

Background: Fibrillar amyloid proteins deposit in plaques and blood vessels in the brain in Alzheimer disease and related disorders.
Results: Early formation of amyloid plaques impedes subsequent amyloid accumulation in blood vessels.
Conclusion: Amyloid deposition in one compartment can impact amyloid accumulation in another compartment in the brain.
Significance: Learning how amyloid proteins influence further formation is important for understanding the progression of pathology in neurodegenerative diseases.
Keywords: Alzheimer Disease, Amyloid, Pathology, Protein Misfolding, Transgenic Mice, Cerebral Vascular, Plaque
Abstract
The fibrillar assembly and deposition of amyloid β (Aβ) protein, a key pathology of Alzheimer disease, can occur in the form of parenchymal amyloid plaques and cerebral amyloid angiopathy (CAA). Familial forms of CAA exist in the absence of appreciable parenchymal amyloid pathology. The molecular interplay between parenchymal amyloid plaques and CAA is unclear. Here we investigated how early-onset parenchymal amyloid plaques impact the development of microvascular amyloid in transgenic mice. Tg-5xFAD mice, which produce non-mutated human Aβ and develop early-onset parenchymal amyloid plaques, were bred to Tg-SwDI mice, which produce familial CAA mutant human Aβ and develop cerebral microvascular amyloid. The bigenic mice presented with an elevated accumulation of Aβ and fibrillar amyloid in the brain compared with either single transgenic line. Tg-SwDI/Tg-5xFAD mice were devoid of microvascular amyloid, the prominent pathology of Tg-SwDI mice, but exhibited larger parenchymal amyloid plaques compared with Tg-5xFAD mice. The larger parenchymal amyloid deposits were associated with a higher loss of cortical neurons and elevated activated microglia in the bigenic Tg-SwDI/Tg-5xFAD mice. The periphery of parenchymal amyloid plaques was largely composed of CAA mutant Aβ. Non-mutated Aβ fibril seeds promoted CAA mutant Aβ fibril formation in vitro. Further, intrahippocampal administration of biotin-labeled CAA mutant Aβ peptide accumulated on and adjacent to pre-existing parenchymal amyloid plaques in Tg-5xFAD mice. These findings indicate that early-onset parenchymal amyloid plaques can serve as a scaffold to capture CAA mutant Aβ peptides and prevent their accumulation in cerebral microvessels.
Introduction
The molecular seeding, assembly, and deposition of fibrillar proteins is a common process of various neurodegenerative disorders (1, 2). In Alzheimer disease (AD)2 and related disorders, the extracellular deposition of fibrillar amyloid β (Aβ) protein in the brain is a prominent pathological feature (3, 4). Aβ is a 39- to 43-amino acid peptide that exhibits a high propensity to self-assemble into β sheet-containing oligomeric forms and fibrils (5, 6). Aβ peptides are derived proteolytically from a large type I integral membrane precursor protein, termed the amyloid β protein precursor (AβPP) that is encoded by a gene located on chromosome 21 (7–9). The amyloidogenic processing of AβPP initially involves a cleavage at the amino terminus of the Aβ peptide sequence by β-secretase, an aspartyl proteinase named BACE (10, 11). Subsequent cleavage of the remaining amyloidogenic membrane-spanning AβPP carboxyl-terminal fragment by γ-secretase liberates the predominant Aβ40 or Aβ42 residue peptides. Presenilin (PS) proteins contain the proteolytic active site as part of a multiprotein γ-secretase complex (12, 13).
In addition to plaques in the AD brain parenchyma, another prominent site of extracellular Aβ deposition is within and along primarily small and medium-sized arteries and arterioles of the cerebral cortex and meninges and in the cerebral microvasculature, a condition known as cerebral amyloid angiopathy (CAA) (14, 15). In AD, the presence of CAA is common and observed in >85% of cases (16, 17). In both AD and sporadic forms of CAA, the vascular amyloid can exist as two prominent forms, known as CAA type 2 and CAA type 1, with an age of onset of generally >60 years (16, 17). In CAA type 2, the more common form, the amyloid deposition is largely restricted within the vessel wall of the cortical and meningeal arterioles and arteries and does not promote surrounding neuroinflammation (15, 16, 18). Accumulation of fibrillar Aβ in CAA type 2 has been shown to cause degeneration and cell death of smooth muscle cells in the affected larger cerebral vessels and to trigger hemorrhage (18–20, 22, 23). On the other hand, CAA type 1 involves amyloid deposition along the brain capillaries and is more often associated with the presence of an apolipoprotein ϵ4 allele (17, 21). In contrast to CAA type 2, where the amyloid is confined within the vessel wall, CAA type 1 results in penetration of the fibrillar amyloid deposits into the surrounding brain parenchyma, promoting a robust localized neuroinflammatory response characterized by strong perivascular microglial activation, accumulation of hyperphosphorylated Tau protein, and neuronal degeneration (18, 20, 22, 23). Furthermore, an increasing number of studies have implicated cerebral microvascular Aβ deposition in promoting neuroinflammation and dementia in AD and in familial and sporadic cases of CAA (18, 24, 25). In particular, cerebral microvascular, but not parenchymal, amyloid deposition is more often correlated with dementia in individuals afflicted with AD and spontaneous CAA disorders as well as in mouse models (26–28).
In addition to the prominent CAA that is found in AD and in sporadic cases of this condition, several monogenic, familial forms of CAA exist that result from mutations that reside within the Aβ peptide sequence of the AβPP gene (29–33). The most recognized example of familial CAA is the Dutch-type E22Q substitution in Aβ that causes early and severe cerebral vascular amyloid deposition, leading to recurrent, and often fatal, intracerebral hemorrhages at midlife (29, 30, 34–36). Similarly, the Iowa-type D23N substitution in Aβ also causes early and severe cerebral vascular amyloid deposition (33). In contrast to the Dutch-type disease, Iowa-type CAA appears more localized to the cerebral microvasculature. In contrast to AD, in both Dutch-type and Iowa-type familial CAA, there is a striking absence of appreciable parenchymal amyloid plaque pathology despite the highly fibrillogenic nature of Dutch and Iowa CAA mutant peptides (37–39). In any case, the impact of parenchymal amyloid plaques on the development of CAA is not well understood.
We previously developed a unique transgenic mouse model, designated Tg-SwDI, that expresses low levels of familial Dutch/Iowa CAA mutant human AβPP in the brain and develops early-onset and progressive accumulation of microvascular CAA (40, 41). Tg-SwDI mice do not develop parenchymal fibrillar Aβ plaque deposits, consistent with the pathology observed in Dutch-type and Iowa-type patients. In close association with cerebral microvascular fibrillar amyloid, there is a robust neuroinflammatory response in Tg-SwDI mice, characterized by highly increased numbers of activated microglia and elevated levels of proinflammatory cytokines (41, 42). Moreover, Tg-SwDI mice exhibit behavioral deficits that coincide with the development of early-onset cerebral microvascular amyloid and neuroinflammation (42, 43). On the other hand, Tg-5xFAD, a commonly used transgenic mouse model that expresses high levels of human AβPP in the brain, produces non-mutated human Aβ peptides and develops progressive parenchymal fibrillar Aβ deposits (44).
Cerebral vascular fibrillar amyloid can occur in the presence or absence of parenchymal fibrillar amyloid pathology, as in AD or in sporadic and familial CAA, respectively. Although parenchymal and cerebral vascular fibrillar amyloid exists in distinct compartments of the brain, the molecular interplay between these two pathologies is understood poorly. Accordingly, in this study, we generated bigenic Tg-SwDI/Tg-5xFAD mice to investigate the impact of early-onset parenchymal plaque formation on the development of microvascular CAA. Here we show that, in the bigenic mice, there is an elevated accumulation of Aβ but a conspicuous absence of microvascular CAA, the prominent pathology in Tg-SwDI mice, and a shift to larger parenchymal fibrillar amyloid plaques compared with Tg-5xFAD mice. This alteration in fibrillar amyloid pathology resulted in more severe neuronal loss and microglial activation in the bigenic Tg-SwDI/Tg-5xFAD mice. Non-mutated Aβ42 amyloid fibril seeds promoted the fibrillar assembly of Dutch/Iowa CAA mutant Aβ in vitro. Lastly, intrahippocampal injection of biotin-labeled Dutch/Iowa CAA mutant Aβ40 strongly deposited on and adjacent to pre-existing parenchymal fibrillar amyloid plaques in Tg-5xFAD mice. Together, these findings show that early-onset parenchymal fibrillar amyloid plaques primarily composed of non-mutated Aβ42 can serve as a scaffold to recruit the codeposition of CAA mutant Aβ peptides and prevent the development of microvascular CAA. This suggests that, in the brain, fibrillar protein deposition in the parenchymal compartment can significantly impact fibrillar protein deposition in the vascular compartment.
EXPERIMENTAL PROCEDURES
Reagents and Chemicals
Aβ peptides were synthesized at the Keck Peptide Synthesis Facility at Yale University using tert-butyloxycarbonyl (tBOC) chemistry. Hydrofluoric acid was used for cleavage and deprotection. The peptides were purified by reverse phase HPLC using linear water-acetonitrile gradients containing 0.1% trifluoroacetic acid. The purity was estimated at >90–95% on the basis of analytical reverse phase HPLC and matrix-assisted laser desorption ionization mass spectrometry. Aβ peptides were initially prepared in 1,1,1,3,3,3-hexafluoro-2-propanol, flash-frozen, lyophilized to remove solvent, and resuspended in pure dimethyl sulfoxide. Thioflavin T was purchased from Sigma-Aldrich (St. Louis, MO).
Animals
All work with animals was approved by the Stony Brook University Institutional Animal Care and Use Committee and was in compliance with the guidelines established by the Public Health Service Guide for the Care and Use of Laboratory Animals. The generation and characterization of Tg-SwDI mice have been described previously (38, 39). Tg-SwDI mice express the human AβPP transgene with the familial AD Swedish K670N/M671L mutations and the familial CAA Dutch E693Q and Iowa D694N mutations in neurons under control of the Thy1 promoter element and develop early-onset CAA mutant Aβ deposition and cerebral microvascular amyloid accumulation in the cortex, thalamus, hippocampus, and subiculum. First, Tg-5xFAD mice were obtained from Jackson Laboratories. Tg-5xFAD mice coexpress transgenes for human AβPP and human presenilin 1 with five familial AD mutations (AβPP K670N/M671L + I716V + V717I and PS1 M146L + L286V) and develop early-onset, non-mutated Aβ deposition and, in this case, parenchymal plaque amyloid accumulation in the cortex, thalamus, hippocampus, and subiculum (44). Heterozygous Tg-SwDI mice on a pure C57/Bl6 background were bred with heterozygous Tg-5xFAD mice on a mixed C57/Bl6/Sjl background to obtain heterozygous Tg-SwDI, heterozygous Tg-5xFAD, and bigenic Tg-SwDI/Tg-5xFAD mice, all on the same mixed background. Eight to 10 mice of each genotype were examined at 3, 6, and 9 months of age for quantitative immunochemical and pathological studies.
Tissue Preparation
Mice were sacrificed at designated time points, and the brains were removed immediately and bisected in the midsagittal plane. One hemisphere was snap-frozen and used for the protein analyses. The other hemisphere was placed in 70% ethanol, followed by xylene treatment and embedding in paraffin. Sagittal sections were cut at 10-μm thickness using a microtome, placed in a flotation water bath at 45 °C, and then mounted on Colorfrost/Plus slides (Fisher Scientific, Houston, TX) for immunohistochemical and histological analyses.
Immunochemical Analysis of Cerebral Aβ Peptides
Soluble pools of Aβ40 and Aβ42 were determined by using specific ELISAs on carbonate-extracted mouse forebrain tissue, and, subsequently, the insoluble Aβ40 and Aβ42 levels were determined by ELISA of guanidine lysates of the insoluble pellets resulting from the carbonate-extracted brain tissue as described previously (45, 46). In the sandwich ELISAs, Aβ40 and Aβ42 were captured using their respective carboxyl terminus-specific antibodies, m2G3 and m21F12, and biotinylated m3D6, specific for human Aβ, was used for detection (45). Each brain lysate was measured in triplicate and compared with linear standard curves generated with known concentrations of human Aβ using a Spectramax M2 plate reader (Molecular Devices, Sunnyvale, CA).
To determine the relative levels of soluble Aβ oligomers, TBS-soluble brain fractions were prepared and analyzed by two different methods. First, a sandwich ELISA was performed using m3D6 to capture Aβ species and biotinylated m3D6 for detection of oligomers of any size (28, 46, 47). Alternatively, soluble Aβ oligomers were analyzed by quantitative dot blot analysis using the polyclonal antioligomer antibody OC11, as described previously (28, 42).
Immunohistochemical Analysis
Procedures were performed as described previously (40, 41). Briefly, sections were cut in the sagittal plane at 10-μm thickness using a microtome, deparaffinated, and rehydrated. Antigen retrieval was performed by treatment with proteinase K (0.2 mg/ml) for 10 min at 22 °C for Aβ, neuronal, and collagen labeling and by 10 mm sodium citrate solution (pH 9.0) for 30 min at 90 °C in a water bath for activated microglia labeling. The following antibodies were used for immunolabeling analysis: rabbit polyclonal antibody directed toward residues 1–5 of human Aβ (1:200, 48); mouse monoclonal 4G8, which recognizes an epitope between residues 17–24 of non-mutated Aβ (1:200, Covance, Princeton, NJ); mouse monoclonal antibody Neural Nuclei (NeuN) to identify neurons (1:500, Chemicon, Temecula, CA); rabbit polyclonal antibody to collagen type IV to identify cerebral microvessels (1:100, Research Diagnostics Inc., Flanders, NJ); and mouse monoclonal antibody 5D4 to identify activated microglia (1:1000, Seikagaku Corp., Tokyo, Japan). Primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (1:1000, Vector Labs, Burlingame, CA) and visualized with a stable diaminobenzidine solution (Invitrogen) as a substrate. Alternatively, deposited fibrillar amyloid was detected with thioflavin S staining, and the primary antibodies to collagen type IV to visualize cerebral microvessels or mAb5D4 for activated microglia were detected with an Alexa Fluor 594-conjugated secondary antibody (1:1000, Invitrogen).
Quantitative Analysis of Regional Parenchymal and Microvascular Amyloid Deposition
The total Aβ and fibrillar amyloid burden in the regions of the cortex, thalamus, hippocampus, and subiculum was quantified on the same set of systematically sampled immunostained or thioflavin S-stained sections, respectively, using National Institutes of Health ImageJ 1.32 software. The percentage of thioflavin S-labeled microvessels in the regions of the frontotemporal cortex, thalamus, hippocampus, and subiculum was quantified, respectively, on the same set of systematically sampled thioflavin S-stained sections using stereological principles, as described previously (49).
Quantitative Analysis of Cortical Neuronal and Microglial Densities
The total numbers of neurons and activated microglia in cortical layer V were estimated using a computerized stereology system (Stereologer, Systems Planning and Analysis, Alexandria, VA). Every tenth section was selected and generated 10–15 sections/reference space in a systematic-random manner. Immunopositive cells were counted using the optical fractionator method with the dissector principle and unbiased counting rules (49).
Thioflavin T Measurements
All Aβ peptides were solubilized in pure dimethyl oxide and diluted in 10 mm phosphate buffer to a concentration of 20 μm at a pH of 7.4. For seeding experiments, non-mutated Aβ42 seeds were grown by incubating the peptide at 37 °C to form mature fibrils. Fibril formation was confirmed by transmission electron microscopy. Aβ42 seeds were sonicated in a glass vial for 10 min before use. Fresh solutions of the Dutch/Iowa CAA mutant Aβ40 peptide were prepared as above, and Aβ42 seeds were added to make a final peptide solution that was 20 μm Dutch/Iowa Aβ40 and either 10 or 20 μm non-mutated Aβ42 (as seeds). Thioflavin T fluorescence was monitored with a SpectraMax M2 microplate reader every 15 s with excitation, emission, and automatic cutoff wavelengths of 446, 490, and 475 nm, respectively. Each experimental point was the mean of the fluorescence signal of three wells containing aliquots of the same solution.
Intrahippocampal Injection of Biotinylated Aβ40DI Peptide in Tg-5xFAD Mice
Six-month-old Tg-5xFAD mice were anesthetized with Avertin (250 mg/kg) and secured in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). A sagittal incision was made caudal-to-rostral, allowing the scalp to be retracted and held in place with microclips to expose the skull surface. To insert the Hamilton syringe (30-gauge), a small burr hole was drilled into the parietal bone. 2 μl of freshly prepared, biotin-labeled Aβ40DI peptide, dissolved in sterile saline at a concentration of 0.5 mg/ml, was injected stereotactically into the hippocampus (bregma, anterior 2.00 mm, lateral 1.50 mm, and 1.80 mm deep) at a rate of 0.3 μl/min using a microinjection unit. The needle was left in place for 5 min post-injection to minimize reflux before being removed slowly. The scalp was closed under sterile conditions using 4-0 nylon sutures, and the animals were placed in a cage warmed with a heating pad and observed until they were alert and mobile. Twenty-four hours later, the mice were sacrificed, and the brains were removed and processed as described above.
Statistical Analysis
The data were analyzed by one-way analysis of variance for each measure at each brain region. Significant analyses of variance (p < 0.05) were followed by Fisher's post hoc tests. The results are reported in the corresponding figure legends.
RESULTS
Bigenic Tg-SwDI/Tg-5xFAD Mice Accumulate Elevated Levels of Cerebral Aβ Peptides
In this study, we sought to determine the influence of early-onset parenchymal amyloid plaque formation on the development of cerebral microvascular amyloid accumulation in transgenic mice. Tg-5xFAD mice, which produce human non-mutated Aβ and develop early-onset parenchymal amyloid plaques, were bred to Tg-SwDI mice, which produce human Dutch/Iowa CAA mutant Aβ and develop cerebral microvascular amyloid. We compared the bigenic mice generated from this cross with the single transgenic animals. After aging 3–9 months, quantitative ELISAs were performed to measure the levels of soluble and insoluble human Aβ40 and Aβ42 in forebrain homogenates prepared from each line of mice. As shown in Fig. 1, A–C, all mice showed a progressive accumulation of soluble and insoluble Aβ species in the brain. At all ages, Tg-5xFAD mice accumulated higher levels of cerebral Aβ than Tg-SwDI mice, consistent with earlier studies (28). The bigenic Tg-SwDI/Tg-5xFAD mice generally accumulated much higher levels of Aβ than each single transgenic line. Notably, at 3 and 6 months of age, there was a significant 44% (p < 0.05) and 67% (p < 0.02) increase, respectively, in total cerebral Aβ in the bigenic mice compared with the additive amounts of cerebral Aβ present in each single transgenic line. Although the levels of Aβ remained somewhat higher at 9 months of age in the bigenic Tg-SwDI/Tg-5xFAD mice compared with the Tg-5xFAD mice, the increase in levels of Aβ42 was no longer significant because this species of Aβ accumulated substantially in the single transgenic line. The ratio of Aβ40:Aβ42 was ≈9:1 in the Tg-SwDI mice, whereas it was reversed in the Tg-5xFAD mice with a ratio of ≈1:6, as reported previously (40, 44). Interestingly, the bigenic Tg-SwDI/Tg-5xFAD mice exhibited an Aβ40:Aβ42 ratio of ≈1:3, reflecting proportionately higher amounts of Aβ40 compared with the Tg-5xFAD mice.
FIGURE 1.

Measurement of Aβ peptide levels in brains of Tg-SwDI, Tg-5xFAD, and bigenic Tg-SwDI/Tg-5xFAD mice. The levels of soluble and insoluble Aβ40 and Aβ42 peptides were measured in mouse forebrain extracts of 3- (A), 6- (B), and 9-month-old (C) Tg-SwDI mice (black bars), Tg-5xFAD mice (blue bars), and bigenic Tg-SwDI/Tg-5xFAD mice (red bars) by ELISA analysis, as described under “Experimental Procedures.” The levels of soluble Aβ oligomers were measured by ELISA (D) and quantitative dot blot analysis using OC antibody (E and F) in the different mice at 3, 6, and 9 months of age, as described under “Experimental Procedures.” Data are mean ± S.D. of eight to 10 mice of each genotype at each age. a, p < 0.05; b, p < 0.01; c, p < 0.001; d, p < 0.0001; ns, not significant.
Soluble Aβ oligomers have been implicated as important pathogenic peptide assemblies (6, 50–52). Therefore, we next determined the levels of soluble Aβ oligomers in the different transgenic mouse lines using two complementary techniques. First, we used a sandwich ELISA technique to measure all forms of oligomers in the soluble brain fraction (28, 46, 47). Alternatively, we performed a quantitative dot blot analysis on soluble brain fractions using the Aβ oligomer-specific antibody OC11 (28, 42, 53). Both of these approaches revealed that, at each of the ages examined, Tg-5xFAD mice possessed much higher levels of soluble Aβ oligomers compared with Tg-SwDI mice (Fig. 1, D–F), as reported previously (28). Although there tended to be higher levels of soluble Aβ oligomers in the bigenic Tg-SwDI/Tg-5xFAD mice compared with Tg-5xFAD mice, this was not significant. Together, these findings indicate that the bigenic mice tended to accumulate more Aβ peptides in their brains that surpassed the additive amounts of each individual transgenic line, particularly at the earlier ages.
We next performed immunolabeling studies to characterize and quantitate the deposition of human Aβ in the brains of the different mice. As shown in Fig. 2, at 3 months of age, Tg-SwDI mice showed early-stage Aβ deposition, most prominently in the subiculum region (Fig. 2, A and M), whereas the Tg-5xFAD mice and bigenic Tg-SwDI/Tg-5xFAD mice exhibited a more widespread deposition of Aβ through the cortex, thalamus, and hippocampus, but it was also highest in the subiculum region (Fig. 2, D, G, and M). As all three lines of mice aged to 6 and 9 months, the amount of deposited Aβ increased markedly and was generally most robust in the bigenic Tg-SwDI/Tg-5xFAD mice, reflecting the Aβ ELISA results obtained in Fig. 1.
FIGURE 2.

Progressive accumulation of Aβ deposition in Tg-SwDI, Tg-5xFAD, and bigenic Tg-SwDI/Tg-5xFAD mouse brains. Shown are representative images of 3- to 9-month-old transgenic mouse brain sections immunostained for Aβ as described under “Experimental Procedures.” A–C, Tg-SwDI mice. D–F, Tg-5xFAD mice. G–I, bigenic Tg-SwDI/Tg-5xFAD mice. Scale bars = 1 mm. Regional cerebral Aβ deposition in the cortex (J), thalamus (K), hippocampus (L), and subiculum (M) of 3-, 6-, and 9-month-old Tg-SwDI mice (black bars), Tg-5xFAD mice (blue bars), and bigenic Tg-SwDI/Tg-5xFAD mice (red bars) was determined by image analysis of Aβ immunostaining. Data are mean ± S.D. of eight mice of each genotype at each age. a, p < 0.05; b, p < 0.01; c, p < 0.001; d, p < 0.0001; ns, not significant.
Bigenic Tg-SwDI/Tg-5xFAD Mice Exhibit Altered Fibrillar Amyloid Deposition
To specifically evaluate fibrillar amyloid deposition in the brain, we performed thioflavin S staining. Overview images revealed that little fibrillar amyloid was observed at 3 months of age in Tg-SwDI mice (Fig. 3A) but that an increase in amyloid was observed as the mice aged from 6 to 9 months (Fig. 3, B and C), mostly in the thalamus, hippocampus, and subiculum regions (Fig. 3, K–M). Although Tg-SwDI mice deposit appreciable parenchymal Aβ in the cortex (Fig. 2J), this is mostly in a diffuse form, whereas fibrillar amyloid is primarily restricted to small microvascular deposits (40–43). In contrast, both Tg-5xFAD mice (Fig. 3, D–F) and Tg-SwDI/Tg-5xFAD mice (Fig. 3, G–I) showed more widespread fibrillar amyloid deposition that was significantly higher in the bigenic animals (Fig. 3, J–M).
FIGURE 3.

Progressive accumulation of fibrillar amyloid deposition in Tg-SwDI, Tg-5xFAD, and bigenic Tg-SwDI/Tg-5xFAD mouse brains. Shown are representative images of 3- to 9-month-old transgenic mouse brain sections stained for fibrillar amyloid using thioflavin S (green) as described under “Experimental Procedures.” A–C, Tg-SwDI mice. D–F, Tg-5xFAD mice. G–I, bigenic Tg-SwDI/Tg-5xFAD mice. Scale bars = 1 mm. Regional fibrillar amyloid deposition in the cortex (J), thalamus (K), hippocampus (L), and subiculum (M) of 3-, 6-, and 9-month-old Tg-SwDI mice (black bars), Tg-5xFAD mice (blue bars), and bigenic Tg-SwDI/Tg-5xFAD mice (red bars) was determined by image analysis of thioflavin S staining. Data are mean ± S.D. of eight mice of each genotype at each age. a, p < 0.05; b, p < 0.01; c, p < 0.001; d, p < 0.0001; ns, not significant.
Subsequent examination of the brain tissues at higher magnification showed that, over the course of 3–9 months, the Tg-SwDI mice exhibited the typical progressive pattern of fibrillar amyloid accumulation, primarily in the form of small cerebral microvascular deposits (Fig. 4, A–C). On the other hand, Tg-5xFAD mice showed a progressive accumulation of parenchymal fibrillar amyloid plaques (Fig. 4, D–F). Interestingly, the bigenic Tg-SwDI/Tg-5xFAD mice (Fig. 4, G–I) exhibited striking differences compared with each single transgenic line. Remarkably, in the bigenic animals, there was essentially a complete loss of cerebral microvascular amyloid deposition, as characteristically observed in Tg-SwDI mice. Quantitative analysis showed progressive and high levels of cerebral microvascular amyloid in the subiculum region from 3- to 9-month-old Tg-SwDI mice that were essentially absent in the Tg-5xFAD mice and bigenic Tg-SwDI/Tg-5xFAD mice (Fig. 4J). The other notable feature was the tendency for larger fibrillar amyloid plaques in the bigenic mice compared with Tg-5xFAD mice. The size distribution of fibrillar amyloid deposits was measured in the subiculum region of the different lines of mice. As shown in Fig. 4, K–M, over the course of 9 months, nearly 90% of the fibrillar amyloid deposits in Tg-SwDI mice were <50 μm2, presenting primarily as small microvascular deposits (Fig. 4, A–C). In contrast, Tg-5xFAD mice presented a range of fibrillar amyloid deposits, in this case as parenchymal plaques with the majority between 50–250 μm2. However, in the bigenic Tg-SwDI/Tg-5xFAD mice, the size distribution of fibrillar plaques changed markedly, especially with age. For example, the relative amounts of small amyloid deposits of <50 μm2 decreased from ≈60% at 3 months to ≈35% at 9 months, whereas the levels in Tg-5xFAD mice remained fairly constant at ≈50% (Fig. 4, K–M). On the other hand, over 9 months in the bigenic mice, there was a 13-fold increase in the amount of very large fibrillar plaques of >500 μm2 compared with Tg-5xFAD mice (p < 0.0001). Together, these results indicate that, in bigenic Tg-SwDI/Tg-5xFAD mice, there is a complete loss of microvascular amyloid and growth of larger parenchymal amyloid plaques.
FIGURE 4.

Altered fibrillar amyloid deposition in bigenic Tg-SwDI/Tg-5xFAD mouse brains. Shown are representative images of 3- to 9-month-old transgenic mouse brain sections stained for fibrillar amyloid using thioflavin S (green) and immunolabeled for cerebral blood vessels using an antibody to collagen IV (red), as described under “Experimental Procedures.” A–C, Tg-SwDI mice. D–F, Tg-5xFAD mice. G–I, bigenic Tg-SwDI/Tg-5xFAD mice. Scale bars = 50 μm. The amount of cerebral microvascular amyloid (J) and the size distribution of all fibrillar amyloid deposits in 3- (K), 6- (L), and 9-month-old (M) Tg-SwDI mice (black bars), Tg-5xFAD mice (blue bars), and bigenic Tg-SwDI/Tg-5xFAD mice (red bars) was determined in the subiculum region using stereological principles, as described under “Experimental Procedures.” Data are the mean ± S.D. of eight mice per group. a, p < 0.05; b, p < 0.01; c, p < 0.001; d, p < 0.0001; ns, not significant.
Increased Parenchymal Fibrillar Amyloid Deposits in Bigenic Tg-SwDI/Tg-5xFAD Mice Enhances Neuronal Loss and Microglial Activation
It was has been reported previously that Tg-5xFAD mice exhibit neuronal loss, specifically in cortical layer V, with increasing age and Aβ pathology (44, 54). Therefore, we examined neuronal loss in the different transgenic lines with respect to fibrillar amyloid deposition and microglial activation. Tg-SwDI mice presented only small amounts of microvascular fibrillar amyloid, sparse microglial activation, and no neuronal loss in cortical layer V (Fig. 5, D–F, M, and N). In contrast, Tg-5xFAD mice had substantial parenchymal fibrillar amyloid plaques and higher accompanying microglial activation that coincided with a significant loss in cortical layer V neurons (Fig. 5, G–I, M, and N). Moreover, the bigenic Tg-SwDI/Tg-5xFAD mice had even higher amounts of parenchymal fibrillar amyloid plaques and increased microglial activation (Fig. 5, L and M). At 6 months of age, this resulted in a near doubling of the amount of neuronal loss in cortical layer V compared with Tg-5xFAD mice (Fig. 5, J, K, and N). However, as the mice aged to 9 months, the loss of cortical layer V neurons further increased in Tg-5xFAD mice and Tg-SwDI/Tg-5xFAD mice but was not significantly different between the two lines as the amyloid pathology and microglial activation rose to high levels in Tg-5xFAD mice.
FIGURE 5.

Increased neuronal loss and microglial activation in bigenic Tg-SwDI/Tg-5xFAD mouse brains. Shown are representative images from 6-month-old transgenic mouse brain sections immunolabeled for neurons using an antibody to NeuN, for activated microglia using mAb5D4, and stained for fibrillar amyloid using thioflavin S (ThS). A, D, G, and J, images of neuronal staining in cortical layers II–VI. Scale bars = 100 μm. B, E, H, and K, images of neuronal staining in cortical layer V. Scale bars = 50 μm. C, F, I, and L, images of thioflavin S staining for fibrillar amyloid (green) and immunolabeling for activated microglia (red). Scale bars = 50 μm. The numbers of activated microglia (M) and neurons (N) in cortical layer V in wild-type mice (gray bars), Tg-SwDI mice (black bars), Tg-5xFAD mice (blue bars), and bigenic Tg-SwDI/Tg-5xFAD mice (red bars) were determined using stereological principles as described under “Experimental Procedures.” Data are mean ± S.D. of five to eight mice per group. a, p < 0.05; b, p < 0.01; c, p < 0.001; d, p < 0.0001; ns, not significant.
Because the parenchymal fibrillar amyloid plaques tended to be larger and were associated with more extensive microglial activation and neuronal loss, we next sought to determine the distribution of non-mutated Aβ and CAA mutant Aβ in the plaques of bigenic Tg-SwDI/Tg5xFAD mice. To discriminate between human non-mutated Aβ and human Dutch/Iowa CAA mutant Aβ peptides in the plaques, we used the monoclonal antibody 4G8, which recognizes a midregion epitope on non-mutated human Aβ (55). The presence of the E22Q Dutch and D23N Iowa CAA mutations abolishes the 4G8 epitope that is normally present in non-mutated human Aβ. Fig. 6A shows a dot blot analysis confirming that the N-terminal human Aβ rabbit polyclonal antibody (pAb-Aβ) recognizes both non-mutated and Dutch/Iowa CAA mutant human Aβ, whereas mAb4G8 only recognizes non-mutated Aβ. Accordingly, as shown in Fig. 6, B–D, Aβ deposits in Tg-SwDI mice are recognized by pAb-Aβ but not mAb4G8 because of the presence of the Dutch E22Q and Iowa D23N mutations in this form of Aβ. On the other hand, parenchymal Aβ deposits in Tg-5xFAD mice are recognized by both pAb-Aβ and mAb4G8 because this Aβ is non-mutated (Fig. 6, E–G). Although the core of the plaques in bigenic Tg-SwDI/Tg-5xFAD mice was labeled with both pAb-Aβ and mAb4G8, the periphery of the plaques contained a halo of small Aβ deposits that were labeled only with pAb-Aβ, suggesting that these satellite plaques were composed primarily of Dutch/Iowa CAA mutant Aβ (Fig. 6J).
FIGURE 6.
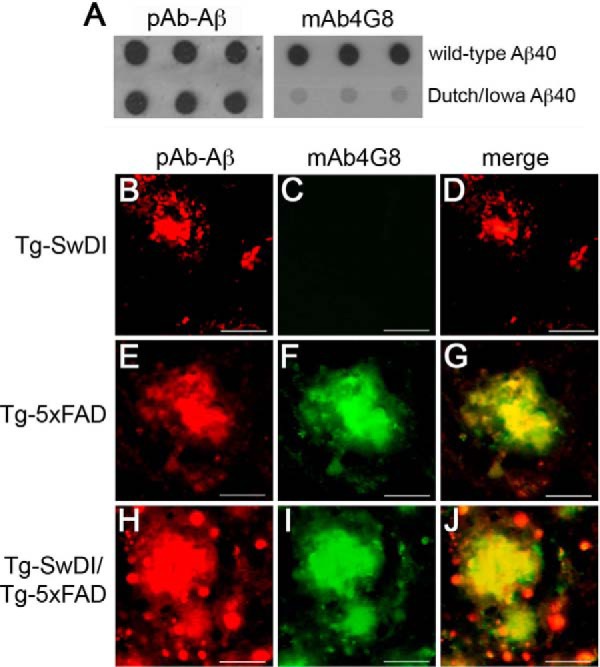
Analysis of non-mutated Aβ and Dutch/Iowa CAA mutant Aβ accumulation in Tg-SwDI, Tg-5xFAD, and bigenic Tg-SwDI/Tg-5xFAD mouse brains. A, a dot blot analysis was performed to demonstrate that rabbit polyclonal antibody to human Aβ (pAb-Aβ) recognizes both non-mutated Aβ and Dutch/Iowa CAA mutant Aβ, whereas mAb4G8 only recognizes non-mutated Aβ. Brain sections obtained from the different transgenic mouse lines at 9 months of age were immunolabeled with pAb-Aβ (red, B, E, and H), mAb4G8 (green, C, F, and I), and merged (D, G, and J). Dutch/Iowa CAA mutant Aβ deposits in Tg-SwDI mice are immunolabeled with pAb-Aβ (B) but not mAb4G8 (C). Non-mutated Aβ deposits were immunolabeled with both pAb-Aβ (E) and mAb4G8 (F). Aβ plaque core deposits in bigenic Tg-SwDI/Tg-5xFAD mice were immunolabeled with both pAb-Aβ (H) and mAb4G8 (I). The merged image shows a halo of Aβ deposits around the periphery of plaques that were not immunolabeled with mAb4G8, suggesting that they are composed of Dutch/Iowa CAA mutant Aβ (J). Scale bars = 50 μm.
Non-Mutated Aβ42 Fibrils Seed Dutch/Iowa CAA Mutant Aβ Fibril Formation and Deposition
The above findings suggest that early-onset parenchymal fibrillar plaques, primarily composed of non-mutated Aβ42, can act as a scaffold to recruit the codeposition of Dutch/Iowa CAA mutant Aβ in the bigenic Tg-SwDI/Tg-5xFAD mice. To directly test whether non-mutated Aβ42 fibrils could promote Dutch/Iowa CAA mutant Aβ40 assembly, we measured the kinetics of fibril formation of the latter in the presence or absence of non-mutated Aβ42 fibril seeds. Fibril seeds were generated by incubating Aβ42 at 37 °C to form mature fibrils, followed by bath sonication to break the fibrils into short segments. The addition of seeds to a population of non-mutated Aβ42 monomers eliminates the lag phase associated with the formation of a fibril nucleus (data not shown). As shown in Fig. 7, the Dutch/Iowa CAA mutant Aβ40, when incubated at a monomer concentration of 20 μm, exhibits a lag phase of 2 h prior to an increase of thioflavin T fluorescence, which is characteristic of fibril formation. The addition of monomeric Dutch/Iowa CAA mutant Aβ40 to non-mutated Aβ42 fibril seeds at concentrations of either 10 μm or 20 μm eliminated the lag phase and resulted in a rapid rise of thioflavin T fluorescence. In contrast, a scrambled, non-mutated Aβ42 peptide did not eliminate this lag phase (data not shown). The increase in fluorescence is attributed to the addition of the Dutch/Iowa CAA mutant Aβ40 to fibril seeds. The small transition in the fluorescence curves at ∼2 h (on top of the rapid initial rise in fluorescence) corresponds to non-seeded fibrillization of the Dutch/Iowa CAA mutant Aβ40 peptide. The non-mutated Aβ42 seeds without added CAA mutant Aβ40 monomer do not result in a change of fluorescence, as expected.
FIGURE 7.
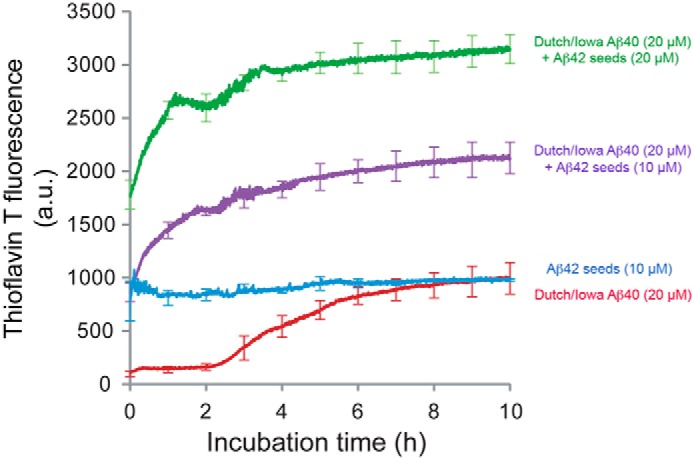
Non-mutated Aβ42 amyloid fibril seeds promote the assembly of Dutch/Iowa CAA mutant Aβ40 fibrils. Freshly prepared solutions of Dutch/Iowa CAA mutant Aβ40 (20 μm) were incubated in the absence (red trace) or presence of 10 μm (purple trace) or 20 μm (green trace) of non-mutated Aβ42 fibrillar seeds. The rate and extent of fibril formation was assessed by thioflavin T fluorescence measurements. The fluorescence curves of the non-mutated Aβ42 fibril seeds with added Dutch/Iowa CAA mutant Aβ40 exhibit two components. The rapid component is attributed to the growth of CAA mutant Aβ40 fibrils on the non-mutated Aβ42 seeds. The slower component is attributed to fibril formation of CAA mutant Aβ40 following nucleation of the monomeric peptide. Non-mutated Aβ42 fibril seeds in the absence of added monomeric CAA mutant Aβ peptide do not exhibit a change in fluorescence (blue trace). Data are mean ± S.D. of triplicate determinations.
We next tested this phenomenon in vivo by injecting biotin-labeled Dutch/Iowa CAA mutant Aβ40 into the hippocampal region of 6-month-old Tg-5xFAD mice, which exhibit prominent parenchymal plaque fibrillar amyloid deposition (Fig. 3H). The injected biotin-labeled Dutch/Iowa CAA mutant Aβ40 showed a strong accumulation on pre-existing parenchymal fibrillar amyloid plaques (Fig. 8, A–D). No accumulations were found when biotin alone was injected into similarly aged Tg-5xFAD mice (Fig. 8,E–H). Interestingly, a higher magnification of the parenchymal plaques (Fig. 8D) showed that biotin-labeled Dutch/Iowa CAA mutant Aβ accumulated around the periphery of the plaques, forming a halo of deposits highly reminiscent of its deposition in the bigenic Tg-SwDI/Tg-5xFAD mice (Fig. 6J). Together, these results indicate that non-mutated Aβ42 fibrils can seed rapid fibrillar assembly and codeposition of Dutch/Iowa CAA mutant Aβ onto parenchymal amyloid plaques.
FIGURE 8.

Non-mutated parenchymal amyloid deposits in Tg-5xFAD mice promote the accumulation of injected biotin-labeled Dutch/Iowa CAA mutant Aβ. Biotin-labeled Dutch/Iowa CAA mutant Aβ40 (A–D) and biotin (E–H) were injected into the hippocampal region of 6-month-old Tg-5xFAD mice. Brain sections were prepared, fibrillar amyloid was visualized by staining with thioflavin S (green), and biotin-labeled Dutch/Iowa CAA mutant Aβ40 or biotin alone was detected using Texas Red-labeled streptavidin (red). A–C and E–G, scale bars = 50 μm. D and H, scale bars = 12.5 μm.
DISCUSSION
The seeding, spreading, and deposition of aggregated proteins in the brain, first associated with prion disorders, is now recognized as a common pathological process of many neurodegenerative diseases, including AD, Parkinson disease, amyotrophic lateral sclerosis, and frontotemporal dementia (1, 2). In the case of AD, the accumulation of fibrillar Aβ occurs in two distinct compartments: in the brain parenchyma in the form of plaques and in cerebral blood vessels as vascular amyloid (5, 14, 15). Although the pathology at both sites involves Aβ deposition, the cross-talk between the parenchymal and cerebral vascular amyloid remains unclear. Here, we addressed this issue by crossing Tg-5xFAD mice, a model of early-onset parenchymal plaque amyloid, with Tg-SwDI mice, a model of cerebral microvascular amyloid, to determine the influence on the amyloid pathology in each compartment. We show that, in the bigenic Tg-SwDI/Tg-5xFAD mice, there is an overall loss of microvascular amyloid and a general shift to larger parenchymal amyloid plaques, accompanied by more severe pathological consequences.
In AD patients and mouse models of Aβ pathology, the emergence of CAA is generally a secondary event and exists in the presence of abundant parenchymal fibrillar amyloid plaques (3, 4). Previous studies using transgenic rodent models have shown that primarily parenchymal, and some cerebral vascular, Aβ pathology can be seeded and spread with exogenous intracerebral administration of amyloid material in a cis manner using fibrillar Aβ seeds (56–59). Alternatively, in certain cases, Aβ pathology could be seeded in a trans manner using distinct amyloid material, such as prion proteins (60).
In contrast to AD, in familial forms of CAA involving mutated forms of Aβ and in corresponding mouse models, the cerebral vascular amyloid is more extensive and occurs in the absence of appreciable parenchymal fibrillar amyloid deposition (29–36, 40, 41, 61). However, genetic analyses of Dutch and Iowa familial CAA patients indicate that they are heterozygous for their respective mutation and, therefore, possess one CAA mutant AβPP allele and one non-mutated AβPP allele (29, 30, 33, 62). Thus, this results in expression of both CAA mutant and non-mutated AβPP and subsequent production of a mixture of non-mutated and CAA mutant Aβ peptides in brain. The influence of early-onset parenchymal fibrillar amyloid plaques on subsequent cerebral vascular amyloid formation is unclear. To address this issue, we chose the Tg-5xFAD mouse model, which aggressively develops amyloid plaque pathology starting at about 2 months of age (28, 44). When crossed with Tg-SwDI mice, the bigenic Tg-SwDI/Tg-5xFAD mice accumulated higher amounts of Aβ and fibrillar amyloid than either single transgenic line. However, the compartmental distribution of fibrillar amyloid was altered significantly altered in the bigenic Tg-SwDI/Tg-5xFAD mice. Notably, there was a complete loss of cerebral microvascular amyloid and a general increase in the size distribution of parenchymal amyloid deposits (Fig. 4).
The composition of the enhanced amyloid plaques in bigenic Tg-SwDI/Tg-5xFAD mice is intriguing. The use of differential antibody recognition indicates that the core of the bigenic amyloid plaques is likely composed of non-mutated Aβ and, perhaps, CAA mutant human Aβ. However, the plaque periphery appears to be composed primarily of CAA mutant Aβ because it lacks recognition by the mAb4G8 antibody (Fig. 6). This finding suggests that, in the bigenic mice, parenchymal fibrillar amyloid plaques can act as a scaffold to capture Dutch/Iowa CAA mutant Aβ and promote its local assembly and deposition into hybrid plaques, thus precluding microvascular amyloid formation. This notion is supported by both our in vitro data demonstrating that non-mutated Aβ42 fibrillar seeds can increase the rate of Dutch/Iowa CAA mutant Aβ fibril assembly and in vivo results showing that exogenously administered Dutch/Iowa CAA mutant Aβ strongly codeposits on and adjacent to fibrillar amyloid plaques in Tg-5xFAD mice. The core/periphery composition of related, but distinct, Aβ peptides in the parenchymal amyloid plaques of the bigenic Tg-SwDI/Tg-5xFAD mice is highly similar to previous findings in mice where one strain of prion protein was shown to form an initial prion plaque core that could act as a scaffold for the aggregation and deposition of another strain of prion protein along the periphery to form hybrid plaques (63). The formation of these small, peripheral satellite plaque deposits of Dutch/Iowa CAA mutant Aβ may further cluster to grow the size of the initial plaque deposit, as shown recently in APP/PS1 mice (64).
In addition to their increase in size, the parenchymal amyloid plaque deposits in the bigenic Tg-SwDI/Tg-5xFAD mice appear to have enhanced pathological consequences. For example, there is a more robust activated microglial response around the amyloid plaques in the bigenic mice compared with the Tg-5xFAD mice, especially at the earlier ages (Fig. 5). We showed previously that the small cerebral microvascular amyloid deposits in Tg-SwDI mice, composed of Dutch/Iowa CAA mutant Aβ, promote a strong neuroinflammatory response and activation of microglia around the affected capillaries (40–43). Thus, the accumulation of Dutch/Iowa CAA mutant Aβ deposits on the periphery of the parenchymal plaques in the bigenic animals may, similarly, lead to enhanced microglial activation. In addition, the presence of Dutch/Iowa CAA mutant Aβ in parenchymal plaques correlates with a stronger loss of cortical neurons in the bigenic mice compared with Tg-5xFAD mice, especially at 6 months of age. This result is consistent with recent studies showing that a marked shift from microvascular amyloid to parenchymal fibrillar amyloid deposits of Dutch/Iowa CAA mutant Aβ in bigenic Tg-SwDI/hApoE mice led to cortical neuronal loss (65). Together, these findings suggest that shifting CAA mutant fibrillar amyloid from the cerebral vascular compartment to the parenchymal compartment in the form of plaques can enhance the surrounding pathology and neuronal loss.
In both Tg-5xFAD mice and Tg-SwDI mice, the source of non-mutated and CAA mutant human Aβ peptides is through neuronal expression and processing of transgene human AβPP (40, 44). A normal clearance route for neuronally derived Aβ peptides in transgenic mice, as well as in humans, is migration through the brain via interstitial fluid to the cerebral capillaries, where it is either transported across the blood-brain barrier into the blood or continues through perivascular drainage pathways into the cerebrospinal fluid (66–69). In Tg-5xFAD mice, the non-mutated Aβ can assemble and deposit as parenchymal amyloid plaques, whereas, in Tg-SwDI mice, the CAA mutant Aβ does not but, instead, migrates to the brain capillaries where, rather than be transported into the blood, it assembles and deposits as microvascular fibrillar amyloid.
It remains uncertain why the Dutch E22Q and Iowa D23N mutations in Aβ lead to preferential accumulation of fibrillar amyloid in the cerebral vessels whereas parenchymal fibrillar amyloid deposits are largely absent. There are, however, several properties of CAA mutant Aβ peptides that may contribute to their affinity for vascular assembly and deposition relative to the brain parenchyma. For example, both the Dutch E22Q and Iowa D23N mutations result in the loss of a negative charge at their respective adjacent sites. Earlier studies have demonstrated that these substitutions increase the fibrillogenic and cytotoxic properties of CAA mutant Aβ compared with non-mutated Aβ (37–39, 70–72). Furthermore, the presence of these substitutions in CAA mutant forms of Aβ may induce conformational changes in the monomeric peptide and/or oligomeric assemblies that suppress or enhance further assembly and deposition in the parenchyma or cerebral vasculature, respectively. Consistent with this idea, it has been shown previously that the GM3 ganglioside, which is present in cultured cerebrovascular cells and in cerebral vessels, preferentially enhances fibrillar assembly of CAA mutant Aβ (73, 74). Lastly, CAA mutant Aβ peptides show significantly impaired clearance from the brain via transport across the blood-brain barrier into the circulatory system and perivascular drainage to the cerebrospinal fluid (66, 75). This reduced clearance could allow for abnormal accumulation of CAA mutant Aβ at the cerebral vessels. These markedly altered properties of CAA mutant forms of Aβ likely provide for its preferential and excessive accumulation as fibrillar amyloid in the cerebral vasculature.
Familial CAA patients and the bigenic Tg-SwDI/Tg-5xFAD mice produce both non-mutated and CAA mutant Aβ in brain. However, in familial CAA patients, the fibrillar amyloid is primarily restricted to the cerebral vasculature, whereas, in the bigenic mice, parenchymal amyloid pathology increased and cerebral microvascular amyloid pathology essentially vanished. This difference may result from the relative amounts of non-mutated Aβ and CAA mutant Aβ in the human and bigenic mouse brain. Familial CAA patients generally harbor one CAA mutant and one non-mutated AβPP allele, thus yielding similar amounts of the corresponding Aβ peptide. One the other hand, Tg-SwDI mice express human AβPP and produce CAA mutant Aβ at physiological levels (40, 67), whereas Tg-5xFAD mice vastly overexpress human AβPP and produce very high levels of non-mutated Aβ (44), which is reflected in the disparate amounts of Aβ in the brains of each transgenic line (Fig. 1). Further, the appearance of fibrillar amyloid plaques is very aggressive in Tg-5xFAD mice, with an onset at about 2 months of age (44). Consequently, the very early appearance of parenchymal fibrillar amyloid plaques in the bigenic Tg-SwDI/Tg-5xFAD mice appears to be sufficient to serve as an effective scaffold to capture and deposit neuronally derived CAA mutant Aβ, preventing its normal migration to the cerebral microvasculature. In conclusion, although fibrillar amyloid can seed and spread subsequent Aβ pathologies, our findings indicate that, in certain situations, fibrillar amyloid can act as an efficient sink to enhance amyloid accumulation in the parenchymal compartment and forego amyloid deposition in the vascular compartment of the brain.
Acknowledgments
We thank Lilly Research Laboratories for the ELISA antibody reagents and Dr. Charles Glabe (University of California, Irvine) for the anti-Aβ oligomer antibody OC.
This work was supported, in whole or in part, by National Institutes of Health Grants AG033209 (to W. E. V. N.) and AG027317 (to S. O. S.). This work was also supported by Alzheimer's Association Grant IIRG-09-15254 and by a gift from the Cowles Charitable Trust.
- AD
- Alzheimer disease
- Aβ
- amyloid β
- AβPP
- amyloid β protein precursor
- CAA
- cerebral amyloid angiopathy
- pAb
- polyclonal antibody.
REFERENCES
- 1. Polymenidou M., Cleveland D. W. (2012) Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 209, 889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eisenberg D., Jucker M. (2012) The amyloid state of proteins in human disease. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 4. Tanzi R. E., Bertram L. (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555 [DOI] [PubMed] [Google Scholar]
- 5. Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Felice F. G., Vieira M. N., Saraiva L. M., Figueroa-Villar J. D., Garcia-Abreu J., Liu R., Chang L., Klein W. L., Ferreira S. T. (2004) Targeting the neurotoxic species in Alzheimer's disease: inhibitors of Aβ oligomerization. FASEB J. 18, 1366–1372 [DOI] [PubMed] [Google Scholar]
- 7. Goldgaber D., Lerman M. I., McBride O. W., Saffiotti U., Gajdusek D. C. (1987) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science 235, 877–880 [DOI] [PubMed] [Google Scholar]
- 8. Kang J., Lemaire H. G., Unterbeck A., Salbaum J. M., Masters C. L., Grzeschik K. H., Multhaup G., Beyreuther K., Müller-Hill B. (1987) The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736 [DOI] [PubMed] [Google Scholar]
- 9. Tanzi R. E., Gusella J. F., Watkins P. C., Bruns G. A., St George-Hyslop P., Van Keuren M. L., Patterson D., Pagan S., Kurnit D. M., Neve R. L. (1987) Amyloid β protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer's locus. Science 235, 880–884 [DOI] [PubMed] [Google Scholar]
- 10. Sinha S., Anderson J. P., Barbour R., Basi G. S., Caccavello R., Davis D., Doan M., Dovey H. F., Frigon N., Hong J., Jacobson-Croak K., Jewett N., Keim P., Knops J., Lieberburg I., Power M., Tan H., Tatsuno G., Tung J., Schenk D., Seubert P., Suomensaari S. M., Wang S., Walker D., Zhao J., McConlogue L., John V. (1999) Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 402, 537–540 [DOI] [PubMed] [Google Scholar]
- 11. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-secretase cleavage of the Alzheimer's precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 12. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 13. Wolfe M. S., Xia W., Ostaszewski B. L., Diehl T. S., Kimberly W. T., Selkoe D. J. (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517 [DOI] [PubMed] [Google Scholar]
- 14. Jellinger K. A. (2002) Alzheimer's disease and cerebrovascular pathology: an update. J. Neural Transm. 109, 813–836 [DOI] [PubMed] [Google Scholar]
- 15. Rensink A. A., de Waal R. M., Kremer B., Verbeek M. M. (2003) Pathogenesis of cerebral amyloid angiopathy. Brain Res. Brain Res. Rev. 43, 207–223 [DOI] [PubMed] [Google Scholar]
- 16. Thal D. R., Ghebremedhin E., Rüb U., Yamaguchi H., Del Tredici K., Braak H. (2002) Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293 [DOI] [PubMed] [Google Scholar]
- 17. Thal D. R., Griffin W. S., de Vos R. A., Ghebremedhin E. (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 115, 599–609 [DOI] [PubMed] [Google Scholar]
- 18. Attems J., Jellinger K., Thal D. R., Van Nostrand W. (2011) Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 37, 75–93 [DOI] [PubMed] [Google Scholar]
- 19. Kawai M., Kalaria R. N., Cras P., Siedlak S. L., Velasco M. E., Shelton E. R., Chan H. W., Greenberg B. D., Perry G. (1993) Degeneration of vascular muscle cells in cerebral amyloid angiopathy of Alzheimer disease. Brain Res. 623, 142–146 [DOI] [PubMed] [Google Scholar]
- 20. Rozemuller A. J., van Gool W. A., Eikelenboom P. (2005) The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer's disease: therapeutic implications. Curr. Drug Targets CNS Neurol. Disord. 4, 223–233 [DOI] [PubMed] [Google Scholar]
- 21. Allen N., Robinson A. C., Snowden J., Davidson Y. S., Mann D. M. (2014) Patterns of cerebral amyloid angiopathy define histopathological phenotypes in Alzheimer's disease. Neuropathol. Appl. Neurobiol. 40, 136–148 [DOI] [PubMed] [Google Scholar]
- 22. Oshima K., Uchikado H., Dickson D. W. (2008) Perivascular neuritic dystrophy associated with cerebral amyloid angiopathy in Alzheimer's disease. Int. J. Clin. Exp. Pathol. 1, 403–408 [PMC free article] [PubMed] [Google Scholar]
- 23. Richard E., Carrano A., Hoozemans J. J., van Horssen J., van Haastert E. S., Eurelings L. S., de Vries H. E., Thal D. R., Eikelenboom P., van Gool W. A., Rozemuller A. J. (2010) Characteristics of dyshoric capillary cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 69, 1158–1167 [DOI] [PubMed] [Google Scholar]
- 24. Thal D. R., Ghebremedhin E., Orantes M., Wiestler O. D. (2003) Vascular pathology in Alzheimer's disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J. Neuropath. Exp. Neurol. 62, 1287–1301 [DOI] [PubMed] [Google Scholar]
- 25. Harkness K. A., Coles A., Pohl U., Xuereb J. H., Baron J. C., Lennox G. G. (2004) Rapidly reversible dementia in cerebral amyloid inflammatory vasculopathy. Eur. J. Neurol. 11, 59–62 [DOI] [PubMed] [Google Scholar]
- 26. Attems J., Jellinger K. A. (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology: a pilot study. Acta Neuropathol. 107, 83–90 [DOI] [PubMed] [Google Scholar]
- 27. Bailey T. L., Rivara C. B., Rocher A. B., Hof P. R. (2004) The nature and effects of cortical microvascular pathology in aging and Alzheimer's disease. Neurol. Res. 26, 573–578 [DOI] [PubMed] [Google Scholar]
- 28. Xu W., Xu F., Anderson M. E., Kotarba A. E., Davis J., Robinson J. K., Van Nostrand W. E. (2014) Cerebral microvascular rather than parenchymal amyloid β-protein pathology promotes early cognitive impairment in transgenic mice. J. Alzheimers Dis. 38, 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levy E., Carman M. D., Fernandez-Madrid I. J., Power M. D., Lieberburg I., van Duinen S. G., Bots G. T., Luyendijk W., Frangione B. (1990) Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126 [DOI] [PubMed] [Google Scholar]
- 30. Van Broeckhoven C., Haan J., Bakker E., Hardy J. A., Van Hul W., Wehnert A., Vegter-Van der Vlis M., Roos R. A. (1990) Amyloid β protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 248, 1120–1122 [DOI] [PubMed] [Google Scholar]
- 31. Hendriks L., van Duijn C. M., Cras P., Cruts M., Van Hul W., van Harskamp F., Warren A., McInnis M. G., Antonarakis S. E., Martin J. J. (1992) Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β-amyloid precursor protein gene. Nat. Genet. 1, 218–221 [DOI] [PubMed] [Google Scholar]
- 32. Tagliavini F., Rossi G., Padovani A., Magoni M., Andora G., Sgarzi M., Bizzi A., Savioardo M., Carella F., Morbin M., Giaccone G., Bugiani O. (1999) A new βPP mutation related to hereditary cerebral haemorrhage. Alz. Rep. 2, S28 [Google Scholar]
- 33. Grabowski T. J., Cho H. S., Vonsattel J. P., Rebeck G. W., Greenberg S. M. (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705 [DOI] [PubMed] [Google Scholar]
- 34. van Duinen S. G., Castaño E. M., Prelli F., Bots G. T., Luyendijk W., Frangione B. (1987) Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 84, 5991–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rozemuller A. J., Roos R. A., Bots G. T., Kamphorst W., Eikelenboom P., Van Nostrand W. E. (1993) Distribution of β/A4 and amyloid precursor protein in hereditary cerebral hemorrhage with amyloidosis-Dutch type and Alzheimer's disease. Am. J. Pathol. 142, 1449–1457 [PMC free article] [PubMed] [Google Scholar]
- 36. Wattendorff A. R., Frangione B., Luyendijk W., Bots G. T. A. M. (1995) Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): clinicopathological studies. J. Neurol. Neurosurg. Psychiatry 59, 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Melchor J. P., McVoy L., Van Nostrand W. E. (2000) Charge alterations of E22 in the amyloid β-protein enhance its pathogenic properties. J. Neurochem. 74, 2209–2212 [DOI] [PubMed] [Google Scholar]
- 38. Miravalle L., Tokuda T., Chiarle R., Giaccone G., Bugiani O., Tagliavini F., Frangione B., Ghiso J. (2000) Substitutions at codon 22 of Alzheimer's Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 275, 27110–27116 [DOI] [PubMed] [Google Scholar]
- 39. Van Nostrand W. E., Melchor J. P., Cho H. S., Greenberg S. M., Rebeck G. W. (2001) Pathogenic effects of D23N “Iowa” amyloid β-protein. J. Biol. Chem. 276, 32860–32866 [DOI] [PubMed] [Google Scholar]
- 40. Davis J., Xu F., Deane R., Romanov G., Previti M. L., Zeigler K., Zlokovic B. V., Van Nostrand W. E. (2004) Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J. Biol. Chem. 279, 20296–20306 [DOI] [PubMed] [Google Scholar]
- 41. Miao J., Xu F., Davis J., Otte-Höller I., Verbeek M. M., Van Nostrand W. E. (2005) Cerebral microvascular amyloid β-protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β-protein precursor. Am. J. Pathol. 167, 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fan R., Xu F., Previti M. L., Davis J., Grande A. M., Robinson J. K., Van Nostrand W. E. (2007) Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J. Neurosci. 27, 3057–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu F., Grande A. M., Robinson J. K., Previti M. L., Vasek M., Davis J., Van Nostrand W. E. (2007) Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid β-protein precursor transgenic mice. Neuroscience 146, 98–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R. (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnson-Wood K., Lee M., Motter R., Hu K., Gordon G., Barbour R., Khan K., Gordon M., Tan H., Games D., Lieberburg I., Schenk D., Seubert P., McConlogue L. (1997) Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 94, 1550–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. DeMattos R. B., O'dell M. A., Parsadanian M., Taylor J. W., Harmony J. A., Bales K. R., Paul S. M., Aronow B. J., Holtzman D. M. (2002) Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10843–10848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang T., Hong S., O'Malley T., Sperling R. A., Walsh D. M., Selkoe D. J. (2013) New ELISAs with high specificity for soluble oligomers of amyloid β-protein detect natural Aβ oligomers in human brain but not in CSF. Alzheimers Dement. 9, 99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davis-Salinas J., Saporito-Irwin S. M., Donovan F. M., Cunningham D. D., Van Nostrand W. E. (1994) Thrombin receptor activation induces secretion and nonamyloidogenic processing of amyloid β-protein precursor. J. Biol. Chem. 269, 22623–22627 [PubMed] [Google Scholar]
- 49. Long J. M., Kalehua A. N., Muth N. J., Calhoun M. E., Jucker M., Hengemihle J. M., Ingram D. K., Mouton P. R. (1998) Stereological estimation of total microglia number in mouse hippocampus. J. Neurosci. Meth. 84, 101–108 [DOI] [PubMed] [Google Scholar]
- 50. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 [DOI] [PubMed] [Google Scholar]
- 51. Glabe C. G. (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575 [DOI] [PubMed] [Google Scholar]
- 52. Ono K., Condron M. M., Teplow D. B. (2009) Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jawhar S., Trawicka A., Jenneckens C., Bayer T. A., Wirths O. (2012) Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol. Aging 33, 196.e29–196.e40 [DOI] [PubMed] [Google Scholar]
- 55. Kim K. S., Miller D. L., Sapienza V. J., Chen C.-M. J., Bai C., Gundke-Iqbal I., Currie J. R., Wisniewski H. M. (1988) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci. Res. Commun. 2, 121–130 [Google Scholar]
- 56. Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A. L., Vigouret J. M., Paganetti P., Walsh D. M., Mathews P. M., Ghiso J., Staufenbiel M., Walker L. C., Jucker M. (2006) Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 [DOI] [PubMed] [Google Scholar]
- 57. Langer F., Eisele Y. S., Fritschi S. K., Staufenbiel M., Walker L. C., Jucker M. (2011) Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J. Neurosci. 31, 14488–14495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rosen R. F., Fritz J. J., Dooyema J., Cintron A. F., Hamaguchi T., Lah J. J., LeVine H., 2nd, Jucker M., Walker L. C. (2012) Exogenous seeding of cerebral β-amyloid deposition in βAPP-transgenic rats. J. Neurochem. 120, 660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hamaguchi T., Eisele Y. S., Varvel N. H., Lamb B. T., Walker L. C., Jucker M. (2012) The presence of Aβ seeds, and not age per se, is critical to the initiation of Aβ deposition in brain. Acta Neuropathol. 123, 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morales R., Estrada L. D., Diaz-Espinoza R., Morales-Scheihing D., Jara M. C., Castilla J., Soto C. (2010) Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J. Neurosci. 30, 4528–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Herzig M. C., Eisele Y. S., Staufenbiel M., Jucker M. (2009) E22Q mutant Aβ peptide (AβDutch) increases vascular but reduces parenchymal Aβ deposition. Am. J. Pathol. 172, 722–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bakker E., van Broeckhoven C., Haan J., Voorhoeve E., van Hul W., Levy E., Lieberburg I., Carman M. D., van Ommen G. J. (1991) DNA diagnosis for hereditary cerebral hemorrhage with amyloidosis (Dutch-type). Am. J. Hum. Genet. 49, 518–521 [PMC free article] [PubMed] [Google Scholar]
- 63. Nilsson K. P., Joshi-Barr S., Winson O., Sigurdson C. J. (2010) Prion strain interactions are highly selective. J. Neurosci. 30, 12094–12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McCarter J. F., Liebscher S., Bachhuber T., Abou-Ajram C., Hübener M., Hyman B. T., Haass C., Meyer-Luehmann M. (2013) Clustering of plaques contributes to plaque growth in a mouse model of Alzheimer's disease. Acta Neuropathol. 126, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu F., Vitek M. P., Colton C. A., Previti M. L., Davis J., Van Nostrand W. E. (2012) Human apolipoprotein E2 promotes parenchymal amyloid deposition and neuronal loss in vasculotropic mutant amyloid β-protein Tg-SwDI mice. J. Alzheimers Dis. 31, 359–369 [DOI] [PubMed] [Google Scholar]
- 66. Zlokovic B. V. (2004) Clearing amyloid through the blood-brain barrier. J. Neurochem. 89, 807–811 [DOI] [PubMed] [Google Scholar]
- 67. Deane R., Wu Z., Sagare A., Davis J., Du Yan S., Hamm K., Xu F., Parisi M., LaRue B., Hu H. W., Spijkers P., Guo H., Song X., Lenting P. J., Van Nostrand W. E., Zlokovic B. V. (2004) LRP-amyloid β-peptide (Aβ) interaction regulates differential brain efflux of Aβ isoforms. Neuron 43, 333–344 [DOI] [PubMed] [Google Scholar]
- 68. Weller R. O., Subash M., Preston S. D., Mazanti I., Carare R. O. (2008) Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 18, 253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weller R. O., Djuanda E., Yow H. Y., Carare R. O. (2009) Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 117, 1–14 [DOI] [PubMed] [Google Scholar]
- 70. Wisniewski T., Ghiso J., Frangione B. (1991) Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem. Biophys. Res. Comm. 179, 1247–1254 [DOI] [PubMed] [Google Scholar]
- 71. Davis J., Van Nostrand W. E. (1996) Enhanced pathologic properties of Dutch-type mutant amyloid β-protein. Proc. Natl. Acad. Sci. U.S.A. 93, 2996–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Verbeek M. M., de Waal R. M., Schipper J. J., Van Nostrand W. E. (1997) Rapid degeneration of cultured human brain pericytes by amyloid β protein. J. Neurochem. 68, 1135–1141 [DOI] [PubMed] [Google Scholar]
- 73. Yamamoto N., Hirabayashi Y., Amari M., Yamaguchi H., Romanov G., Van Nostrand W. E., Yanagisawa K. (2005) Assembly of hereditary amyloid β-protein variants in the presence of favorable gangliosides. FEBS Lett. 579, 2185–2190 [DOI] [PubMed] [Google Scholar]
- 74. Yamamoto N., Van Nostrand W. E., Yanagisawa K. (2006) Further evidence of local ganglioside-dependent Aβ assembly in brain. Neuroreport 17, 1735–1737 [DOI] [PubMed] [Google Scholar]
- 75. Monro O. R., Mackic J. B., Yamada S., Segal M. B., Ghiso J., Maurer C., Calero M., Frangione B., Zlokovic B. V. (2002) Substitution at codon 22 reduces clearance of Alzheimer's amyloid-β peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol. Aging 23, 405–412 [DOI] [PubMed] [Google Scholar]