfamily of GPCRs that participates in the control of pain relief and emotional responses in the nervous system (22). The human δOR (hδOR) carries two N-glycans in its extracellular N-terminal domain at Asn18 and Asn33 (23), flanking a polymorphic site at position 27 (24). Unlike the Phe27 variant, the less common Cys27 variant (allelic frequency in Caucasians is ∼10% (24, 25)) matures inefficiently and is partially targeted for premature ERAD (26–28). Removal of the two N-glycosylation sites of the hδOR-Cys27 variant by site-directed mutagenesis leads to a further decrease in receptor cell surface expression (23). This was found to result mostly from the prominent internalization and lysosomal degradation of plasma membrane receptors (23). Furthermore, the non-N-glycosylated hδOR-Cys27 was found to be exported from the ER with enhanced kinetics and reach the cell surface partially in a nonnative conformation, unable to bind a ligand (23). This suggests that the variant is normally retained in the ER by the glycoprotein QC machinery but is able to escape the compartment when N-glycosylation sites have been disrupted. The present study was undertaken to characterize the glycoprotein ERQC mechanisms that monitor newly synthesized hδORs in more detail, focusing on its major component CNX that acts early in the folding/degradation pathway. Furthermore, we wanted to investigate whether the targeting of newly synthesized hδORs to ERAD requires N-glycosylation, i.e. whether they are substrates of N-glycan-independent ERQC.
EXPERIMENTAL PROCEDURES
DNA Constructs
The hδOR-Cys27-pFT-SMMF and hδOR-Phe27-pFT-SMMF constructs encoding WT hδOR Cys27 and Phe27 variants, respectively, have been described previously (28, 29). These receptor constructs include a cleavable influenza HA signal peptide, an N-terminal Myc tag, and a C-terminal FLAG tag (see Fig. 1A). The hδOR-Cys27(N18E,N33E) mutant in the pFT-SMMF vector was prepared by site-directed mutagenesis as described (23), and the corresponding Phe27 mutant was prepared accordingly. The construct encoding the WT hδOR-Cys27-pcDNA3.1 with a C-terminal FLAG tag has been described earlier (26). The pRK5-HA-Ubiquitin plasmid encoding the HA-tagged WT ubiquitin was acquired from Addgene (plasmid 17608).
FIGURE 1.
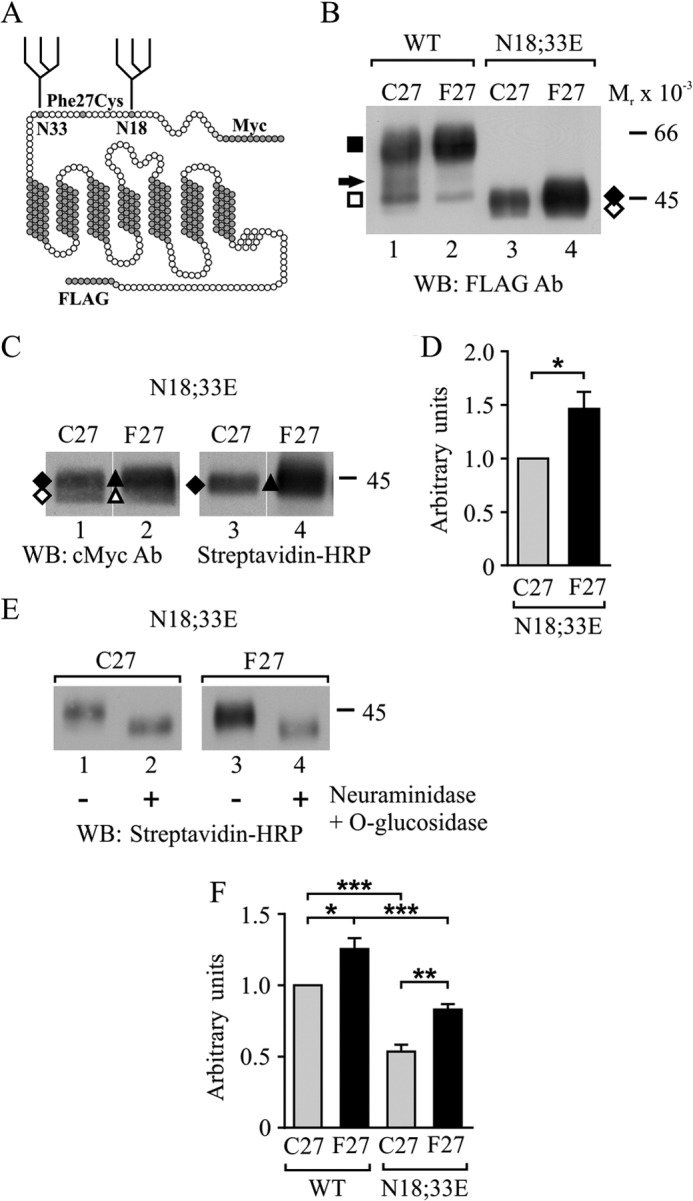
Removal of N-glycosylation sites decreases hδOR variant cell surface levels in stably transfected HEK293i cells. A, receptor model highlighting the extracellular N terminus with the F27C polymorphic site located between the two N-glycosylation sites at Asn18 and Asn33. The N- and C-terminal epitope tags are indicated. B–F, stably transfected HEK293i cells were induced with tetracycline for 24 h to express the WT or the N18E,N33E mutant hδOR variants. Cells were harvested immediately (B and F) or after labeling cell surface proteins with sulfo-NHS-biotin (C–E). Receptors were purified from cellular lysates (B–E) by immunoprecipitation with FLAG M2 antibody and analyzed by SDS-PAGE and Western blotting using c-Myc (A-14) or FLAG M2 antibodies or HRP-conjugated streptavidin, as indicated. In E, purified receptors were deglycosylated with neuraminidase and O-glycosidase before SDS-PAGE. Molecular weight markers are indicated on the right. The precursor and mature receptor forms are depicted with open and closed symbols, respectively, and the mature receptor carrying only one N-glycan is indicated with an arrow. For D, blots corresponding to lanes 1 and 2 in C were analyzed by densitometric scanning. The values are means ± S.E. from five independent experiments normalized to values obtained for the Cys27 variant. The data were analyzed with the one-sample t test. F, cell surface receptors were analyzed by flow cytometry using c-Myc 9E10 antibody. The values are means ± S.E. of three independent experiments performed in triplicate or quadruplicate. The GeoMean values were normalized to those obtained for the WT Cys27 variant. The data were analyzed with the Tukey's multiple comparison test after the one-way analysis of variance. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Ab, antibody; WB, Western blot.
Cell Culture and Transfections
Inducible stably transfected HEK293i cells expressing the WT hδOR Cys27 and Phe27 variants and the Cys27(N18E,N33E) mutant have been described elsewhere (23, 28, 29). The cell line expressing the Phe27(N18Q,N33E) mutant was prepared following the same protocol under blasticidin S (4 μg/ml; Invivogen) and hygromycin (400 μg/ml; Invivogen) selection. The stably transfected HEK293S cell line constitutively expressing the WT hδOR Cys27 variant has been described previously (26). Cells were maintained in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 0.1 mg/ml streptomycin (complete DMEM), and the appropriate selection antibiotics at 37 °C in a humidified atmosphere of 5% CO2. For experiments, cells were plated on 24-well plates (0.1–0.2 × 106 cells), 75-cm2 culture flasks (5–6 × 106 cells), 25-cm2 culture flasks (2 × 106 cells), or 100-mm plates (5–6 × 106 cells) and cultured for 3 days in complete DMEM. The receptor expression in HEK293i cell lines was induced by supplementing the culture medium with tetracycline (500 ng/ml, Invitrogen) for 24 h, unless otherwise stated. For Western blotting and metabolic labeling experiments, cells were incubated in PBS, 20 mm N-ethylmaleimide for 10 min before harvesting, quick frozen in liquid nitrogen, and stored at −70 °C. For transient transfections, 4 × 106 cells were seeded on 100-mm plates and cultured for 24 h before transfection with HA-ubiquitin (5 μg). Lipofectamine 2000 (Invitrogen) was used according to the manufacturer's instructions (15 μl). After 4 h, cells were trypsinized and divided on two 25-cm2 culture flasks and cultured overnight for metabolic labeling.
Metabolic Labeling with [35S]Methionine/Cysteine
Cells were incubated in methionine- and cysteine-free DMEM for 60 min, and labeling was performed in fresh medium with 75–150 μCi/ml of [35S]methionine/cysteine (Easytag Express 35S-protein labeling mix, 1175 Ci/mmol; PerkinElmer Life Sciences) for 30 or 60 min. The labeling was stopped by washing, and cells were chased in complete DMEM, supplemented with 5 mm methionine, for various periods of time as specified in the figures. The proteasomal inhibitor lactacystin (LA, 10 μm; Enzo Life Sciences or Merck Millipore) was added into the depletion medium and was maintained throughout labeling and chase. The glucosidase I/II inhibitor castanospermine (CST, 150 μg/ml; Enzo Life Sciences) was added into the depletion medium and was maintained thereafter. Alternatively, it was removed after labeling or added only to the chase medium, as specified in the figures. The opioid antagonists naltriben (10 μm; Tocris) and naltrexone (NTX; 10 μm; Tocris) were added to the pulse and chase medium or only to the chase medium, respectively. Control samples were treated with the corresponding vehicles.
Cell Surface Biotinylation
Cell surface proteins in stably transfected HEK293i cells were biotinylated with sulfo-NHS-biotin (Thermo Fischer Scientific) as described previously (26). For the chase, the residual biotin was quenched with 1 m Tris-HCl, pH 7.4, and cells were incubated in complete DMEM at 37 °C before harvesting.
Preparation of Whole Cell Extracts and Immunoprecipitation
Total cellular lysates were prepared by mixing thawed cells for 30 min in buffer A (0.5% (w/v) n-dodecyl-β-d-maltoside (Enzo Life Sciences), 25 mm Tris-HCl, pH 7.4, 140 mm NaCl, 2 mm EDTA, 2 μg/ml aprotinin, 0.5 mm PMSF, 2 mm 1,10-phenanthroline, 5 μg/ml leupeptin, 5 μg/ml soybean trypsin inhibitor, and 10 μg/ml benzamidine). For CNX immunoprecipitation, the detergent was replaced with 0.5% (w/v) digitonin (Merck Millipore), and the buffer contained 10 mm CaCl2 and lacked 1,10-phenanthroline and EDTA (buffer B). The insoluble material was removed by centrifugation at 16,000 × g for 30 min, and the supernatants were supplemented with 0.1% (w/v) BSA before immunoprecipitation.
Receptors and HA-ubiquitin were purified with one- or two-step immunoprecipitation using FLAG M2 or HA antibody resins (Sigma), respectively. The antibody-coupled resin (15–20 μl) was added to the lysates and incubated under gentle agitation for 1–16 h. For CNX co-immunoprecipitation, samples were first precleared with 20 μl of protein G-Sepharose (GE Healthcare) for 1 h and then incubated with polyclonal rabbit CNX antibody (1:100; Thermo Fisher Scientific) for 1 h before adding protein G-Sepharose for 2 h. Preimmune serum was used as a control. Immunoprecipitates were washed twice with buffer A/B and four times with buffer C/D (like buffer A/B but containing 0.1% detergent). Precipitated proteins were eluted with SDS sample buffer (62.5 mm Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 0.001% (w/v) bromphenol blue) or FLAG peptide (0.2 mg/ml) in buffer C, followed by concentration with Microcon-30 concentrators (Merck Millipore). For two-step immunoprecipitation, proteins were first eluted by incubating in 25 mm Tris-HCl, pH 7.5, 1% (w/v) SDS, at 95 °C for 5 min and thereafter diluted 10-fold with buffer A for reimmunoprecipitation. For deglycosylation, the receptors were eluted in 1% (w/v) SDS, 50 mm sodium phosphate, pH 5.5. All steps during lysate preparation and immunoprecipitation were performed at 4 °C unless otherwise indicated.
Deglycosylation of Immunoprecipitated Receptors
Purified receptors were subjected to enzymatic deglycosylation with neuraminidase and O-glycosidase or with endoglycosidase H, as described previously (all 50 millunits/ml; Roche Applied Science) (23, 29).
Cell Fractionation
Cells were homogenized with a Dounce homogenizer and subjected to differential centrifugation as described earlier (27) with an additional 30-min 100,000 × g clearing centrifugation for the cytosolic fraction. Receptors were solubilized from the membrane fraction (27), and the soluble fraction was supplemented with 0.5% n-dodecyl-β-d-maltoside before immunoprecipitation.
SDS-PAGE, Western Blotting, and Fluorography
Samples reduced with 50 mm DTT for 2 min at 95 °C were analyzed by SDS-PAGE using 10% SDS-polyacrylamide gels and reagents from Bio-Rad or Amresco. Separated proteins were electroblotted onto Immobilon-P membranes (Millipore) and probed with FLAG M2 (0.5 μg/ml; Sigma), c-Myc A-14 (1:1000; Santa Cruz Biotechnology), or HA (HA-7, 1:10,000; Sigma) antibodies or with HRP-conjugated streptavidin (1:40,000; Thermo Fisher Scientific) as described (29). Gels containing radiolabeled samples were treated with EN3HANCE (PerkinElmer Life Sciences), dried, and exposed at −70 °C for 1–21 days using the Biomax MR film (Eastman Kodak) and intensifying screens. The films were analyzed by densitometric scanning with the Umax PowerLook 1120 color scanner (GE Healthcare) or the Agfa Arcus II laser scanner, and quantified as described (30).
Radioligand Binding Assays
Membranes were prepared, and ligand binding assays were performed as described (29) using [15,16-3H]diprenorphine (50.0–54.9 Ci/mmol; PerkinElmer Life Sciences) as a radioligand.
Flow Cytometry
The total pool of cell surface receptors was analyzed by flow cytometry as described earlier (31) using c-Myc 9E10 antibody (1 μg/ml; Santa Cruz Biotechnology) and phycoerythrin-conjugated rat anti-mouse IgG1 (2 μg/ml; BD Biosciences). The ligand-binding competent pool of receptors was labeled with FITC-conjugated NTX; Invitrogen) at a saturating concentration, as described (23). The fluorescence was measured with the Becton-Dickinson FACSCalibur flow cytometer, and the data were analyzed with the CellQuestPro 6.0 software (BD Biosciences). The mean fluorescence of live induced cells minus the mean fluorescence of live induced cells stained with only the secondary antibody was used for calculations for c-Myc antibody-stained cells. The mean fluorescence of live stained cells minus the mean fluorescence of live stained cells with added displacing ligand (0.1 μm NTX) was used for FITC-NTX labeled cells.
Data Analysis
The data were analyzed with the GraphPad Prism 6.0 software (GraphPad Software). The one-phase decay and sigmoidal dose response models of the nonlinear regression analysis were used for receptor precursor turnover and maturation data in the metabolic labeling experiments, respectively. The one-site binding (hyperbola) model was used to analyze the ligand binding data. Statistical analyses were performed using unpaired or one-sample t tests for comparisons between two groups or the one- or two-way analysis of variance followed by Tukey's or Bonferroni's multiple comparison post hoc tests, respectively, for multiple comparisons. The limit of significance was set as at p < 0.05. The data are presented as means ± S.E.
RESULTS
Removal of N-Glycosylation Sites Decreases Expression Level but Does Not Prevent Cell Surface Delivery of hδOR F27C Variants
To assess the N-glycan-dependent and -independent QC of hδOR variants, the two N-glycosylation sites were eliminated, and epitope-tagged mutants (N18E,N33E) (Fig. 1A) were expressed in stably transfected HEK293i cells along with the corresponding WT variants. All constructs were expressed from the same genomic locus under tetracycline induction (32), permitting direct comparisons in expression levels. Membranes were prepared after 24 h of receptor expression, and solubilized receptors were immunoprecipitated and analyzed by SDS-PAGE and Western blotting (Fig. 1B). As we have reported earlier (28), both the Cys27 and Phe27 WT hδOR variants migrated as two main forms with Mr values of 47,000 and Mr 61,000 (Fig. 1B, lanes 1 and 2), representing core-N-glycosylated receptor precursor and mature fully N- and O-glycosylated cell surface forms, respectively. A faint band of Mr 51,000 was also apparent, corresponding to mature receptors that carry only one N-glycan (23). In comparison, the N18E,N33E mutants lacking the N-glycosylation sites migrated faster, showing two very closely spaced species with Mr values of ∼42,000–45,000 (Fig. 1, B, lanes 3 and 4, and C, lanes 1 and 2) (23). Thus, both variants appear to be modified by N-glycosylation in a similar manner.
The two closely migrating species of the hδOR-N18E,N33E mutants were characterized further by cell surface biotinylation and deglycosylation assays. Cells were labeled with membrane-impermeable sulfo-NHS-biotin, and immunoprecipitated receptors were analyzed by Western blotting using either FLAG M2 antibody or HRP-conjugated streptavidin (Fig. 1C). The latter was able to bind only to the slower migrating forms (Fig. 1C, lanes 3 and 4), whereas the former recognized both forms (Fig. 1C, lanes 1 and 2). Thus, the Mr 42,000 and Mr 45,000 receptor species represent intracellular precursors and cell surface receptors, respectively. Following enzymatic deglycosylation with neuraminidase and O-glycosidase, both variants migrated faster as a Mr 42,000 species (Fig. 1E, lanes 2 and 4). The difference in size of the two forms can therefore be explained by O-glycosylation that takes place in the Golgi complex (26). Densitometric analysis (Fig. 1D) revealed that the level of the cell surface Mr 45,000 form of the Phe27 variant was 1.47-fold higher than that of the corresponding Cys27 variant. Thus, although both non-N-glycosylated mutants are able to reach the cell surface, the level of the Phe27 variant is significantly higher.
To assess receptor levels at the cell surface more precisely, flow cytometry assays were performed (Fig. 1F), using c-Myc antibody that recognizes the receptor N-terminal Myc epitope (Fig. 1A). As expected, the Phe27 variant was present at the cell surface at a significantly higher level than the Cys27 variant. This was apparent whether the variants were N-glycosylated or not (Fig. 1F). However, the cell surface level of the N18E,N33E mutants was significantly lower compared with the corresponding WT forms. These results indicate that the differing plasma membrane level of the variants is an intrinsic property of the polypeptide chain and is not determined by N-glycosylation. The lack of N-glycans, however, decreases the level of both variants, supporting the role of N-glycans in the processing of the receptor.
The Non-N-glycosylated hδOR-Phe27 Is More Stable at the Cell Surface than the Corresponding Cys27 Variant
The cell surface expression level of GPCRs is governed by two phenomena, namely the targeting of plasma membrane receptors to degradation and the transport of newly synthesized receptors from the ER to the cell surface. In our previous study, both of these phenomena were found to contribute to the cell surface level of WT hδOR variants (28). Furthermore, it was observed that the lack of N-glycosylation destabilizes the Cys27 variant and that a fraction of the mutant is in a ligand-binding incompetent form at the plasma membrane (23). Thus, we assessed whether the absence of N-glycans would lead to similar destabilization of the Phe27 variant. First, the ligand binding ability of the cell surface receptors was assessed by flow cytometry (Fig. 2A). HEK293i cells were induced for 24 h to express the WT variants or the corresponding non-N-glycosylated mutants, and aliquots of the intact cells were incubated with c-Myc antibody to label the total receptor pool or with the FITC-conjugated opioid antagonist NTX (23) to label the ligand binding competent receptors. The non-N-glycosylated Cys27 variant stood out from the other receptor constructs (Fig. 2A): the ligand-binding competent pool was only 70% of the total pool. In contrast, no differences in the two receptor pools were detected for the other receptor constructs tested.
FIGURE 2.
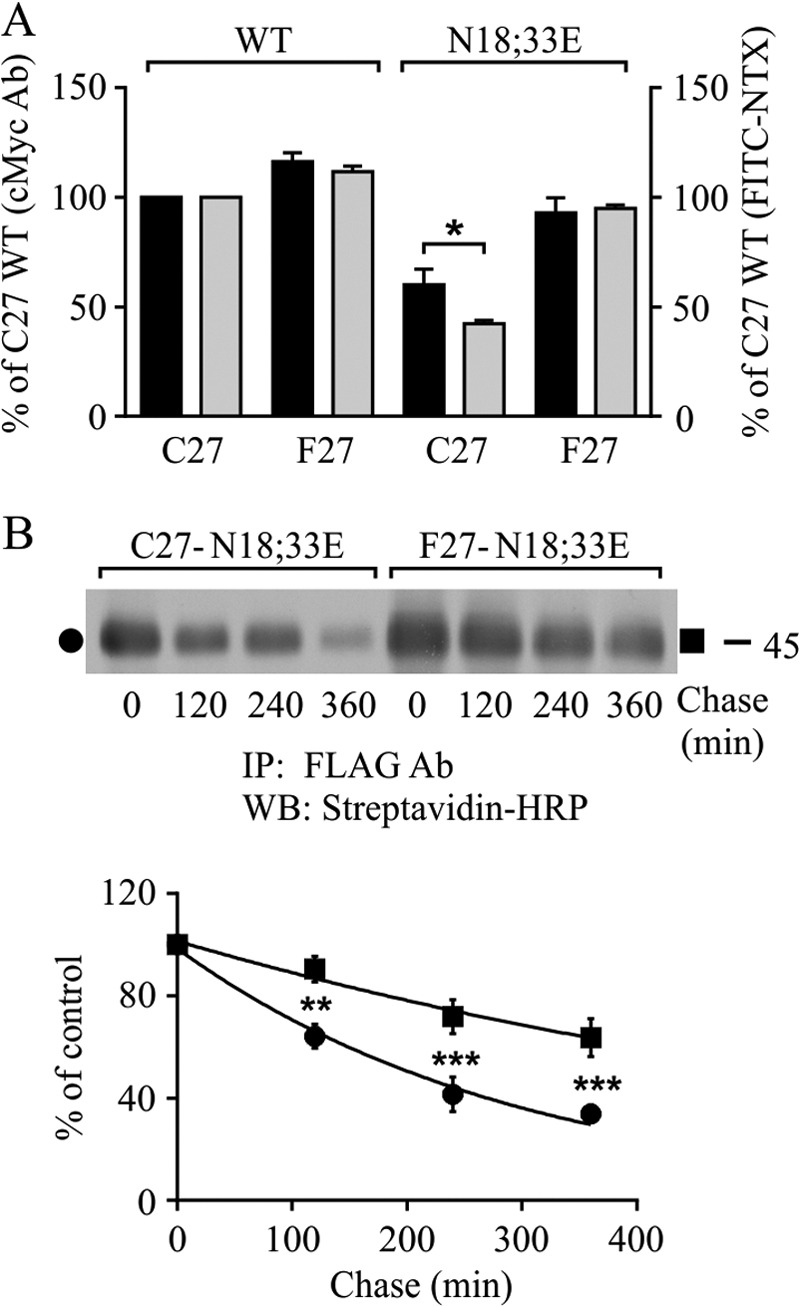
Lack of N-glycosylation destabilizes hδOR variants at the cell surface, but only the Cys27 variant shows compromised ligand binding ability. Stably transfected HEK293i cells were induced for 24 h to express WT receptor variants and their non-N-glycosylated mutants, and samples were subjected to flow cytometry (A) or cell surface biotinylation chase (B) assays. For A, cells were incubated with c-Myc (9E10) antibody followed by phycoerythrin-conjugated secondary antibody to detect the total receptor pool at the cell surface (black bars) or with the FITC-conjugated NTX (gray bars) to detect the ligand binding competent receptors. The GeoMean values were normalized to those obtained for the WT Cys27 variant. The data shown are from three to five independent experiments performed in triplicate or quadruplicate. In B, cells were labeled with sulfo-NHS-biotin and chased for the indicated periods of time at 37 °C. Receptors were immunoprecipitated from cellular lysates with FLAG M2 antibody and analyzed by Western blotting with HRP-streptavidin. The data from densitometric analysis from five independent experiments are shown. The values are means ± S.E., are normalized to the 0-h chase samples, and were fitted to the one-phase decay equation. The data were analyzed with the two-way analysis of variance and the Bonferroni's multiple comparison test. **, p < 0.01; ***, p < 0.001. Ab, antibody; IP, immunoprecipitation; WB, Western blot.
The results described above suggest that the non-N-glycosylated Phe27 variant, unlike the Cys27 one, is in a fully native form at the cell surface. To assess whether this property might be related to differences in receptor stability, biotinylation chase assays were performed (Fig. 2B). Cells were labeled with sulfo-NHS-biotin, and the disappearance of plasma membrane receptors was assessed by removing the label and chasing the cells in culture medium without biotin. The immunoprecipitated receptors were then analyzed by Western blotting with HRP-conjugated streptavidin. As seen in Fig. 2B, the variants behaved differently. The Mr 45,000 cell surface form of the non-N-glycosylated Phe27 variant was significantly more stable than the corresponding Cys27 variant, and the turnover was slower (t½ was 530 and 210 min for the Phe27 and Cys27 variants, respectively). After 6-h chase, 64% of the Phe27 variant remained, whereas only 34% of the Cys27 variant was still detectable. Thus, altered stability at the plasma membrane can explain, at least partially, the dissimilar cell surface level of the hδOR-N18E,N33E mutants.
Next, the functionality of the hδOR-N18E,N33E mutants was assessed further by testing the ligand binding properties in isolated membrane preparations. Saturation binding assays with the radiolabeled opioid ligand [3H]diprenorphine showed that at steady state conditions after 24-h tetracycline induction, the maximal binding capacity, Bmax, for the Phe27 variant was significantly higher compared with the Cys27 variant (58.8 ± 3.8 and 17.3 ± 1.1 pmol/mg for Phe27 and Cys27, respectively, p < 0.01, n = 6–8). The binding affinity, Kd, for the two mutants was in the similar low nanomolar range as was detected earlier for the corresponding WT forms (23, 28): 1.51 ± 0.1 and 0.68 ± 0.1 nm for the mutant Phe27 and Cys27 variants, respectively, (p < 0.01, n = 6–8).
The Non-N-glycosylated hδOR Mutants Are Exported from the ER with Enhanced Kinetics, but Only the Cys27 Variant Matures Inefficiently
Differences in the maturation kinetics and efficiency of export from the ER may also contribute to the divergent cell surface level of the non-N-glycosylated hδOR variants. To test this alternative possibility, metabolic pulse-chase labeling experiments were performed using HEK293i cells expressing the non-N-glycosylated variants and their WT counterparts (Fig. 3). After 2-h induction, cells were labeled with [35S]methionine/cysteine and chased for various periods of time, and immunoprecipitated receptors were analyzed by SDS-PAGE and fluorography. In the case of the WT variants, two precursor forms of Mr 44,000 and Mr 47,000 were apparent at the end of the pulse, in accordance with our previous studies (23, 28) (Fig. 3, A and B). During the chase, these forms, carrying one or two N-glycans, respectively (23), were converted to the corresponding mature receptor forms of Mr 51,000 and Mr 61,000. The one-N-glycan forms were more abundant for the Cys27 variant (28). The maturation was slow for both variants, reaching completion after 2 h (Fig. 3C). This was in contrast to what was observed for the N18E,N33E mutants (Fig. 3, D–F). At the end of the pulse, a substantial fraction of the mutants was already in the O-glycosylated mature Mr 45,000 form, and the half-time for maturation was short, only 27 and 24 min for the Phe27 and Cys27 variants, respectively. The efficiency of maturation was, however, significantly different between the two variants. Virtually all mutant non-N-glycosylated Phe27 precursors reached the mature form within 60 min, but only ∼50% of the corresponding Cys27 variant matured during this time (Fig. 3F). Thereafter, both variants started to disappear, and after 4 h of chase, approximately half of the mature receptors remained (58 and 45% for the Phe27 and Cys27 variants, respectively; Fig. 3F). The N18E,N33E mutant variants showed a significant difference also in the turnover of the precursors, because the half-life was 34 and 54 min for the Phe27 and Cys27 precursors, respectively (Fig. 3F). A similar difference in the turnover rate and maturation efficiency was observed for the WT variant precursors (Fig. 3, A–C). Thus, differences in maturation efficiency are contributing to the divergent amount of the mature variants. Furthermore, the slower turnover and lower maturation efficiency of the Cys27 precursors in comparison with the corresponding Phe27 ones indicate that the former is partially retained in the ER, whether it is N-glycosylated or not.
FIGURE 3.
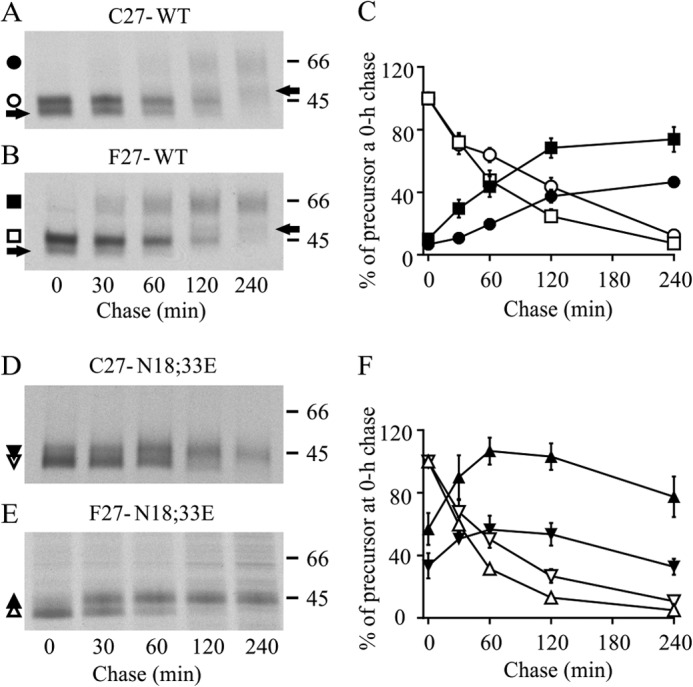
Lack of N-glycosylation enhances ER export of hδOR variants but only the Cys27 variant matures inefficiently. Stably transfected HEK293i cells expressing WT or non-N-glycosylated mutant receptors were induced for 16 h, pulse-labeled with [35S]methionine/cysteine for 30 min, and chased for the indicated periods of time. Cellular lysates were prepared, and receptors were subjected to two-step immunoprecipitation with FLAG M2 antibody and analyzed by SDS-PAGE, fluorography, and densitometric scanning of the fluorograms. Precursor and mature receptor forms are indicated with open and closed symbols, respectively, and one-N-glycan forms of the WT receptors with an arrow. C and F represent the quantitative analysis of the fluorograms, showing time-dependent changes in the intensity of the labeled receptor forms. The data of the two-N-glycan forms are shown for the WT receptors. The values are means ± S.E. of seven or eight independent experiments, normalized to the corresponding precursor at the end of the pulse.
Both WT hδOR Variants Interact with CNX, and the Interaction Is Involved in ER Retention of the Cys27 Variant
The substantially faster ER export of hδOR-N18E,N33E mutants compared with the corresponding WT receptors indicates that the latter interact with glycoprotein QC components that retain glycoproteins in the ER. Thus, we investigated this retention, focusing on CNX that is known to interact with membrane-bound glycoproteins (33, 34). We first assessed the interaction by performing sequential co-immunoprecipitation (Fig. 4A). The WT and non-N-glycosylated variant precursors from pulse-labeled HEK293i cells were subjected to immunoprecipitation with CNX antibody in native conditions. Following denaturation, the CNX-bound receptors were then immunoprecipitated with FLAG M2 antibody. As expected, both WT variant precursors, but not the N18E,N33E mutants, co-immunoprecipitated with CNX antibody (Fig. 4A). The interaction was more prominent for the two-N-glycan form of the Cys27 variant than for the one-N-glycan form (Fig. 4A, compare lane 1 in the first and second panel), in line with our previous observations (23).
FIGURE 4.

CNX mediates ER retention of the WT hδOR-Cys27. A, stably transfected HEK293i cells were induced to express WT or non-N-glycosylated mutant receptors for 2 h and labeled with [35S]methionine/cysteine for 30 min. Aliquots of cellular lysates were first immunoprecipitated with FLAG M2 (10% of the sample) or CNX (40% of the sample) antibodies or with preimmune serum (40% of the sample), as indicated. After elution with SDS, samples were subjected to a second immunoprecipitation with FLAG M2 antibody and analyzed by SDS-PAGE and fluorography. B–E, stably transfected HEK293i cells expressing WT hδORs were induced for 2 or 16 h, labeled with [35S]methionine/cysteine for 30 min, and chased for the indicated time periods. Receptors were purified from cellular lysates by sequential immunoprecipitation to recover the CNX-bound receptors (red circles and squares) and the total pool of receptors (black circles and squares). Samples were analyzed by SDS-PAGE, fluorography, and densitometric scanning of the fluorograms. The values for the two-N-glycan precursors shown in C and E are means ± S.E. of three independent experiments that were normalized to the corresponding 0-h chase samples, and were fitted to the one-phase decay equation. The data from the 16-h induced cells are depicted with open symbols. The arrows indicate precursors carrying only one N-glycan. Ab, antibody; IP, immunoprecipitation; WB, Western blot.
The kinetics of hδOR-CNX association was then investigated in pulse-chase labeling experiments using HEK293i cells that were induced to express the WT variants. As seen in Fig. 4 (B–E), the CNX-bound precursors started to disappear immediately after the pulse, and the turnover was faster than that of the total pool of precursors. Thus, both variants interact transiently with CNX during or shortly after synthesis. However, there was a significant difference between the variants because the half-life of the CNX-bound Cys27 precursors was significantly longer (43 and 32 min for Cys27 and Phe27, respectively) (Fig. 4, B–E). This is in line with the notion that CNX is involved in ER retention and ERAD of the WT Cys27 variant. This was further supported by the observation that after long term receptor expression that was previously found to lead to extensive ER retention of the Cys27 variant, but not the Phe27 variant (28), the Cys27 precursors showed significantly slower dissociation from CNX. The half-life of CNX-bound precursors lengthened 22 min for the Cys27 variant but only 2 min for the Phe27 variant after increasing the time of receptor expression from 2 to 16 h (Fig. 4, C and E).
Impairment of Glucose Trimming of hδOR-Cys27 N-Glycans Attenuates Receptor Maturation
The functional role of CNX interaction in the prominent ER retention of the WT Cys27 variant was assessed in more detail using the glucosidase I/II inhibitor CST. CST is known to either favor or inhibit CNX interaction (1, 2). When it is added while proteins are being synthesized, it can inhibit the co-translational glucosidase I-mediated removal of the outermost glucose from nascent N-glycans and thus inhibit CNX interaction (35, 36). On the other hand, when CST is present post-translationally, it can inhibit the glucosidase II-mediated removal of the innermost glucose from core N-glycans (35, 36). In this case, glycoproteins retain the ability to interact with CNX and potentially remain in the CNX/CRT cycle for longer periods of time than in normal conditions.
The addition of CST to the chase medium slowed down the turnover of CNX-bound WT Cys27 precursors in HEK293S cells constitutively expressing the receptor (Fig. 5, compare E and F with A and B). This was accompanied with impaired conversion of receptor precursors to the mature form (Fig. 5G) and a significant increase in the relative amount of precursors after 4 h of chase (Fig. 5H). These results are in line with the notion that long term CNX interaction attenuates the ability of precursors to exit the ER. Interestingly, a similar impairment in receptor maturation was detected when CST was added before the labeling pulse and was maintained thereafter (Fig. 5, C and D). In these conditions, CST first inhibited receptor-CNX interaction, because only a small fraction of precursors co-purified with CNX after the labeling pulse (Fig. 5, C and D). A tendency toward a small transient increase in the minute CNX-bound pool of precursors during the chase (Fig. 5, C and D) suggests that also the glucosidase II activity was inhibited, thus favoring the post-translational receptor-CNX interaction. Similar findings were obtained when CST was present only during the pulse (data not shown). Importantly, the total pool of receptors that were detected after 4 h of chase did not increase in cells treated with CST during the entire pulse-chase experiment, in contrast to what was observed when CST was present only during the chase (Fig. 5, I and K, lanes 1, 3, and 5). These findings imply that the newly synthesized WT Cys27 precursors that are not able to interact with CNX co-translationally are targeted to degradation.
FIGURE 5.

CNX-hδOR-Cys27 interaction in CST-treated cells. Stably transfected HEK293S cells constitutively expressing the WT hδOR-Cys27 were labeled with [35S]methionine/cysteine for 60 min and chased for the indicated periods of time. The glucosidase inhibitor CST (200 μg/ml) was added to the culture medium at the beginning of depletion and was maintained thereafter or, alternatively, was only added to the chase medium, as indicated. The opioid receptor antagonist NTX (10 μm) was added to the chase medium (J), and the proteasomal inhibitor LA (10 μm) was added to the depletion, pulse, and chase media (K). Receptors were purified from cellular lysates by sequential immunoprecipitation to recover the total (top panels of A, C, and E; K and J) and CNX-bound receptors (bottom panels of A, C, and E). Samples were analyzed by SDS-PAGE, fluorography, and densitometric scanning of the fluorograms. Precursor and mature receptor forms are indicated with open and closed symbols, respectively. The precursor carrying only one N-glycan is indicated with an arrow, and the degradation intermediate is indicated with an asterisk. The arrowhead in J points to an unknown protein co-purifying with the receptor in LA-treated cells. B, D, and F represent quantitative analysis of fluorograms in A, C, and E, respectively, showing time-dependent changes in the intensity of total and CNX-bound pools of precursors. G shows corresponding changes in the amount of mature receptors. The values in B, D, and F are normalized to the precursor of the 0-h chase control samples, and the values in G are normalized to the corresponding precursor at the end of the pulse. The data shown are representative of three independent experiments. H depicts changes in the ratio of precursor and mature receptor forms in the 4-h chase samples, and I shows the relative changes in the total receptor amount compared with the corresponding nontreated control samples. The values are means ± S.E. of six independent experiments. The data in H were analyzed with the Tukey's multiple comparison test after the two-way analysis of variance, and the data in I were analyzed with the one-sample t test. *, p < 0.05; ***, p < 0.001. C, chase; P, pulse; Ab, antibody; IP, immunoprecipitation.
Inhibition of hδOR-Cys27-CNX Interaction with CST Leads to Receptor Targeting to ERAD
The hypothesis that nascent WT Cys27 variant precursors that are incapable of CNX interaction shortly after or during synthesis are targeted to ERAD was tested by treating the Cys27 variant expressing HEK293S cells simultaneously with CST and the proteasomal inhibitor LA (Fig. 5J). We reasoned that the inhibition of proteasomal degradation would stabilize receptor precursors that would otherwise be degraded. As expected, LA was able to stabilize the precursors when CST was added to the pulse and chase medium (Fig. 5J, compare lanes 3 and 5) but had no effect when it was present only during the chase (Fig. 5J, compare lanes 4 and 6). Thus, it can be concluded that in conditions in which CNX interaction of nascent receptors is inhibited, the Cys27 precursors are mainly targeted to degradation rather than being transported along the secretory pathway. It can be hypothesized that other ER factors in the N-glycan-dependent QC pathway are capable of targeting the receptors to ERAD if CNX interaction is inhibited. Furthermore, these results give support to the notion that the correct folding of the Cys27 variant requires CNX interaction.
A Pharmacological Chaperone Can Rescue hδOR-Cys27 Precursors Forced to Remain in the CNX Cycle
Because CST treatment appeared to impair the dissociation of hδOR-Cys27 precursors from CNX during the chase and led to an increase in their total amount, we next assessed whether these precursors could be retargeted to ER export. For this purpose, Cys27 variant expressing HEK293S cells were treated simultaneously with CST and an opioid antagonist NTX. This membrane-permeable ligand acts as a pharmacological chaperone for the δOR and enhances its maturation (13, 29). As seen in Fig. 5K, the antagonist that was added to the chase medium increased the relative amount of mature receptors substantially (Fig. 5K, compare lanes 1 and 2). This also occurred in cells treated with CST, whether added before the pulse (Fig. 5K, lane 4) or at the beginning of the chase (Fig. 5K, lane 6). Thus, receptors appear to retain their competency for ER exit and are not permanently bound to CNX in conditions in which the glucosidase II-mediated removal of the innermost glucose of nascent N-glycans is inhibited.
N-Glycan-independent QC Targets the Non-N-glycosylated Cys27 Variant to ERAD
The results presented above are in line with the notion that the Cys27 variant is subject to both N-glycan-dependent and -independent QC. To investigate the latter in more detail, we assessed whether the Cys27-N18E,N33E mutant is targeted to ERAD and degraded in proteasomes in a similar manner as the corresponding WT receptor (27) (Fig. 6A). When LA was added to the culture medium during pulse-chase labeling experiments, a significant 1.4 ± 0.2-fold increase in the amount of receptors was detected at the end of the pulse (p < 0.05, n = 9), and concomitantly the amount of mature receptors increased during the chase (Fig. 6A). The mutant is thus rapidly degraded already during the 30-min labeling pulse. This is the most likely explanation for the lower relative amount of Cys27-N18E,N33E precursors detected at the end of the pulse in Fig. 4A compared with the corresponding Phe27 precursors.
FIGURE 6.
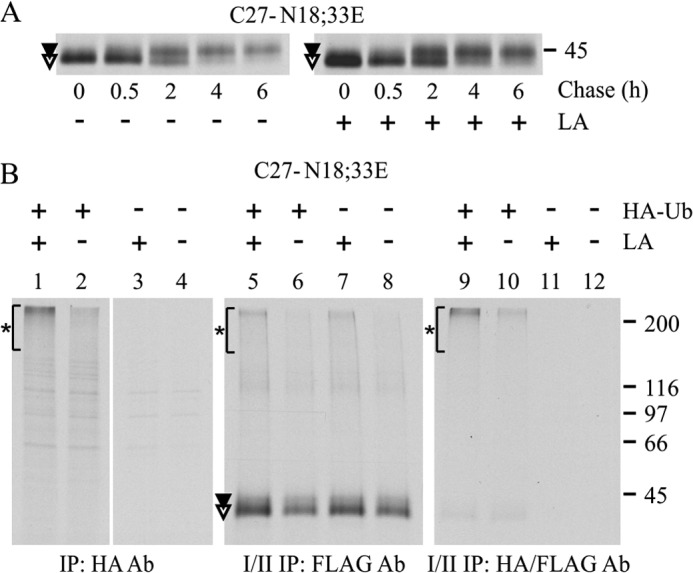
The non-N-glycosylated hδOR-Cys27 is degraded via the proteasome-ubiquitin pathway. A, stably transfected HEK293i cells were induced to express the non-N-glycosylated Cys27 variant for 2 h, labeled with [35S]methionine/cysteine for 30 min, and chased as indicated. The proteasomal inhibitor LA or vehicle was added during depletion and was maintained thereafter. Receptors were purified from cellular lysates by two-step immunoprecipitation with FLAG M2 antibody. B, HEK293i cells stably transfected with Cys27-N18E,N33E were transiently transfected with HA-ubiquitin (HA-Ub) or empty vector, divided onto two plates 4 h after transfection, and cultured overnight. Receptor expression was induced for 4 h, and cells were subjected to 30-min labeling with [35S]methionine/cysteine. The proteasomal inhibitor (LA; 10 μm) was added to the depletion and labeling medium. Equal aliquots of cellular lysates were subjected to one-step immunoprecipitation with HA antibody to detect HA-ubiquitinated proteins (lanes 1–4), with two-step immunoprecipitation with FLAG M2 antibody to detect the total pool of receptors (lanes 5–8) or with sequential immunoprecipitation with HA and FLAG M2 antibodies to detect HA-ubiquitinated receptors (lanes 9–12). Samples were analyzed by SDS-PAGE and fluorography. Precursor and mature receptor forms are indicated with open and closed symbols, respectively, and ubiquitinated proteins are indicated with asterisks. Ab, antibody; IP, immunoprecipitation.
Next, we tested whether the non-N-glycosylated hδOR-Cys27 is ubiquitinated prior to degradation, which is typical for proteins targeted to ERAD (20, 21). This was investigated by transiently transfecting the receptor expressing HEK293i cells with HA-tagged ubiquitin and following receptor ubiquitination in LA and mock-treated pulse-labeled cells (Fig. 6B). The HA antibody was able to immunoprecipitate proteins that migrated as a smear at the top of the gel from transfected cells (Fig. 6B, lanes 1 and 2) but not from control cells (Fig. 6B, lanes 3 and 4). The smear represents polyubiquitinated proteins (27) that, as expected, became more abundant in LA-treated cells (Fig. 6B, compare lanes 1 and 2). When aliquots of denatured HA antibody-immunoprecipitated samples were subjected to a second immunoprecipitation with FLAG M2 antibody, a similar smear at the top of the gel was detected, in addition to a weak band co-migrating with the receptor precursor (Fig. 6B, lanes 9 and 10). Thus, the non-N-glycosylated hδOR-Cys27 precursors are subjected to polyubiquitination. This finding was further supported by the observation that a similar smear at the top of the gel was detected when an aliquot of labeled receptors was subjected to two-step FLAG M2 antibody immunoprecipitation (Fig. 6B, lanes 5 and 6). Furthermore, the same smear was apparent in mock-transfected cells (Fig. 6B, lanes 7 and 8), most likely representing receptors polyubiquitinated with endogenous ubiquitin.
Retrotranslocation of the WT and Non-N-glycosylated Cys27 Variant to the Cytosol Can Be Inhibited with a Pharmacological Chaperone
Proteasomal degradation of misfolded ER-targeted proteins, including hδORs, takes place after they have been retrotranslocated to the cytosol (27, 28). Thus, we next assessed whether the same process is involved in the degradation of the non-N-glycosylated Cys27 mutant. For this purpose, HEK293i cells expressing the WT or the N18E,N33E mutant Cys27 variant were pulse-labeled and chased for 4 h in the absence or presence of LA. Thereafter, the cells were subjected to subcellular fractionation involving differential centrifugation. The 10,000-g membrane pellet and the 100,000-g supernatant, representing the cytosolic fraction (27), were collected, and receptors were immunoprecipitated from the two fractions. As seen in Fig. 7 (A and C), a significant increase in the amount of receptors was detected in the 10,000-g membrane fraction of LA-treated cells (compare lanes 1 and 3; see also Fig. 7, B and D). This concerned the two N- and O-glycosylated mature WT receptor forms as well as the Mr 45,000 O-glycosylated mature form of the N18E,N33E mutant. Thus, proteasomal blockade appears to redirect some receptor precursors from degradation to ER export. Importantly, receptors also became apparent in the cytosolic fraction after LA treatment. A Mr 42,000 receptor form was detected in the cytosolic fraction in cells expressing either the WT receptor or the N18E,N33E mutant (Fig. 7, A and B, compare lanes 7 and 5). It co-migrated with the enzymatically deglycosylated precursor of the WT receptor (Fig. 7E). This is in line with previous findings showing that hδOR retrotranslocation to the cytosol occurs concomitantly with N-glycan removal (27). Taken together, these data show that the non-N-glycosylated Cys27 variant is retrotranslocated and degraded by the ubiquitin-proteasomal pathway in a similar manner as the WT receptor.
FIGURE 7.

Retrotranslocation of the WT and non-N-glycosylated hδOR-Cys27 to the cytosol is inhibited with a pharmacological chaperone. Stably transfected HEK293i cells were induced to express the WT or non-N-glycosylated hδOR-Cys27 for 16 h, labeled with [35S]methionine/cysteine for 30 min, and chased as indicated. The proteasomal inhibitor (LA; 10 μm) was added to the culture medium during depletion, and the opioid antagonist naltriben (NTB; 10 μm) was added during labeling; both were maintained thereafter. Cells were homogenized and subjected to cellular fractionation by differential centrifugation. Membrane-bound receptors from the solubilized 10,000-g pellet (P) and cytosolic receptors from the 100,000-g supernatant (S) fraction were purified by two-step immunoprecipitation with FLAG M2 antibody. Samples were subjected to SDS-PAGE and fluorography immediately (A and C) or after enzymatic deglycosylation with endo-β-N-acetylglucosaminidase H (E). Precursor and mature receptor forms are indicated with open and closed symbols, respectively, and the one-N-glycan form of the WT receptor with an arrow. The degradation intermediate of the WT receptor in the LA-treated cells co-migrating with the endo-β-N-acetylglucosaminidase H-digested precursor is indicated with an asterisk. B and D represent quantitative analysis of fluorograms in A and C, respectively, and the data are shown as means ± S.E. of three or four independent experiments, normalized to the corresponding nontreated control samples. The analysis was done with the Tukey's multiple comparison test after the two-way analysis of variance (pellet samples) or with the one-sample t test (supernatant samples). *, p < 0.05; **, p < 0.01.
Finally, we investigated whether the membrane-permeable opioid receptor pharmacological chaperone naltriben could rescue the receptors from proteasomal degradation. As seen in Fig. 7 (A and C, lane 2), adding the ligand to the culture medium significantly increased the amount of mature hδOR-Cys27 in the membrane fraction whether it carried N-glycans or not. The increase was 2.0- and 2.5-fold in the case of the WT and the mutant receptors, respectively (Fig. 7, B and D). A simultaneous LA treatment increased the amount of receptors further, but the additional effect did not reach statistical significance (Fig. 7, A; C, lane 4; B; and D). Importantly, the combined effect of naltriben and LA decreased the amount of polyubiquitinated receptors and furthermore resulted in a significant decrease in the amount of receptors in the cytosolic fraction compared with cells treated only with LA (Fig. 7, A; C, lanes 7 and 8; B, and D). These results are in line with the notion that pharmacological chaperones can help the precursors, whether N-glycosylated or not, to attain proper conformation and rescue them from polyubiquitination, retrotranslocation, and degradation in the cytosol.
DISCUSSION
Most GPCRs are known to contain N-glycans in their extracellular domains, but the functional role of this modification has remained largely unknown. Most studies investigating the disruption of N-glycosylation sequons have reported decreased cell surface receptor levels (23, 37–40), implying a role for the modification in the early steps of GPCR biosynthesis in the ER. The present study highlights the importance of the N-glycan-dependent ERQC in preventing the export of functionally compromised hδORs from the ER to the cell surface. In addition, we demonstrate that there is an alternative, although less precise, backup ERQC mechanism that can survey the competence of newly synthesized receptors in an N-glycan-independent manner and target the misfolded/incompletely folded ones to ERAD. Both systems were found to discriminate a single amino acid difference in the hδOR variants, underlining the sensitivity of the cellular QC machinery.
The two hδOR variants showed distinct differences in their reliance on N-glycosylation in the early secretory pathway. The N-glycans and N-glycan-dependent QC were dispensable for the hδOR-Phe27 variant and the protein folded in the ER and was transported to the cell surface efficiently whether it carried N-glycans or not. The receptor was fully in its native conformation at the cell surface even when the mutation of the two N-glycosylation sites caused its rapid export from the ER. In contrast, the hδOR-Cys27 variant that differs from the more common Phe27 variant by only one amino acid in the N terminus was critically dependent on the N-glycan-dependent ERQC. The lack of N-glycosylation resulted in the expression of ligand binding incompetent receptors at the cell surface and their rapid disappearance. Furthermore, the non-N-glycosylated receptor precursors were targeted extensively to ERAD. The targeting for degradation took place rapidly, and the receptors were polyubiquitinated and retrotranslocated to the cytosol. Thus, an alternative backup QC mechanism was able to inspect the receptors and target them for degradation independent of their N-glycosylation. Importantly, because pharmacological chaperones were able to rescue both WT and non-N-glycosylated Cys27 precursors from ERAD, both the N-glycan-dependent and -independent QC appear to target receptors for degradation prematurely, although they are still folding competent. This apparent “overactivity” of QC mechanisms may be typical for cellular QC, because it has been shown that more than 30% of all proteins are degraded shortly after they have been synthesized (41).
The ER lectin chaperone CNX was found to be the key component of the N-glycan-dependent QC responsible for retaining the newly synthesized hδORs in the ER. When the two N-glycosylation sites of the receptor were disrupted, the mutants were exported from the ER with enhanced kinetics, suggesting that the WT receptors are retained in the ER via a mechanism involving N-glycosylation. The involvement of CNX in the retention was verified by co-immunoprecipitation. Only the WT variants, but not the non-N-glycosylated mutants, co-immunoprecipitated with CNX antibody. In addition, the interaction between CNX and the WT receptors was almost completely abolished when the removal of the two outermost glucoses from the receptor core N-glycans was inhibited during receptor synthesis with the glucosidase I/II inhibitor CST. These results indicate that the interaction is mediated by receptor N-glycans, and argue against the possibility that it involves direct protein-protein contacts. This has been suggested to occur in the case of a few other membrane proteins, including the angiotensin II type 1 receptor (42) and the D1 and D2 dopamine receptors (43). In these studies, the interaction of CNX with mutant non-N-glycosylated receptors was shown by one-step co-immunoprecipitation using native conditions. In these circumstances, nonspecific interactions between membrane proteins are likely. These problems can be avoided using a two-step protocol, denaturing the samples before the second immunoprecipitation step as was done in the present study.
The interaction between CNX and the hδOR variants was only transient. The turnover of the CNX-bound precursors was, however, significantly longer for the Cys27 variant, indicating that it was engaged in the CNX cycle for a more extended period of time than the Phe27 variant. This was probably caused by the fact that CNX does not act alone but recruits other factors to its substrates. One of these factors is the protein-disulfide isomerase ERp57 that interacts with the CNX P-domain and is known to assist in folding and to form mixed disulfides with CNX substrates (44). It is therefore a likely candidate to be specifically involved in the folding of the Cys27 variant while being dispensable for the Phe27 variant. The cysteine at position 27 is the sole cysteine in the receptor N terminus and is thus very prone to form transient mixed disulfides. Only two other cysteines are found in δOR extracellular domains: Cys121 and Cys198 that form a conserved disulfide bond linking the third transmembrane domain with the second extracellular loop (45). It can be envisioned that the N-terminal cysteine might interfere with the formation of the conserved disulfide bond. In this case, the isomerase activity of the oxidoreductase would be required for the correct receptor folding. The involvement of mixed disulfides in the Cys27 variant ER retention is a very tempting idea and would explain the importance of CNX interaction for the correct receptor folding, as well as for slowing down of the dissociation of CNX-receptor complexes during long term receptor expression. The latter phenomenon can be explained by futile folding attempts and repeated formation of mixed disulfides with the oxidoreductase. The dynamic dissociation and reassociation of CNX-receptor complexes is supported by the observation that a pharmacological chaperone was able to target the receptor precursors toward ER exit and maturation even in conditions that favored association with CNX, i.e. when glucosidase II was post-translationally inhibited with CST, increasing the relative amount of receptors carrying monoglucosylated N-glycans. The role of mixed disulfides in the hδOR-Cys27 ER retention needs to be verified in future studies. So far, attempts to show interaction of ERp57 with the receptor have been unsuccessful, most likely because of the poor quality of antibodies used for co-immunoprecipitation and the transient nature of the interaction.
Another component that could be involved in the stalled CNX cycle and impaired targeting of the WT Cys27 variant to ERAD after long term receptor expression is another ER lectin, XTP3-B/erlectin. In a recent study, this lectin was suggested to inhibit premature degradation of folding intermediates by interacting with its substrate proteins via N-glycans that contain nine mannoses, i.e. glucose-trimmed core N-glycans that have not yet been demannosylated. Overexpression of XTP3-B impaired ERAD of mutant misfolded α1-antitrypsin, whereas knockdown of the endogenous protein in HEK293 cells caused enhancement in its degradation (46). Because nonnative proteins carrying glucose-trimmed N-glycans are also substrates for UDP-glucose:glycoprotein glucosyltransferase, it can be hypothesized XTP3-B could impair degradation indirectly via extending and favoring the CNX/CRT cycle.
Receptor oligomerization in the ER is also a factor contributing to the QC of GPCRs (47). It can be speculated that the unpaired Cys residue of the hδOR Cys27 variant and the interactions that it mediates could hinder receptor dimerization in the ER. This would give an explanation to our recent finding that in co-expressed cells, the Cys27 variant was found to target part of the Phe27 variant to ERAD in a dominant negative manner (48). The heterodimers might be less likely to oligomerize properly compared with the hδOR-Phe27 homodimers, thus failing to pass the QC checkpoints.
In addition to the CNX-mediated QC, the hδOR-Cys27 was shown to undergo scrutiny by N-glycan-independent ERQC machinery that was able to target the precursors to ERAD. The mutant receptors that were devoid of N-glycans were substrates of this machinery. This was demonstrated by the ability of the proteasomal inhibitor LA to stabilize the precursors. The proteasomal blockade also resulted in polyubiquitination and cytosolic accumulation of the non-N-glycosylated mutant precursors, both phenomena that are characteristic for the WT receptor ERAD pathway (27, 28). Thus, the two QC pathways appear to target their substrates ultimately to the same destination, cytosolic proteasomes. However, significant differences were observed in the early steps of the two QC pathways. Whereas the N-glycan-independent QC delivered receptors to ERAD swiftly, the CNX-mediated pathway was slow and started to stall in time. This apparent kinetic difference is most likely explained by the fact that the N-glycan-independent QC chooses its substrates for destruction earlier than the N-glycan-dependent QC that is specifically tuned to assist in repeated folding attempts of its substrates. Another difference between the two pathways was the efficiency by which they were able to distinguish correctly folded forms from misfolded and incompletely folded ones. The N-glycan-independent QC was a less accurate system, because it let misfolded/incompletely folded receptors escape from the ER. Alternatively, the system was incapable of recognizing the imperfections in these escaped receptor forms.
The N-glycan-independent QC is thus clearly a backup system for the more efficient N-glycan-dependent pathway for the hδOR. In a recent study, Ushioda et al. (49) proposed that BiP together with ERdj5 could act as a backup QC system for N-glycosylated proteins in conditions of cellular stress, although in normal conditions these ER factors target non-N-glycosylated substrates. Another ER component that has been proposed to act in the N-glycan-independent QC is EDEM1. It has an important role in accepting substrates from CNX and targeting them for degradation (2) but has recently been shown to be also capable of protein-protein interactions involving non-N-glycosylated misfolded proteins, targeting them to ERAD (50).
Taken together, the present study demonstrates that there are alternative ERQC mechanisms that survey newly synthesized δORs and possibly also other GPCRs and polytopic membrane proteins in general. The glycosylated receptors are likely to rely mainly on the N-glycan-dependent QC. However, they may also be subjected to scrutiny by pathways that function independently of the glycosylation status of the substrate proteins, probably in altered cellular conditions. These alternative pathways may constitute backup systems that improve the overall fidelity of QC mechanisms that maintain cellular homeostasis.
Acknowledgments
We thank Dr. Tarja Leskelä for expert technical assistance and members of the GPCR research team in Oulu for fruitful discussions. We are grateful for Prof. Ted Dawson for the pRK5-HA-Ubiquitin plasmid.
This work was supported by the Sigrid Jusélius Foundation, the Medical Research Center Oulu and Grant 127199 from the Academy of Finland (to U. E. P.-R.).
- ER
- endoplasmic reticulum
- CNX
- calnexin
- CST
- castanospermine
- CRT
- calreticulin
- GPCR
- G protein-coupled receptor
- ERAD
- ER-associated degradation
- hδOR
- human δ opioid receptor
- LA
- lactacystin
- NTX
- naltrexone
- QC
- quality control.
REFERENCES
- 1. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 2. Aebi M., Bernasconi R., Clerc S., Molinari M. (2010) N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82 [DOI] [PubMed] [Google Scholar]
- 3. Hebert D. N., Molinari M. (2012) FLAGging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci. 37, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aridor M. (2007) Visiting the ER: the endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv. Drug Deliv. Rev. 59, 759–781 [DOI] [PubMed] [Google Scholar]
- 5. Bichet D. G. (2006) Nephrogenic diabetes insipidus. Adv. Chronic Kidney Dis. 13, 96–104 [DOI] [PubMed] [Google Scholar]
- 6. Tzekov R., Stein L., Kaushal S. (2011) Protein misfolding and retinal degeneration. Cold Spring Harb. Perspect. Biol. 3, a007492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. René P., Le Gouill C., Pogozheva I. D., Lee G., Mosberg H. I., Farooqi I. S., Valenzano K. J., Bouvier M. (2010) Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J. Pharmacol. Exp. Ther. 335, 520–532 [DOI] [PubMed] [Google Scholar]
- 8. Morello J. P., Petäjä-Repo U. E., Bichet D. G., Bouvier M. (2000) Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol. Sci. 21, 466–469 [DOI] [PubMed] [Google Scholar]
- 9. Bernier V., Bichet D. G., Bouvier M. (2004) Pharmacological chaperone action on G-protein-coupled receptors. Curr. Opin. Pharmacol. 4, 528–533 [DOI] [PubMed] [Google Scholar]
- 10. Conn P. M., Ulloa-Aguirre A. (2010) Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol. Metab. 21, 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valenzano K. J., Benjamin E. R., René P., Bouvier M. (2010) G protein-coupled receptors. In GPCR Molecular Pharmacology and Drug Targeting: Shifting Paradigms and New Directions (Gilchrist A., ed) John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 12. Boyd R. E., Lee G., Rybczynski P., Benjamin E. R., Khanna R., Wustman B. A., Valenzano K. J. (2013) Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 56, 2705–2725 [DOI] [PubMed] [Google Scholar]
- 13. Petäjä-Repo U. E., Hogue M., Bhalla S., Laperrière A., Morello J. P., Bouvier M. (2002) Ligands act as pharmacological chaperones and increase the efficiency of δ opioid receptor maturation. EMBO J. 21, 1628–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pietilä E. M., Tuusa J. T., Apaja P. M., Aatsinki J. T., Hakalahti A. E., Rajaniemi H. J., Petäjä-Repo U. E. (2005) Inefficient maturation of the rat luteinizing hormone receptor. A putative way to regulate receptor numbers at the cell surface. J. Biol. Chem. 280, 26622–26629 [DOI] [PubMed] [Google Scholar]
- 15. Lukacs G. L., Verkman A. S. (2012) CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 18, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petäjä-Repo U. E., Lackman J. J. (2014) Targeting opioid receptors with pharmacological chaperones. Pharmacol. Res. 10.1016/j.phrs.2013.12.001 [DOI] [PubMed] [Google Scholar]
- 17. Pearse B. R., Hebert D. N. (2010) Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim. Biophys. Acta 1803, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Määttänen P., Gehring K., Bergeron J. J., Thomas D. Y. (2010) Protein quality control in the ER: the recognition of misfolded proteins. Semin. Cell Dev. Biol. 21, 500–511 [DOI] [PubMed] [Google Scholar]
- 19. Braakman I., Bulleid N. J. (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 20. Smith M. H., Ploegh H. L., Weissman J. S. (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brodsky J. L. (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waldhoer M., Bartlett S. E., Whistler J. L. (2004) Opioid receptors. Annu. Rev. Biochem. 73, 953–990 [DOI] [PubMed] [Google Scholar]
- 23. Markkanen P. M., Petäjä-Repo U. E. (2008) N-Glycan-mediated quality control in the endoplasmic reticulum is required for the expression of correctly folded δ-opioid receptors at the cell surface. J. Biol. Chem. 283, 29086–29098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gelernter J., Kranzler H. R. (2000) Variant detection at the δ opioid receptor (OPRD1) locus and population genetics of a novel variant affecting protein sequence. Hum. Genet. 107, 86–88 [DOI] [PubMed] [Google Scholar]
- 25. Sarajärvi T., Tuusa J. T., Haapasalo A., Lackman J. J., Sormunen R., Helisalmi S., Roehr J. T., Parrado A. R., Mäkinen P., Bertram L., Soininen H., Tanzi R. E., Petäjä-Repo U. E., Hiltunen M. (2011) Cysteine 27 variant of the δ-opioid receptor affects amyloid precursor protein processing through altered endocytic trafficking. Mol. Cell. Biol. 31, 2326–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petäjä-Repo U. E., Hogue M., Laperriere A., Walker P., Bouvier M. (2000) Export from the endoplasmic reticulum represents the limiting step in the maturation and cell surface expression of the human δ opioid receptor. J. Biol. Chem. 275, 13727–13736 [DOI] [PubMed] [Google Scholar]
- 27. Petäjä-Repo U. E., Hogue M., Laperriere A., Bhalla S., Walker P., Bouvier M. (2001) Newly synthesized human δ opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J. Biol. Chem. 276, 4416–4423 [DOI] [PubMed] [Google Scholar]
- 28. Leskelä T. T., Markkanen P. M., Alahuhta I. A., Tuusa J. T., Petäjä-Repo U. E. (2009) Phe27Cys polymorphism alters the maturation and subcellular localization of the human δ opioid receptor. Traffic 10, 116–129 [DOI] [PubMed] [Google Scholar]
- 29. Leskelä T. T., Markkanen P. M., Pietilä E. M., Tuusa J. T., Petäjä-Repo U. E. (2007) Opioid receptor pharmacological chaperones act by binding and stabilizing newly synthesized receptors in the endoplasmic reticulum. J. Biol. Chem. 282, 23171–23183 [DOI] [PubMed] [Google Scholar]
- 30. Hakalahti A. E., Khan H., Vierimaa M. M., Pekkala E. H., Lackman J. J., Ulvila J., Kerkelä R., Petäjä-Repo U. E. (2013) β-Adrenergic agonists mediate enhancement of β1-adrenergic receptor N-terminal cleavage and stabilization in vivo and in vitro. Mol. Pharmacol. 83, 129–141 [DOI] [PubMed] [Google Scholar]
- 31. Petäjä-Repo U. E., Hogue M., Leskelä T. T., Markkanen P. M., Tuusa J. T., Bouvier M. (2006) Distinct subcellular localization for constitutive and agonist-modulated palmitoylation of the human δ opioid receptor. J. Biol. Chem. 281, 15780–15789 [DOI] [PubMed] [Google Scholar]
- 32. Apaja P. M., Tuusa J. T., Pietilä E. M., Rajaniemi H. J., Petäjä-Repo U. E. (2006) Luteinizing hormone receptor ectodomain splice variant misroutes the full-length receptor into a subcompartment of the endoplasmic reticulum. Mol. Biol. Cell 17, 2243–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hebert D. N., Zhang J. X., Chen W., Foellmer B., Helenius A. (1997) The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J. Cell Biol. 139, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Molinari M., Eriksson K. K., Calanca V., Galli C., Cresswell P., Michalak M., Helenius A. (2004) Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol. Cell 13, 125–135 [DOI] [PubMed] [Google Scholar]
- 35. Hebert D. N., Foellmer B., Helenius A. (1995) Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81, 425–433 [DOI] [PubMed] [Google Scholar]
- 36. Hebert D. N., Foellmer B., Helenius A. (1996) Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. EMBO J. 15, 2961–2968 [PMC free article] [PubMed] [Google Scholar]
- 37. Lanctôt P. M., Leclerc P. C., Escher E., Leduc R., Guillemette G. (1999) Role of N-glycosylation in the expression and functional properties of human AT1 receptor. Biochemistry 38, 8621–8627 [DOI] [PubMed] [Google Scholar]
- 38. Hawtin S. R., Davies A. R., Matthews G., Wheatley M. (2001) Identification of the glycosylation sites utilized on the V1a vasopressin receptor and assessment of their role in receptor signalling and expression. Biochem. J. 357, 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Compton S. J., Sandhu S., Wijesuriya S. J., Hollenberg M. D. (2002) Glycosylation of human proteinase-activated receptor-2 (hPAR2): role in cell surface expression and signalling. Biochem. J. 368, 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J. G., Chen C., Liu-Chen L. Y. (2007) N-Glycosylation of the human κ opioid receptor enhances its stability but slows its trafficking along the biosynthesis pathway. Biochemistry 46, 10960–10970 [DOI] [PubMed] [Google Scholar]
- 41. Schubert U., Antón L. C., Gibbs J., Norbury C. C., Yewdell J. W., Bennink J. R. (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774 [DOI] [PubMed] [Google Scholar]
- 42. Lanctôt P. M., Leclerc P. C., Escher E., Guillemette G., Leduc R. (2006) Role of N-glycan-dependent quality control in the cell-surface expression of the AT1 receptor. Biochem. Biophys. Res. Commun. 340, 395–402 [DOI] [PubMed] [Google Scholar]
- 43. Free R. B., Hazelwood L. A., Cabrera D. M., Spalding H. N., Namkung Y., Rankin M. L., Sibley D. R. (2007) D1 and D2 dopamine receptor expression is regulated by direct interaction with the chaperone protein calnexin. J. Biol. Chem. 282, 21285–21300 [DOI] [PubMed] [Google Scholar]
- 44. Ellgaard L., Ruddock L. W. (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Granier S., Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Weis W. I., Kobilka B. K. (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fujimori T., Kamiya Y., Nagata K., Kato K., Hosokawa N. (2013) Endoplasmic reticulum lectin XTP3-B inhibits endoplasmic reticulum-associated degradation of a misfolded α1-antitrypsin variant. FEBS J. 280, 1563–1575 [DOI] [PubMed] [Google Scholar]
- 47. Bulenger S., Marullo S., Bouvier M. (2005) Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 26, 131–137 [DOI] [PubMed] [Google Scholar]
- 48. Leskelä T. T., Lackman J. J., Vierimaa M. M., Kobayashi H., Bouvier M., Petäjä-Repo U. E. (2012) Cys-27 variant of the human δ opioid receptor modulates maturation and cell surface delivery of Phe-27 variant via heteromerization. J. Biol. Chem. 287, 5008–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ushioda R., Hoseki J., Nagata K. (2013) Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 24, 3155–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shenkman M., Groisman B., Ron E., Avezov E., Hendershot L. M., Lederkremer G. Z. (2013) A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J. Biol. Chem. 288, 2167–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]